

1 Introduction
Les biopolymères sont des macromolécules synthétisées par des organismes vivants. Ils sont composés de glucides lorsqu'il s'agit de polysaccharides, d'acides aminés dans le cas des protéines ou encore d'alkanoates dans le cas des biopolymères d'origine bactérienne. Une autre famille de biopolymères regroupe des molécules issues de la polymérisation de monomères naturels renouvelables, tels que l'acide lactique ou des triglycérides. Chez les polysaccharides, on distingue deux catégories : les homopolysaccharides, constitués du même ose comme le glucose dans la cellulose ou l'amidon, et les hétéropolysaccharides, formés de différents oses (pectines, hémicelluloses). Ces macromolécules sont les éléments structuraux majeurs des parois cellulaires des végétaux (cellulose, pectines, hémicelluloses), contribuant aux propriétés mécaniques des organes et aux mécanismes de défense contre des agressions biotiques et abiotiques [1]. Les polysaccharides comme l'amidon ont également un rôle essentiel dans le stockage de l'énergie [2]. Ces biopolymères se trouvent généralement sous la forme de composites complexes formés via des interactions avec différents types de macromolécules (différentes structures de polysaccharides, de protéines ou de polyphénols). La caractérisation structurale de ces macromolécules isolées et l'identification de leurs interactions au sein de ces systèmes végétaux complexes sont essentielles pour une meilleure compréhension de leurs fonctions au sein des plantes. Elles sont également nécessaires pour une meilleure maîtrise de leurs propriétés (propriétés mécaniques ou de texture, susceptibilité à des enzymes par exemple) dans l'objectif de contrôler et d'améliorer leurs usages industriels.
Notre approche consiste à utiliser différentes techniques de RMN afin de caractériser, à différentes échelles, les biopolymères isolés ou sous forme de composites dans les organes végétaux. La RMN haut champ est d'un apport précieux pour étudier la structure fine, le polymorphisme ou encore la cristallinité de ces macromolécules isolées. La description de la topologie d'assemblages complexes comme les parois végétales aux échelles microscopiques (quelques nanomètres) et macroscopiques (quelques microns) est également possible à travers, respectivement, des mesures de relaxométrie des protons de l'eau à bas champ et de coefficients d'auto-diffusion de sondes moléculaires de tailles variées sous l'influence de gradients de champ. Quelques exemples d'application sont décrits ici, concernant notamment la structure fine de l'amidon ou de la cellulose, la composition des parois végétales en cours de maturation d'un fruit et finalement, à un niveau supramoléculaire, la porosité des parois végétales, démontrant la complémentarité des différentes techniques RMN pour la description structurale des biopolymères aux différentes échelles.
2 Matériels et méthodes
2.1 Préparation des résidus de parois insolubles à l'alcool
Les fruits de tomate (Lycopersicon esculentum Mill. Cv. Tradiro), aux stades de maturation vert (35 fruits) et rouge mature (38 fruits), nous ont été fournis par le Centre technique interprofessionnel des fruits et légumes (CTIFL-Carquefou, France). Les tissus du péricarpe des fruits verts et rouges ont été isolés et mis en suspension pendant 20 min dans un volume d'éthanol à 95° correspondant à une concentration finale de 70%. Les résidus ont ensuite été lavés par de l'éthanol à 70%, jusqu'à ce que les lavages ne contiennent plus aucun ose détectable par colorimétrie [3]. Ces matériels insolubles à l'alcool (MIA) ont ensuite été déshydratés par des lavages dans de l'acétone et de l'éthanol 95°, puis séchés sous vide à 40 °C en présence de P2O5. Les MIA secs ont été finalement tamisés à travers un tamis de 2 mm. Le polyéthylène glycol (PEG), d'un poids moléculaire moyen de 6000 g mol−1 (Merck).
2.2 Préparation de l'amylose enrichie en 13C
L'amylose enrichie en 13C a été préparée à partir d'amylosaccharase et de saccharose enrichie en 13C selon la méthode décrite par Rondeau-Mouro et al. [4].
2.3 Spectroscopie par RMN
Les expériences de RMN du solide ont été effectuées sur un spectromètre Bruker DMX-400 opérant à la fréquence du 13C à 100,62 MHz et équipé d'une sonde 4 mm CPMAS ou HRMAS double résonance H/X, selon l'expérience. La vitesse de rotation a été fixée à 7 kHz pour les résidus de parois et pour la tunicine et à 10 kHz pour l'amylose enrichie en 13C. Chaque expérience à été réalisée à température ambiante (294 ± 1 K). La séquence CPMAS (Fig. 1) [5] a été utilisée avec une impulsion 90° proton de 4 μs, un temps de contact de 1 ms à 62,5 kHz et un temps de recyclage de 7 s pour un temps d'acquisition de 52 ms, pendant lequel un découplage dipolaire (TPPM [6]) a été appliqué. Les déplacements chimiques ont été référencés par rapport à la glycine, en attribuant le signal de la fonction carbonyle à 176,03 ppm.

Schéma de la séquence CPMAS (Cross-Polarisation Magic-Angle Spinning).
Le logiciel utilisé pour décomposer les spectres de la tunicine est basé sur la méthode d'ajustement Simplex (dmfit2003, Massiot et al, 1994), suivant la stratégie décrite par Rondeau-Mouro et al. [7].
Les spectres de détection directe des carbones des résidus de tomate ont été enregistrés avec une impulsion 90° comprise entre 5 and 6 μs et un découplage proton de 3 kHz (WALTZ16) pendant l'acquisition (1,6 s), mais également pendant le temps de recyclage (2 s), afin d'augmenter l'intensité des signaux carbone par les effets NOE (Nuclear Overhauser Enhancement) [8]. Les spectres protons ont été enregistrés en utilisant une impulsion 90° de 4,5 μs avec une pré-saturation du pic de l'eau. L'acquisition des spectres 2D-TOCSY a été effectuée sur une largeur spectrale de 4006 Hz dans les deux dimensions, avec 2 K points, 128 scans et 512 incréments.
La séquence DARR [9] a été utilisée avec un découplage dipolaire hétéronucléaire de 10 kHz pendant le temps de mélange τ (voir séquence de la Fig. 2). L'expérience DARR a été appliquée entre chaque carbone glucosidique de l'amylose, excepté pour le carbone C4, qui ne présentait pas de résonance isolée sur le spectre 1D. Les cinétiques d'évolution k de l'intensité des signaux en fonction du temps de mélange ont été déterminées en ajustant les données mesurées par une courbe théorique de la fonction exponentielle suivante (équation (1)) :
| (1) |

Schéma de la séquence DARR (Dipolar-Assisted Rotational Resonance).
Les mesures de relaxométrie ont été menées sur les résidus finaux de parois réhydratés, sur un spectromètre bas-champ opérant à la fréquence des protons de 20 MHz (Minispec Bruker PC 120, 0,47 T). Les temps de relaxation spin–spin (T2) ont été mesurés à partir du signal de précession libre (free induction decay – FID) et de la séquence CPMG (Carr Purcell Meiboom Gill [10]), avec, respectivement pour le PEG et pour l'eau dans les résidus de parois, un délai de recyclage de 20 s ou de 5 s, un délai entre les impulsions 90° et 180° de 1 ms ou de 0,1 ms en utilisant 10 000 ou 3000 points pour l'acquisition. Les courbes de relaxation ont été ajustées par la méthode du maximum d'entropie (MEM [11]) suivie de la méthode discrète de Levemberg–Marquardt [12]. Les mesures de diffusion moléculaire ont été effectuées à 294 K à l'aide d'une sonde dédiée DIFF30 (Bruker) émettant un gradient de champ de 1180 (±0,2) G/cm. La séquence PFG–STE LED [13] a été utilisée avec un délai de diffusion de 30 ms, une impulsion de gradient de forme trapézoïdale d'une durée totale de 900 μs.
3 Résultats et discussion
3.1 Approche moléculaire
3.1.1 Composition des parois cellulaires d'origine végétale
Les études par RMN du solide des biopolymères représentent, depuis plusieurs années, un véritable challenge. En effet, ces molécules à l'état solide présentent des interactions dipolaires et une anisotropie de déplacements chimiques telles que leur spectre RMN est souvent ininterprétable, composé de raies très larges et superposées. Ces interactions anisotropes ont rendu pendant très longtemps l'analyse des biopolymères par RMN du 1H très difficile par les techniques classiques. Cependant, depuis le développement de la technique de la rotation à l'angle magique (MAS, Magic-Angle Spinning [14a,14b]), il est possible d'enregistrer des spectres RMN sur lesquels on est capable de distinguer des raies à des déplacements chimiques différents, attribuables aux molécules étudiées. La technique MAS est couramment utilisée pour étudier des mélanges de biopolymères comme les parois cellulaires des végétaux (c'est-à-dire mélanges de cellulose, hémicelluloses, pectines, polymères phénoliques tels que lignine et subérine) [15–19]. Leur composition et leur structure tridimensionnelle varient selon l'espèce végétale, la maturité, le type de cellules, etc. [1].
Chez la tomate, plante modèle des fruits charnus, les cellules du péricarpe présentent une paroi de type primaire, hydrophile, quasiment dépourvue de lignine et riche en polysaccharides pectiques. Leur caractérisation par des méthodes physicochimiques modernes est facilitée par des méthodes d'extraction douces, permettant ainsi de préserver la structure et les interactions de ces polysaccharides.
La Fig. 3 représente les spectres de RMN 1H du matériel insoluble à l'alcool (MIA) des cellules du péricarpe d'une tomate verte (TV) et d'une tomate rouge mature (TR). L'attribution des résonances a été réalisée à partir de spectres 1H et 13C de haute résolution (Figs. 4 et 5) en utilisant la technique HRMAS (High-Resolution Magic-Angle Spinning), combinée à des recherches bibliographiques sur les données spectrales déjà publiées (voir les références dans [20]). Le Tableau 1 regroupe les déplacements chimiques 1H et 13C et l'attribution des signaux détectés sur les spectres des Figs. 3 et 5.

Spectres de détection directe 1H des résidus de parois de tomates vertes (TV) et tomates rouges (TR), St : amidon, Ara : α-1,5 arabinanes, Gal : β-1,4 galactanes, AGal : α-1,4 galacturonanes, Rha : α-1,2 rhamnoses, FattA : acides gras.
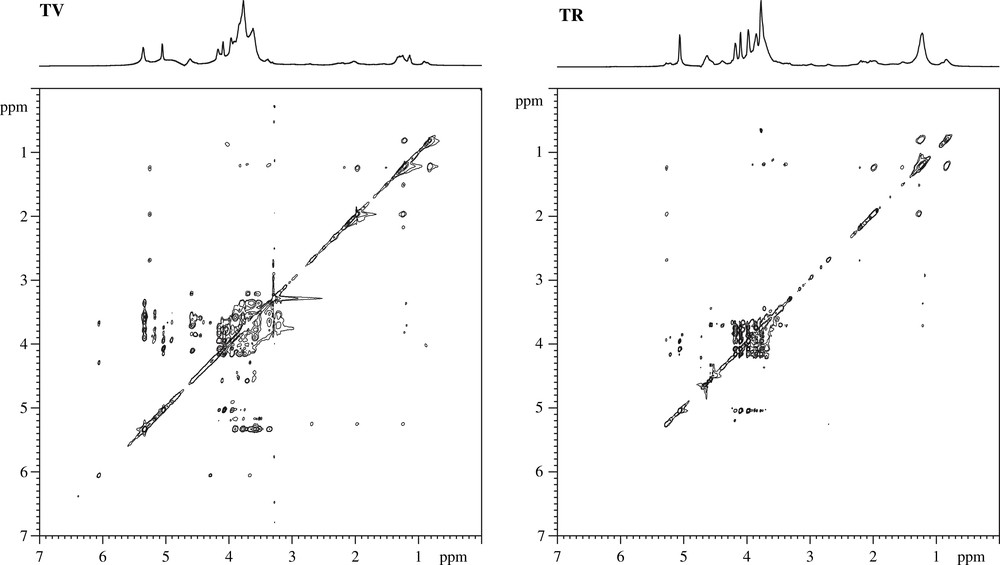
Spectres 1H 2D-TOCSY des résidus de parois de tomates vertes (TV) et de tomates rouges (TR).

Spectres de détection directe 13C des résidus de parois de tomates vertes (TV) et tomates rouges (TR), St : amidon, Ara : α-1,5 arabinanes, Gal : β-1,4 galactanes, AGal : α-1,4 galacturonanes, Rha : α-1,2 rhamnoses.
Déplacements chimiques et attributions des spectres 1H et 13C d'un matériel insoluble à l'alcool (MIA) de cellules du péricarpe de tomate
| Position | δ 1H (ppm) | δ13C (ppm) | |
| α-1,2 Rhamnoses (Rha) | 1 | 5,34 | nd |
| 2 | 3,54 | nd | |
| 3 | 3,78 | nd | |
| 4 | 3,64 | nd | |
| 5 | 3,38 | nd | |
| 6 | 1,20 | 17,97 | |
| Amidon (St) | 1 | 5,34 | 100,79 |
| 2 | 3,59 | 72,79 | |
| 3 | 3,91 | 74,44 | |
| 4 | 3,66 | 77,86 | |
| 5 | 3,77 | 72,66 | |
| 6-6′ | 3,80–3,86 | 61,86 | |
| Acides gras (FattA) | 5,26 | nd | |
| 2,70 | nd | ||
| 2,19 | nd | ||
| 1,98 | nd | ||
| 1,50 | nd | ||
| 1,25 | nd | ||
| 0,82 | nd | ||
| α-1,4 Galacturonanes (AGal) | 1 | 5,18 | 100,88 |
| 2 | 3,50 | nd | |
| 3 | 3,59 | nd | |
| 4 | 3,77 | 79,90 | |
| 5 | 3,88 | nd | |
| 6* | 3,52 | 171,9 or 176,00 | |
| 7* | nd | 54,00 | |
| α-1,5 Arabinanes (Ara) | 1 | 5,04 | 108,63 |
| 2 | 4,08 | 81,92 | |
| 3 | 3,96 | 78,06 | |
| 4 | 4,16 | 83,56 | |
| 5-5′ | 3,75–3,83 | 68,00 | |
| β-1,4 Galactanes (Gal) | 1 | 4,58 | 105,47 |
| 2 | 4,11 | 72,88 | |
| 3 | 3,71 | 74,44 | |
| 4 | 3,84 | 78,80 | |
| 5 | 3,22 | 75,67 | |
| 6-6′ | 3,55–3,63 | 61,61 |
* Signaux correspondant aux fonctions acides carboxyliques (176,0 ppm) et esters méthyliques (171,9 ppm), avec le groupement méthyl résonant à 54 ppm.
Les signaux détectés sur les spectres des résidus de tomates vertes correspondent à l'amidon et aux constituants majoritaires des parois : les pectines et plus précisément, des α-1,5 arabinanes (Ara), β-1,4 galactanes (Gal), α-1,4 galacturonanes (AGal) et α-1,2 rhamnoses (Rha). Le pic observé à 1,25 ppm sur les spectres 1H (Fig. 3) donne des taches de corrélation sur les spectres 2D-TOCSY (Fig. 4), avec des signaux à 0,82, 1,5, 1,98, 2,19, 2,17 et 5,26 ppm, suggérant son attribution à des groupements méthylène (CH2)n de lipides insaturés. Les signaux notés α et β sur le spectre 13C en détection directe (Fig. 5) sont attribués aux terminaisons réductrices des oses.
Les différents spectres 1H et 13C indiquent clairement des modifications de composition liées aux phénomènes de maturation de la tomate. Les changements majeurs entre les résidus de tomates vertes et ceux extraits des tomates rouges concernent les chaînes de β-1,4 galactanes et l'amidon. Ce dernier, contaminant les préparations pariétales, est utilisé comme source d'énergie par la plante et diminue en quantité pendant la maturation. La perte des signaux carbone caractéristiques des pectines (β-1,4 galactanes (Gal), α-1,4 galacturonanes (AGal) et α-1,2 rhamnoses (Rha)) reflète leur décomposition pendant la maturation. Ce phénomène s'accompagne de l'élimination d'esters méthyliques présents au niveau des acides galacturoniques pectiques, comme on peut l'observer sur les spectres 13C à 176 ppm, signal caractéristique de la fonction acide dé-estérifiée des pectines (Fig. 5). Ces travaux permettent de montrer que la RMN est un outil adéquat pour suivre des changements structuraux dûs à des phénomènes biochimiques complexes, comme ceux qui sont liés à la maturation des fruits charnus. Il faut toutefois souligner que les expériences bidimensionnelles haute résolution utilisées pour ces travaux peuvent durer jusqu'à 48 h. Ce type d'acquisition, réalisée sur une poudre réhydratée, ne peut se faire que sur des échantillons peu fragiles et n'évoluant pas dans le temps à température ambiante.
3.1.2 Structure de la cellulose
La caractérisation structurale et dynamique des biopolymères en utilisant la RMN du solide du 13C est possible depuis le développement de la technique de rotation à l'angle magique couplée à la polarisation croisée (CPMAS, Fig. 1). Grâce à cette méthode, un transfert de cohérence entre un noyau abondant (proton en général) et un autre peu abondant (dit rare, comme le 13C) permet d'augmenter la sensibilité du noyau rare. La séquence consiste à irradier les deux noyaux simultanément (spin-lock ou contact) et de façon contrôlée (condition de Hartmann–Hahn) [5a] et d'acquérir le signal RMN du noyau rare en appliquant un découplage dipolaire sur les protons [5b].
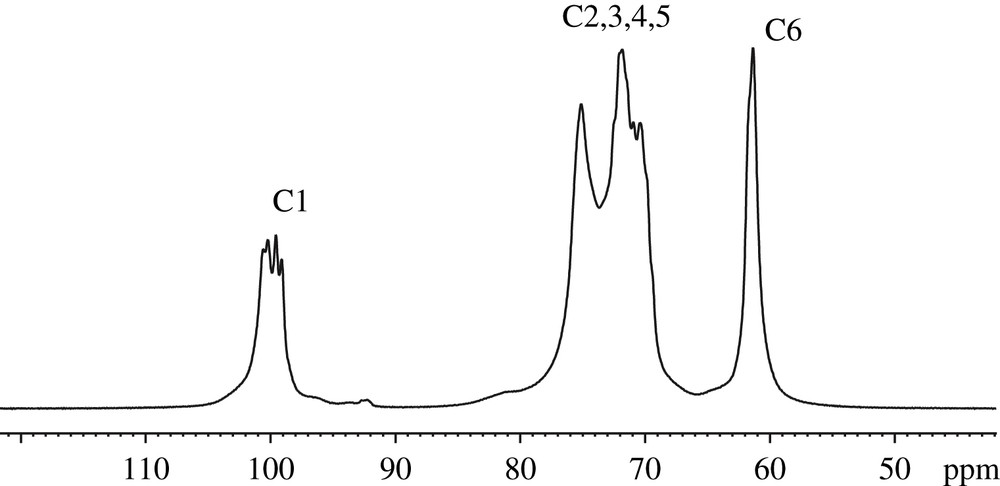
L'application de la technique CPMAS pour les études structurales et conformationnelles des constituants des parois cellulaires date de 1980, lors de la découverte par Atalla et al. [21] d'un spectre RMN 13C de cellulose présentant des signaux distincts prouvant l'existence de deux allomorphes Iα et Iβ. Il a été montré que la forme cristalline Iα correspond à un arrangement d'une simple chaîne de glucose dans une maille triclinique, alors que la phase Iβ est monoclinique et caractérisée par deux chaînes parallèles [22a,22b]. Les celluloses natives sont en fait composées d'un mélange de ces deux phases cristallines, le rapport Iα/Iβ dépendant de la source botanique des fibres de cellulose, mais également des conditions environnementales lors de la croissance de la plante ayant produit ces fibres [23a,23b]. Il a également été démontré que la phase Iα est métastable, pouvant être convertie en Iβ par des traitements hydrothermiques à 260 °C [24]. D'autres études par RMN haut champ ont permis de prouver son efficacité pour l'estimation du taux de cristallinité et de la taille latérale des microfibrilles de cellulose [25a,25b]. Un exemple de spectre CPMAS de cellulose est présenté sur la Fig. 6. Il s'agit du spectre de la tunicine (cellulose de tunicier, organisme marin) sur lequel on observe quatre massifs correspondant à la résonance des six carbones des cycles de glucose de la cellulose. Le premier massif situé à haut champ entre 60 et 70 ppm est attribué au carbone C6, alors que le large pic situé entre 70 et 80 ppm représente la résonance des carbones C2, C3 et C5. Le massif du carbone C4 résonne entre 80 et 90 ppm, et finalement le pic situé autour de 104 ppm correspond au carbone C1. Pour cet échantillon, le carbone C1 présente deux pics distincts à 105,7 et 103,8 ppm attribués à la phase cristalline Iβ. Afin d'évaluer le rapport allomorphique Iα/Iβ et le taux de cristallinité de la tunicine étudiée, nous avons procédé à une décomposition spectrale des signaux C1 et C4. La décomposition du C1 a permis d'estimer le rapport Iα/Iβ à 0,3 (23/76). Le carbone C4, quant à lui, présente la caractéristique de résonner sur une large gamme spectrale (environ 10 ppm) avec une raie large entre 80 et 86 ppm attribuée aux C4 des chaînes de glucose amorphes (en surface et dans les défauts cristallins des microfibrilles), et un deuxième massif entre 86 et 90 ppm, caractéristique du cœur cristallin des microfibrilles de cellulose. Le rapport entre les intégrales de ces deux raies permet de calculer un taux de cristallinité de 84%. Cet exemple permet de démontrer l'intérêt de la RMN du solide pour la caractérisation structurale des celluloses. Cette méthode est complémentaire de la diffraction des rayons X, qui ne permet pas de distinguer entre les formes cristallines Iα et Iβ. Cependant, la quantification des spectres CPMAS est fortement dépendante, d'une part, de la mobilité des noyaux de la molécule étudiée et, d'autre part, de l'opérateur qui décompose les spectres en raies élémentaires afin de mettre en évidence les composantes des massifs complexes. En effet, la dynamique de la polarisation croisée doit être bien maîtrisée avant toute quantification. Pour chaque échantillon, des spectres enregistrés à différents temps de contact permettent de reconstruire une courbe d'aimantation et ainsi d'estimer le temps de contact nécessaire pour un maximum de transfert d'aimantation entre les protons et les carbones des molécules étudiées. En ce qui concerne les logiciels de décomposition spectrale, la plupart sont basés sur des algorithmes d'optimisation de la distance quadratique entre les données expérimentales et le modèle en variant les paramètres de chaque raie (largeur à mi-hauteur, intensité, déplacement chimique et allure –gaussienne ou lorentzienne). Cette méthode exige une bonne connaissance des molécules analysées afin d'imposer des contraintes d'ajustement à l'algorithme de décomposition. En général, le taux de cristallinité est connu par ailleurs (mesurée par DRX) ce qui permet d'imposer des surfaces de raies caractéristiques des structures cristallines. Le deuxième inconvénient de la méthode provient du fait qu'elle n'assure pas l'unicité de la solution proposée. Le nombre de raies d'un massif n'est pas limité et dépend seulement du choix de l'opérateur.

Spectre 13C CPMAS décomposé de la tunicine.
3.1.3 Structure de l'amylose
Malgré un gain en signal et en temps non négligeable, la méthode d'acquisition CPMAS permet d'enregistrer un spectre de polysaccharides qui peut prendre plusieurs heures. La préparation d'échantillons enrichis en 13C devient alors un véritable enjeu. Nous avons, pour la première fois, synthétisé in vitro de l'amylose du type B enrichi en 13C (à 100%), présentant un taux de cristallinité de 90% [4]. La Fig. 7 présente un spectre CPMAS de l'amylose enrichie en 13C enregistré en moins de 2 min au lieu de plusieurs heures pour l'amylose en abondance naturelle en 13C. Le spectre est caractéristique d'un polymère de glucose présentant trois massifs, l'un attribuable au carbone anomérique C1 à 100 ppm, l'autre autour de 72 ppm, correspondant aux carbones C2, C3, C4, C5 et le dernier à 60 ppm attribué au C6. Le signal du carbone C1 se présente en quadruplet, alors que l'amylose de type B est caractérisé par une symétrie des chaînes de glucose, donnant un doublet du C1 en RMN [26–30]. Il a été montré que la multiplicité des raies, et en particulier de celle du carbone C1 de la liaison glucosidique, était liée à la symétrie et à l'arrangement en doubles hélices des chaines d'amylose dans la maille cristalline. Pour l'allomorphe de type A, l'empilement est de type monoclinique. Chaque brin de double hélice est généré par un axe d'ordre 2, l'unité répétitive étant le maltotriose. Trois conformations différentes des liaisons glucosidiques sont nécessaires pour définir la double hélice constituant le type A, ce qui explique l'observation d'un triplet en RMN pour le carbone C1. Dans l'allomorphe B, les doubles hélices sont générées par un axe d'ordre 31, l'unité répétitive est le maltose possédant deux conformations différentes des liaisons glucosidiques ce qui explique l'observation d'un doublet en RMN pour le carbone C1. Ici, le dédoublement du carbone C1 en quadruplet (Fig. 7) est dû aux couplages scalaires entre les carbones C1 et C2, observables pour des molécules enrichies en 13C à ces positions.
Spectre RMN 13C CPMAS de l'amylose de type B enrichie en 13C.
C'est depuis 1988, avec l'étude des effets de la résonance rotationnelle dans les solides, qu'un certain nombre de séquences couplant la rotation à l'angle magique et des impulsions de radiofréquence ont été développées dans le but de réintroduire les informations relatives aux couplages dipolaires et ainsi de mesurer des distances entre noyaux (voir les références dans [31]). Ces techniques sont connues sous le terme générique de méthodes de recouplage dipolaire. Une d'entre elles, appelée DARR pour Dipolar-Assisted Rotational Resonance [9], a été démontrée comme étant très efficace pour l'estimation de longues distances 13C–13C, même en présence de forts couplages entre carbones directement liés. De plus, cette séquence (Fig. 2) semble être moins sensible aux inhomogénéités de radiofréquence comparée à une autre expérience largement utilisée dans le domaine, la séquence RFDR (Radiofrequency-Driven Recoupling) [32]. La séquence DARR fonctionne sous la condition de résonance rotationnelle modifiée. La résonance rotationnelle est une méthode robuste qui permet de réintroduire des couplages dipolaires homonucléaires de façon sélective [33a,33b]. Pour cela, il faut régler la vitesse de rotation à l'angle magique de telle sorte qu'elle soit égale à un multiple entier de la différence de fréquence de résonance entre deux spins sélectionnés (équation (2)). Pour atteindre ces conditions, le découplage dipolaire C–H pendant le temps de mélange de l'expérience doit être complètement éliminé. Dans la séquence DARR, on applique la condition de résonance rotatoire pour laquelle la puissance de découplage hétéronucléaire C–H est égale à un multiple entier de la vitesse de rotation (équation (3)). Dans les conditions où n = 1 ou 2, les couplages dipolaires C–H notés D ne sont pas annulés et modifient la condition rotationnelle selon l'équation (4). C'est le recouplage dipolaire assisté à la condition de résonance rotationnelle (Dipolar-Assisted Rotational Resonance Recoupling).
| (2) |
| (3) |
| (4) |

Courbes d'évolution de l'intensité des pics croisés en fonction du temps de mélange de l'expérience DARR.

Corrélation entre le facteur k, caractéristique de l'efficacité des transferts dipolaires dans l'expérience DARR, et les distances mesurées sur le modèle d'Imberty and Perez [35] ; « intra » désigne les distances entre carbones dans un même cycle glucosidique, « inter » correspond aux distances entre les carbones de deux cycles glucosidiques consécutifs, et « bb » indique les distances entre les brins d'une double hélice.
3.2 Organisation supramoléculaire : porosité des parois végétales
Les parois cellulaires peuvent être considérées comme des systèmes poreux avec une interface solide distribuée dans les trois dimensions de l'espace et composée de chaînes de polysaccharides et/ou de protéines isolés ou en composite. En fait, la porosité d'une paroi peut être définie comme étant le rapport entre le volume de liquide remplissant les trous et le volume total de l'échantillon de paroi considéré. Dans ce type de matériau, la porosité dépend de la nature des constituants, de leur structure et des interactions mises en jeu. Un de nos objectifs est de relier les propriétés physicochimiques des parois végétales à des propriétés mécaniques telles que des changements de texture, par exemple. La porosité des parois cellulaires est donc une des variables que l'on cherche à évaluer. La RMN est une technique qui peut mesurer des tailles de pore dans des matériaux solides à mous, de quelques nanomètres à plusieurs millimètres. En effet, plusieurs méthodes de RMN permettent de mesurer des phénomènes de transport dans les trois dimensions de l'espace à travers un système poreux.
Il a été montré par Hills et al. [36a,36b] que, dans les systèmes aqueux de biopolymères, la contribution de l'eau liée à la relaxation transversale globale des protons de l'eau est négligeable par rapport à la contribution des protons de l'eau en échange avec les protons des biopolymères. Par conséquent, la mesure des temps de relaxation transversale des protons de l'eau (à bas champ et pour des délais d'écho courts) permet de décrire la topologie des réseaux de biopolymères, autrement dit leur porosité à l'échelle nanométrique [37]. Les techniques fondées sur les gradients de champ pulsés sont, quant à elles, applicables à l'évaluation de macropores à l'échelle de plusieurs microns (mesures de coefficients d'auto-diffusion), voir millimètres s'il s'agit d'imagerie par RMN.
Afin d'estimer des variations de taille de pores entre des résidus de parois cellulaires de tige d'une plante modèle dicothylédone (Arabidopsis thaliana), nous avons mesuré, à bas champ, les temps de relaxation T2 de l'eau présente dans les échantillons ainsi que son coefficient d'auto-diffusion (mesuré à court délai de diffusion, à haut champ) et celui d'une sonde moléculaire, le polyéthylène glycol. Nous avons choisi d'effectuer nos premiers tests avec du PEG 6000 dont le diamètre moléculaire se situe autour de 5 nm, ce qui correspond théoriquement à la taille maximale des pores de paroi [38]. Plusieurs travaux sur des systèmes biomimétiques semblent indiquer que les pectines et les hémicelluloses des parois ont un rôle régulateur sur la formation du réseau cellulose/hémicellulose et moduleraient ainsi la porosité des parois. Afin de vérifier ces hypothèses, nous avons effectué nos mesures sur des modèles biologiques extraits de parois naturelles d'A. thaliana, sur la plante sauvage et sur un mutant présentant une déficience en pectines (QUASIMODO-1 ou QUA-1) [39a,39b]. La plante mutée apparaît affectée dans la synthèse d'homogalacturonanes (squelette plus ou moins méthyle-estérifiés des pectines) et présente un défaut de cohésion cellulaire observé au niveau des feuilles, des hypocotyles et des racines ainsi qu'au niveau de suspensions cellulaires en culture [39a,39b].
Les résultats présentés dans le Tableau 2 indiquent diverses composantes de T2 et de coefficients d'auto-diffusion (D), les mesures ayant été effectuées sur des échantillons présentant la même masse solide et le même taux d'hydratation. Ces mesures reflètent une grande hétérogénéité dans la taille des pores des résidus de parois. Les résidus de parois issus des tiges de la plante mutée (QS) montrent des T2 plus faibles, avec trois composantes caractéristiques de diverses dynamiques d'échanges des protons de l'eau avec ceux des surfaces solides. Ces trois dynamiques d'échange sont liées à des tailles de pores, à l'échelle nanométrique, plus faibles que dans le cas des résidus de parois issues de la plante sauvage (WS). Ces derniers présentent deux composantes de coefficients d'auto-diffusion mesurées pour le PEG 6000, avec une composante majoritaire (97%) deux fois plus faible que la valeur D du PEG libre (0,72 × 10−10 m2/s). La deuxième composante est beaucoup moins importante (0,018 × 10−10 m2/s) et proche de l'unique valeur mesurée pour les résidus de la plante mutée (QS, 0,015 × 10−10 m2/s). Pour l'eau, une seule valeur de D, proche de celle de l'eau libre (14,9 × 10−10 m2/s) est mesurée pour la plante sauvage (WS), alors que l'eau des résidus de parois de la plante mutée présente trois coefficients de diffusion. Ces résultats corroborent les mesures de T2 indiquant une diminution de la porosité chez la plante mutée, déficiente en homogalacturonanes.
Valeurs discrètes des temps de relaxation T2 et des coefficients d'auto-diffusion de l'eau et du polyéthylène glycol mesurés dans des résidus de hampes d'Arabidopsis thaliana sauvage (WS) et du mutant QUA-1 (QS), n. o. signifie non observé, T2* correspond aux T2 des protons de la phase solide
| QS | WS | |
| Relaxation time (ms) | ||
| T2∗ | 0,025 | 0,019 |
| T2a | 4,00 | 7,90 |
| int | 4,6 | 5,7 |
| T2b | 27,78 | 37,21 |
| int | 39,1 | 54,5 |
| T2c | 70,74 | 102,28 |
| int | 56,3 | 39,8 |
| Diffusion (10−10 m2/s) | ||
| DH2Oa | 13,78 | 13,05 |
| int | 83,3 | 100 |
| DH20b | 7,12 | n. o. |
| int | 12,5 | |
| DH20c | 1,22 | n. o. |
| int | 4,2 | |
| DPEGa | n. o. | 0,46 |
| int | 96,7 | |
| DPEGb | 0,015 | 0,018 |
| int | 100 | 3,3 |
Afin de mieux comprendre d'un point de vue phénoménologique, le mode de diffusion du PEG à travers les parois cellulaires en utilisant un modèle approprié (modèle de la reptation dans un gel de polymère selon de Gennes [40a], ou modèle plus « universel » de diffusion des macromolécules proposées par Phillies [40b], par exemple), il est envisagé de procéder aux mêmes expériences en utilisant diverses concentrations de biopolymères (ou de teneur en eau) et diverses tailles de PEG. De futurs travaux portant sur le dosage des oses par des méthodes biochimiques et sur l'analyse de la structure fine des pectines devront également permettre de mieux comprendre les effets de la mutation QUASIMODO-1 sur la porosité des parois cellulaires.
4 Conclusion
Nous avons montré que la spectroscopie par RMN offrait une diversité de méthodes permettant de caractériser, d'une part, la structure des biopolymères d'origine végétale et, d'autre part, leur organisation dans des systèmes composites complexes, comme les parois cellulaires. Ces travaux combinent l'utilisation des techniques de la RMN du solide basées sur la rotation à l'angle magique, couplée à la polarisation croisée dans le cas des acquisitions 13C, et les mesures de dynamiques moléculaires en RMN du liquide par les techniques de relaxométrie à bas champ et celles basées sur la mesure des coefficients d'auto-diffusion à haut champ.