

1 Introduction
Clostridium difficile, a strict anaerobic human pathogenic bacterium, can thrive from fermenting 3 mol l-leucine to 1 mol isovalerate (3-methylbutyrate) and 2 mol isocaproate (4-methylpentanoate) via an oxidative and a reductive pathway branch, respectively [1,2]. The dehydration of 2-hydroxyisocaproyl-CoA to 2-isocaprenoyl-CoA found in the reductive branch is the chemically most difficult step, due to the elimination of the non-acidic β-H (pK ca. 40). We have shown that this reaction involves radical intermediates, which therefore classifies the dehydratase as a radical enzyme as defined previously [3]. In the proposed mechanism (Fig. 1), a one-electron transfer from the activated enzyme to 2-hydroxyisocaproyl-CoA generates the substrate-derived ketyl radical anion I, which as a nucleophile removes the α-hydroxyl group and yields an enoxy radical. The next step, the deprotonation in the β-position, is now possible because enoxy radical formation lowers the pK by about 26 units [4,5]. This leads to the resonance stabilised product-related ketyl radical anion II, which is oxidised to the product whereby the electron is recycled for at least 10,000 turnovers. The electron stems from a one [4Fe–4S]1+/2+ cluster-containing homodimeric activator (HadI), which transfers it to the iron–sulfur cluster of 2-hydroxyisocaproyl-CoA dehydratase (HadBC) in an ATP-dependent manner. The genes coding for both enzymes have been expressed in Escherichia coli and the produced enzymes have been purified and characterised [6]. The crystal structure of the closely related activator from Acidaminococcus fermentans (60% sequence identity) showed the [4Fe–4S] cluster facing the N-termini of two α-helices, each from one subunit, and thus bridging the subunits at the interface with an helix–cluster–helix angle of 105° [7]. Each subunit contains an ATP/ADP binding site with the ASKHA motif [8] at which ADP was detected in the crystal structure. The activator is functionally very similar to the phylogenetically unrelated nitrogenase Fe-protein, which is a member of the G-protein family. Both proteins transfer an electron from the [4Fe–4S] cluster to their main enzyme coupled with ATP hydrolysis [9]. In both cases the ATP/ADP binding sites are ∼20 Å apart from the [4Fe–4S] clusters, which makes it difficult to assess how ATP hydrolysis is coupled to electron transfer. It was shown that MgATP binding at the Fe-protein of nitrogenase caused a conformational change by exposing the metal cluster towards the main enzyme MoFe-protein to induce uni-directional electron transfer [10]. Considering the functional similarity of both proteins, it has been proposed that MgATP binding to the activator opens the helix–cluster–helix angle from 105° to 180° and thereby exposes the [4Fe–4S] cluster for the electron transfer to the dehydratase [11]. The so-called switch II region, positioned between the ATP/ADP site and the [4Fe–4S] cluster, was assumed to interact with the γ-phosphate of the bound nucleotide and to change its conformation upon ATP binding [7]. Another interesting feature of the activator is the relatively high redox potential (E0′ ≥ −350 mV versus the normal hydrogen electrode) because it is almost completely reduced by ferredoxin (E0′ = −450 mV), dithionite or Ti(III)citrate [11]. As the reduction of the dehydratase probably requires the very low redox potential of ca. −900 mV, it has been assumed that ATP hydrolysis should overcome the redox barrier of ca. 550 mV between the two enzymes, which is equivalent to 106 kJ mol−1 or 2 ATP under physiological conditions [12].

Proposed mechanism for the dehydration of 2-hydroxyisocaproyl-CoA to 2-isocaprenoyl-CoA. The activator (HadI) transfers an electron to the 2-hydroxyisocaproyl-CoA dehydratase (HadBC) with the coupled ATP hydrolysis. The electron reduces the substrate (R)-2-hydroxyisocaproyl-CoA forming the substrate-derived ketyl radical anion I. The following removal of the α-hydroxyl group yields the enoxy radical and the β-proton can be removed forming the product-related ketyl radical anion II. Return of the electron to the dehydratase yields the product (E)-2-isocaprenoyl-CoA and the enzyme is ready for the next turnover.
Similarly to the case of the nitrogenase Fe-protein [13] we analysed the conformational changes of the activator by chelation of the iron–sulfur cluster with bathophenanthroline disulfonate (BPT). We found that reduction of the activator as well as addition of MgATP induced large conformational changes. Upon interaction of the reduced activator with the dehydratase, ATP is hydrolysed faster than with the oxidised form.
2 Materials and methods
All experiments were performed under anoxic conditions in the Coy Anaerobic Chamber using an atmosphere of 95% N2 and 5% H2 at 20 °C [6]. Recombinant Strep-tagged activator and recombinant 2-hydroxyisocaproyl-CoA dehydratase were obtained from E. coli by heterologous gene over-expression and purified [6]. The activator (10 μM in 50 mM Mops pH 7.0, 1 mM ADP and 1 mM MgCl2) was reduced by incubation with 100 μM dithionite for 10 min at 20 °C.
Fe(II) chelation was measured in 100 mM Tris/HCl pH 8.0, 10 mM MgCl2, 10 mM dithiothreitol, 2 mM bathophenanthroline disulfonate (BPT), activator containing ADP and ATP, when indicated. The formation of the Fe(II)–BPT complex was followed by the absorbance increase at 535 nm (ɛ = 22.1 mM−1 cm−1) [13]. Protein concentration was determined with the Bio-Rad Protein Assay. Bovine serum albumin was used as standard [14].
ATPase activity was measured in a cuvette (d = 1 cm), which contained in a total volume of 0.5 ml 50 mM Mops pH 7.0, 5 mM dithiothreitol, 5 mM MgCl2, 1 mM phosphoenolpyruvate, 0.2 mM NADH, and 2 U each of pyruvate kinase and lactate dehydrogenase [6]. The reaction was started by addition of the activator following the absorbance decrease of NADH at 340 nm (ɛ = 6.3 mM−1 cm−1) [15]. One unit (U) of ATPase activity is defined as 1 μmol NADH oxidised per minute. In order to determine the ATPase activity of the reduced activator, 1 mM Ti(III)citrate [16] was added. Whereas dithionite reduced NAD+ non-enzymatically, in the presence of Ti(III)citrate the coenzyme remained in the oxidised state, but only at pH ≤ 7.0 (E. Jayamani and W. Buckel, unpublished observation).
3 Results
Bathophenanthroline disulfonate (BPT) has been used as iron(II) chelation reagent forming the Fe(II)–BPT complex with a maximum absorbance at 535 nm. In the presence of oxidised activator as purified and stabilised with 0.1–0.2 mM ADP and 1 mM MgCl2 an initial chelation rate, v = 0.34 ± 0.05 mol Fe min−1 mol−1 activator (0.34 ± 0.05 min−1) was observed. Addition of 200 μM ATP decreased the initial chelation rate to a constant value, v = 0.132 ± 0.005 min−1 (data not shown). After about 2 h the chelation of the [4Fe–4S]2+ cluster was finished, as indicated by the complete release of 4.0 ± 0.4 mol Fe from 1 mol activator, regardless whether ADP or ATP was present. No increase of the Fe(II)–BPT complex formation could be observed by heating the reaction mixture at 95 °C for 10 min and removing insoluble precipitants by centrifugation. Since a [4Fe–4S]2+ cluster contains 2 Fe(III) and 2 Fe(II), one would expect to detect with BPT only the ferrous part. Probably the dithiothreitol present in the incubations (10 mM) and the concomitant released sulfides reduced the two ferric ions during destruction of the cluster.
In contrast, only ATP initiated Fe release from the reduced activator (v = 0.128 ± 0.004 min−1 at 200 μM ATP), whereas ADP had almost no effect (v < 0.001 min−1, Fig. 2). Addition of an equivalent amount of 2-hydroxyisocaproyl-CoA dehydratase to the activator (1.0 mol/mol) increased the initial chelation rate by 23% (v = 0.13–0.16 min−1). The chelation rate of the reduced activator was determined as a function of the ATP concentration. We expected that the plots of the rates (v) versus ATP concentrations would be either sigmoid following the rate law v = Vmax {[ATP]/(Km + [ATP])}2 [13] or parabolic v = Vmax [ATP]2/(Km + [ATP ]2) [17], as observed for nitrogenase Fe-protein, considering that the activator contains two identical ATP binding sites. However, simple Michaelis–Menten kinetics were detected, v = Vmax [ATP]/(Km + [ATP]) (Fig. 3A), which could be linearized by plotting v−1 versus [ATP]−1, yielding Km = 21 ± 1 μM (Fig. 3B). It seems that already the binding of 1.0 mol ATP to 1.0 mol activator suffices to change the conformation giving BPT access to the iron–sulfur cluster, although the homodimeric activator contains two ATP binding sites.
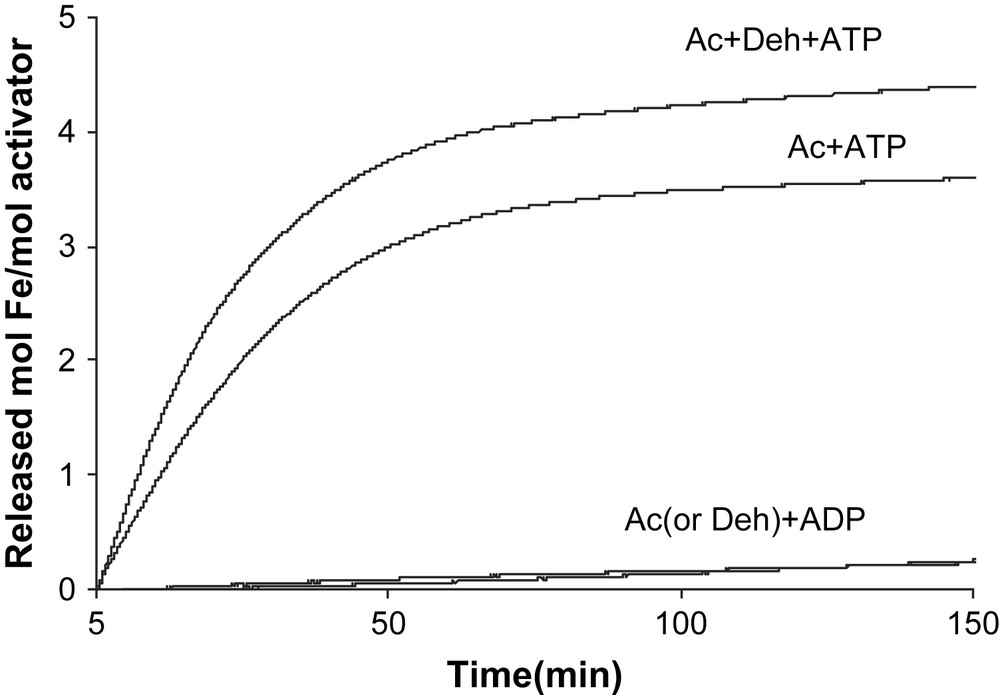
Chelation of the [4Fe–4S]1+ cluster of the reduced activator. The reactions (total volume 1.0 ml) contained 7.8 μM reduced activator (Ac), 100 mM Tris/HCl pH 8.0, 10 mM MgCl2, 5 mM dithiothreitol, 1 mM dithionite and 2 mM BPT. Addition of 10 mM ATP or ADP started the reaction. In the upper curve, the assay was supplemented with 7.8 μM 2-hydroxyisocaproyl-CoA dehydratase (Deh).
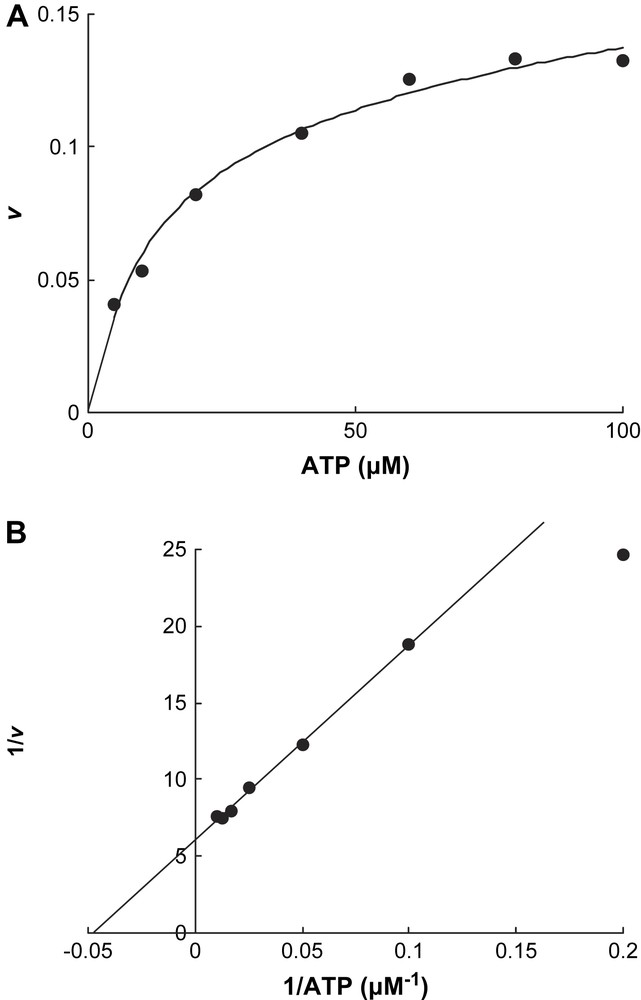
Chelation rate at various ATP concentrations. Each assay contained 4.3 μM reduced activator in 0.5 ml of the same incubation as in Fig. 2 and was started by the addition of ATP. The reaction rate v is given as mol Fe released min−1 mol−1 activator. A, initial slope as a function of [ATP]; B, Lineweaver–Burk plot of A.
The oxidised or the reduced activator alone exhibited very low ATPase activity (0.04 U mg−1). Addition of an excess of dehydratase to the oxidised ‘as purified’ activator (activator/dehydratase = 1:150 mol/mol) increased the activity about 12-fold (0.5 U mg−1 activator), whereas the dehydratase alone was devoid of ATPase activity. The large excess of dehydratase was necessary to get measurable rates. As expected, the highest ATPase activity could be observed with the reduced activator in the presence of dehydratase (2.2 U mg−1 activator). The dependence of the ATPase activity on the concentration of ATP measured with the reduced activator in the presence of Ti(III)citrate approximately obeyed Michaelis–Menten kinetics, Km = 0.5 ± 0.1 μM. Due to the extremely low ATP concentrations, a more accurate value could not be obtained.
4 Conclusions
The crystal structure of the oxidised ADP-containing activator from A. fermentans revealed two sides of the [4Fe–4S] cluster, both of which could be accessible from the solvent. On the ‘top side’, the cluster is located between the two hydrophobic helices, whereas at the ‘bottom side’ predominantly hydrophilic amino acids are located [7]. We conclude therefore that in the presence of ADP the iron chelator removes the metal ion from the bottom (Fig. 4). Reduction of the cluster causes a decrease of the chelation rate to almost zero, indicating that the bottom side has been closed. Addition of MgATP most likely opens the angle between the two cluster helices from 105° to 180°, which gives the chelator access to the cluster from the top side. This hypothesis was deduced from the structure of the complex between nitrogenase and its iron protein stabilised by ADP–AlF4− [10]. It should be mentioned that a similar complex has been obtained between activator and dehydratase, but its crystal structure remains to be established (J. Kim and W. Buckel, unpublished). An alternative explanation that ATP reduces the stability of [4Fe–4S]1+ cluster in the reduced activator by a mode different from a conformational change of the protein is not corroborated by the EPR spectra of the closely related activator from Clostridium sporogenes (58% sequence identity) in the presence of ATP or ADP. Under both conditions the spin state of the cluster was mainly S = 3/2 (93%) and only 7% S = 1/2 [18]. Any change in the structure or stability of the cluster would have affected the spin state. Addition of ATP to the oxidised activator diminishes the chelation rate by 61%, almost equal to that of the reduced activator in the presence of ATP. This could be due to the opening of the helix–cluster–helix angle, whereby the bottom side becomes closed. In summary, ATP and/or reduction of the cluster close the most readily accessible bottom side, whereas only ATP opens the top side, in order to enable the electron transfer from the cluster of the activator to that of the dehydratase. As soon as the complex between reduced activator and dehydratase has been formed, ATP is hydrolysed.
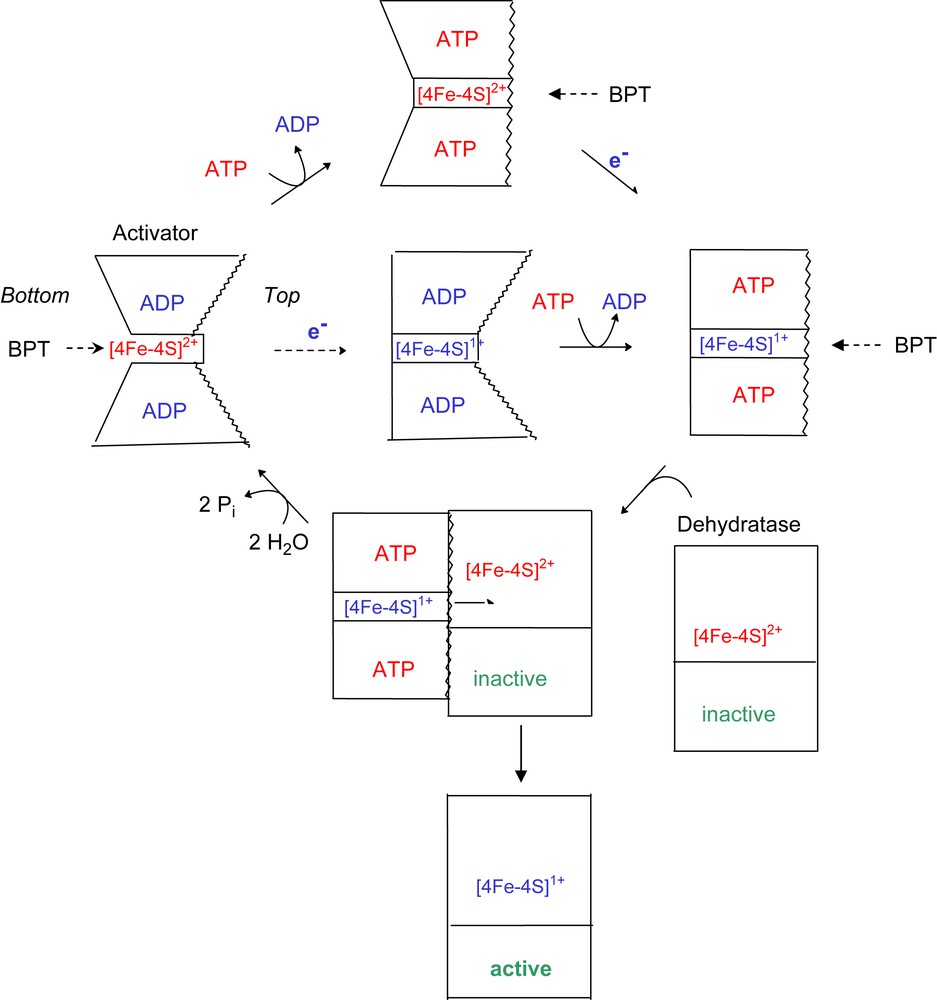
Proposed scheme for the conformational change of the activator depending on the redox state and bound nucleotides. In the oxidised and ADP-bound activator, BPT can reach the [4Fe–4S] cluster from the bottom. Reduction of the [4Fe–4S]2+ cluster (red colour) to [4Fe–4S]+ (blue) closes the bottom and ATP opens the top by increasing the helix–cluster–helix angle from 105° to 180° (central pathway; the helices are indicated by wavy lines). In this state, the dehydratase can bind and the electron is transferred to its cluster concomitant with ATP hydrolysis. The oxidised ADP-bound activator dissociates from the active dehydratase, which now is ready to catalyse about 10,000 turnovers (lower pathway). The upper pathway shows that ATP induces the conformational change also in the oxidised activator (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
It should be noted that these experiments apparently contradict earlier observations with the activator from A. fermentans with respect to chelation rate and ATPase activity [11]. At this time the activator was stabilised with 1 mM ATP, which had partially hydrolysed, but the actual concentrations were not analysed. Since the apparent Kms for chelation and hydrolysis are very low, 22 and 0.5 μM, respectively, any residual ATP could have interfered the measurements. Furthermore, in the experiments described here always fresh activator from C. difficile was used, prepared on the same or previous day.
Inside an actively growing anaerobic cell the ATP concentration of 2–5 mM is about 100 × Km of the ATP-induced chelation of the reduced activator and the ratio of oxidised and reduced ferredoxin probably equals about 1.0 (E′ = −420 mV). Thus the activator is always loaded with ATP and – due to its relatively high redox potential (E0′ ≥ −350 mV), vide supra – always in the reduced form ready to transfer one electron to the dehydratase.
Acknowledgements
This work was funded by grants from the Deutsche Forschungsgemeinschaft (DFG) and the Fonds der Chemischen Industrie. We thank Mohamed Baani (Max-Planck-Institut für terrestrische Mikrobiologie, Marburg, Germany) for translation of the title and the abstract into French.