

1 Introduction
Le développement de l'industrie chimique est étroitement lié à celui de la catalyse [1]. En effet, près des deux tiers des grands intermédiaires nécessitent l'utilisation d'un catalyseur métallique au cours de leur élaboration. La catalyse homogène par complexes de métaux de transition représente 15% de ces procédés, qui font en permanence l'objet de recherches destinées à les rendre plus efficaces et plus économiques. C'est la raison pour laquelle de nombreuses études sont aujourd'hui encore consacrées à l'élaboration de nouveaux catalyseurs moléculaires. Parmi ceux-ci figurent notamment des composés organométalliques du nickel et du titane capables de polymériser ou oligomériser des oléfines [2,3]. Les produits de ces réactions donnent lieu à des applications multiples [4,5].
Notre laboratoire étant impliqué dans la thématique « métallocalixarènes », nous avons orienté une partie de nos recherches vers l'obtention de catalyseurs d'intérêt industriel construits sur des plateformes du type calix[4]arène [6]. Ces macrocycles, qui comportent quatre entités phénoliques liées entre elles par des ponts méthyléniques tout en définissant un espace cavital, possèdent de nombreux points d'ancrage facilitant la confection de coordinats préorganisés. La fonctionnalisation des calixarènes permet, non seulement d'accéder à des ligands sophistiqués, mais également de fixer ces macrocycles dans une conformation définie (Schéma 1). Il faut préciser que seuls les calix[4]arènes dans la conformation cône peuvent efficacement fonctionner comme récepteurs, mais cette dernière propriété n'est pas nécessairement recherchée.

Les quatre conformations extrêmes d'un calix[4]arène.
Dans cette micro-revue, nous décrivons les propriétés de plusieurs complexes calix[4]aréniques capables de polymériser ou d'oligomériser de l'éthylène, et montrons comment les diverses propriétés du châssis macrocyclique (flexibilité, taille, dynamique intrinsèque) peuvent être mises à profit pour contrôler ces réactions.
2 Dimérisation de l'éthylène
Les calixarènes 1 et 2 [7] contiennent deux groupements diphénylphosphanyle greffés sur des positions para-phénoliques distales. Ces phosphines sont thermiquement très robustes. De fait, elles sont assimilables à des (tris-aryle)-phosphines, ce qui les rend particulièrement attractives pour des applications catalytiques. Ces deux ligands forment facilement et sélectivement des complexes chélate avec de nombreux précurseurs métalliques, et en particulier des sels de nickel (II) [8,9]. Les complexes 3–5 en sont des exemples (Schéma 2).

Synthèse des complexes 3–5.
L'analyse structurale de ces derniers révèle, pour chacun d'eux, une pseudo-symétrie Cs à l'état solide, l'atome de nickel étant orienté vers l'un des bords du calixarène [9–11]. Comme le montre une étude RMN à température variable (Fig. 1), réalisée avec le complexe 3 (NB : les complexes 4 et 5 sont paramagnétiques), cette molécule n'est pas rigide en solution. En fait, une double dynamique s'y produit (Schéma 3).
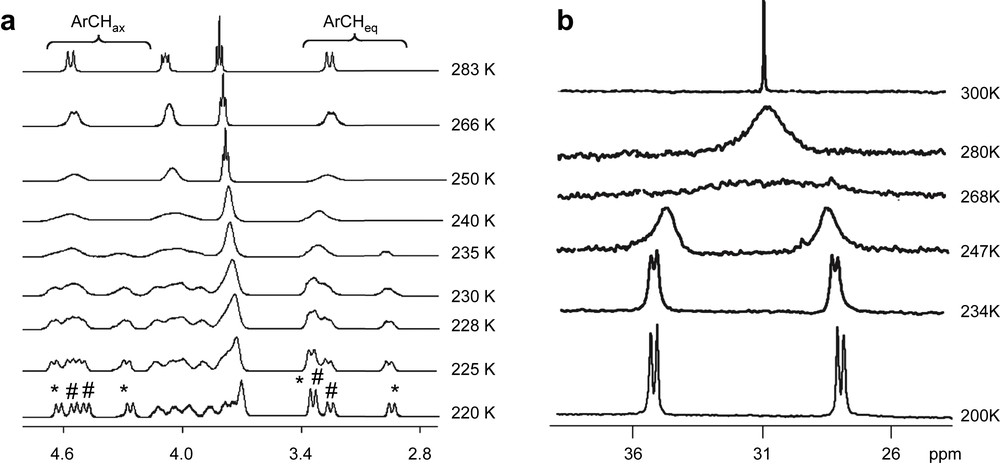
Spectres RMN à température variable 1H (a) et 31P (b) du complexe 3.
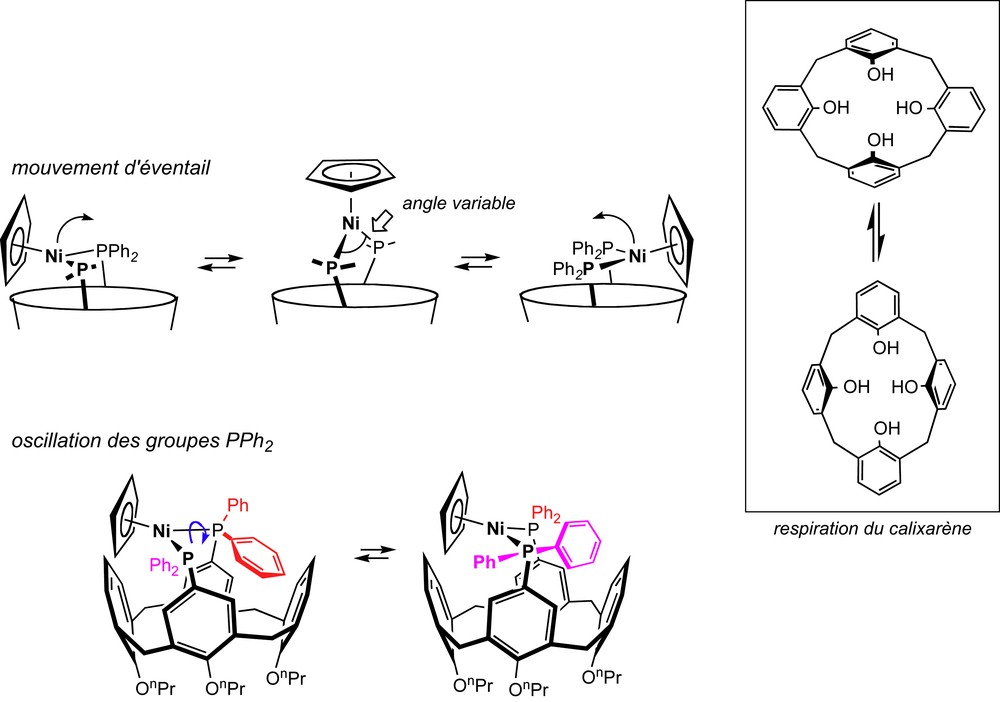
Dynamiques du complexe 3.
La première dynamique correspond à un mouvement d'éventail du plan métallique, qui bascule de part et d'autre de l'axe du calixarène. Ce mouvement est étroitement lié à la respiration « naturelle » du cœur calixarénique, un mouvement caractéristique des calix[4]arènes qui, en alternance, rapproche puis écarte chacune des paires de groupes phénoxy se faisant face (Schéma 3). Au cours de ce mouvement périodique, l'angle PNiP subit une variation d'amplitude substantielle (environ 20°). C'est lorsque le métal croise l'axe du calixarène que cet angle atteint sa valeur maximale. La seconde dynamique est une oscillation de chacun des groupes PPh2 autour de la liaison P–Ni correspondante (rotation partielle autour de l'axe). Ce mouvement résulte probablement d'une répulsion stérique entre les deux noyaux PPh endo-orientés, qui viennent, à tour de rôle, occuper l'entrée de la cavité macrocyclique. Des mouvements identiques ont été mis en évidence dans les complexes plan carré [MX2L] (M = Pd, Pt ; L = 1, 2).
Les complexes [NiBr2·1] (4) et [NiBr2·2] (5) deviennent, après activation avec du méthylaluminoxane (MAO), des catalyseurs de dimérisation de l'éthylène (formation de but-1-ène et but-2-ènes) avec des sélectivités en C4 supérieures à 95% (Eq. (1)). Ces catalyseurs se sont avérés nettement plus efficaces que leur homologue [NiBr2(dppe)] (dppe : bis(diphénylphosphino)éthane) (Tableau 1, entrées 1 et 6).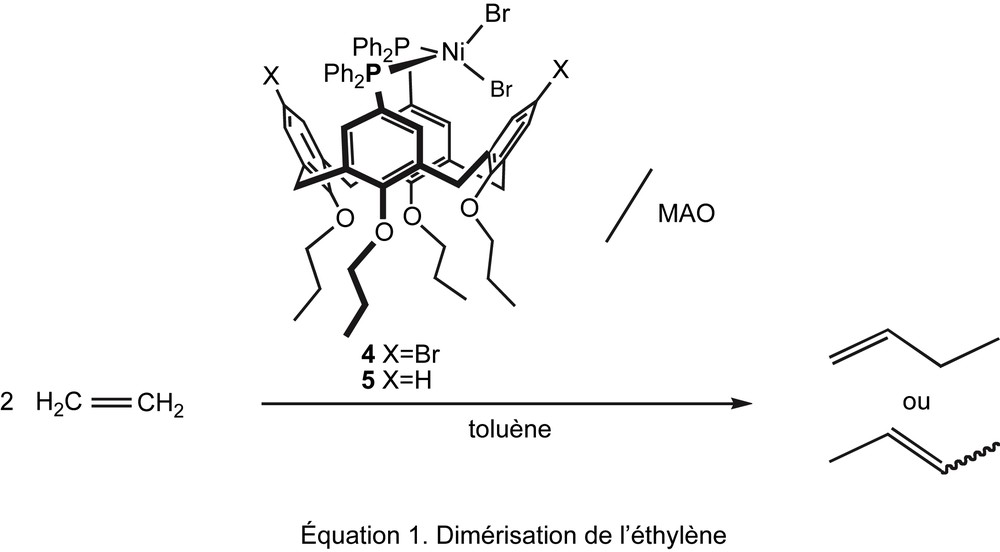
Dimérisation de l'éthylène catalysée par les complexes 4 et 5a
| Entrée | Précurseur | n(Ni) (μmol) | t (h) | P(C2H4) (bar) | T (°C) | ΔTb (°C) | rendementc (g) | TOFd/10−4 |
| 1 | NiBr2(dppe) | 4,5 | 0,25 | 30 | 40 | 13 | 1,07 | 3,39 |
| 2 | 4 | 4,5 | 0,25 | 30 | 25 | 5 | 4,27 | 13,54 |
| 3 | 4 | 4,5 | 0,5 | 30 | 25 | 5 | 5,94 | 9,42 |
| 4 | 4 | 4,5 | 1 | 30 | 25 | 5 | 7,00 | 5,55 |
| 5 | 4 | 4,5 | 0,25 | 20 | 25 | 5 | 1,90 | 6,03 |
| 6 | 4 | 4,5 | 0,25 | 30 | 40 | 40 | 2,63 | 8,34 |
| 7 | 4 | 0,09 | 0,25 | 20 | 25 | 3 | 0,76 | 120,54 |
| 8 | 5 | 4,5 | 0,25 | 20 | 25 | 57 | 3,70 | 11,74 |
| 9 | 5 | 0,09 | 0,25 | 20 | 25 | 3 | 0,52 | 83,53 |
a Toluène 22 mL, MAO 400 équiv./Ni, agitation mécanique 500 tours par min.
b Élévation maximale de la température pendant la catalyse.
c Rendement déterminé par la masse finale du milieu réactionnel vs masse de contrôle de 22 mL de toluène saturé d'éthylène.
d mol(C2H4 converti) mol(Ni)−1 h−1.
Des différences importantes de réactivité ont été mises en évidence entre les complexes 4 et 5. Ainsi, lorsque les réactions de dimérisation sont réalisées dans un réacteur de 100 mL avec 4,5 μmol de nickel, l'activité du complexe 5 est environ deux fois plus importante que celle du complexe 4 (Tableau 1, entrées 5 et 8). La plus faible réactivité de 4 est probablement due à une réaction du MAO avec les atomes de brome du calixarène 1, transformant celui-ci en un nouveau ligand très encombré, et réduisant ainsi la facilité d'accès de l'éthylène au centre catalytique. Il est aussi intéressant de noter qu'à la concentration en nickel utilisée, une rapide et forte exothermie se produit, en particulier avec le précatalyseur 5. Avec ce dernier, la température du milieu réactionnel atteint 82 °C, ce qui désactive le centre actif après quelques minutes. La forte montée en température observée avec 5 est responsable de l'isomérisation du but-1-ène formé en but-2-ènes. Si la température du milieu est maintenue à 30°, la proportion de but-2-ènes reste importante (env. 75% dans les conditions de l'entrée 5, Tableau 1). Pour assurer au catalyseur 5/MAO une activité sur une période plus importante et pour former préférentiellement du but-1-ène, la quantité d'espèce active a été divisée par 50. Avec ces nouvelles conditions, la température du milieu reste proche de 25 °C et l'activité du catalyseur est effectivement maintenue durant tout le temps de la dimérisation. La proportion de but-1-ène s'élève alors à 91%, démontrant que le contrôle de l'exothermie permet de réduire drastiquement l'isomérisation du but-1-ène. Les activités atteignent, respectivement, 1,20 × 106 et 0,83 × 106 mol(C2H4) mol(Ni)−1 h−1 (!!!) pour les précatalyseurs 4 et 5 (Tableau 1, entrées 7 et 9), ce qui correspond aux plus fortes activités observées avec des précatalyseurs du type [NiX2(diphosphine)] [12–14]. À haute dilution, et en appliquant le même rapport MAO/Ni que pour les expériences précédentes (MAO/Ni = 400), le précatalyseur 4 est le plus actif, laissant supposer que, dans ces conditions, une proportion importante du MAO mis en œuvre (2 mg) sert à purifier le milieu réactionnel plutôt qu'à réagir avec les atomes de brome du calixarène, évitant ainsi une modification de l'encombrement stérique du complexe 4.
Pour expliquer les fortes activités enregistrées, qui sont comparables à celles du meilleur système connu ([NiCl2(PCy3)2]/MAO), il faut se rappeler qu'en raison du mouvement d'éventail du plan PMP discuté plus haut, l'angle de chélation du ligand subit une variation d'amplitude périodique. Il se trouve qu'une augmentation de l'angle PMP entraîne une diminution sensible de l'angle entre les deux liaisons opposées aux liaisons M–P, et ce à la fois pour des raisons électroniques que stériques (on suppose ici que les intermédiaires catalytiques ont une structure plan carré). C'est précisément le rapprochement des fragments éthyle et éthylène dans l'intermédiaire clé [Ni(diphos)(C2H5)(éthylène)]+ qui favorise alors l'étape d'insertion (Schéma 4).

Variation périodique de l'angle de pince.
3 Oligomérisation de l'éthylène
Nous nous sommes également intéressés à des calixarènes porteurs de substituants phosphine fixés sur des atomes d'oxygène phénoliques. Le calixarène 6, par exemple, dont deux positions proximales sont substituées par des fragments –CH2PPh2, forme avec des halogénures de nickel des complexes chélatés de type NiX2L. Tous ces complexes ont une structure tétraédrique. On pourra noter que les métallomacrocycles formés sont très grands, chacun d'eux comportant 12 liaisons. La disparité des angles PMP dans ces complexes (158,4 (7) ; 158,6 (8) et 118,6° (9)) illustre la grande flexibilité du chélateur (Fig. 2) [15,16].

Formule de la diphosphine 6 et structure RX des complexes 7–9 contenant 6. Seuls les atomes de carbone Cipso des unités PPh ont été représentés.
Les structures révèlent par ailleurs que, dans tous ces complexes, l'encombrement stérique autour du nickel est sensiblement supérieur à ce qu'il est dans les complexes de nickel décrits dans le paragraphe précédent. C'est probablement cette propriété qui induit avec le système 7/MAO la formation de chaînes légèrement plus longues que celles obtenues avec 4 ou 5/MAO. Avec 7, on forme en effet des oligomères C4–C12 suivant une distribution de Schulz–Flory (α = 0,22). Précisons que l'activité du système (9800 mol(C2H4) mol(Ni)−1 h−1, 30 bar) est comparable à celle obtenue avec [NiCl2(Ph2PCH2PPh2)] ou [NiCl2(Ph2PCH2CH2CH2PPh2)] [17].
4 Oligomérisation et polymérisation avec des « calix-ylures de phosphore »
Les complexes de nickel de formule générale [NiPh(P,O)L](L = phosphine tertiaire ; P,O = ligand chélatant de type « phosphinoénolate »), communément appelés catalyseurs SHOP, constituent des précurseurs très efficaces pour la réaction d'oligomérisation de l'éthylène [1–3,18–20]. Ils sont généralement formés in situ par réaction de Ni(0) avec un ylure de phosphore de type Ph3PCR2C(O)R3 en présence d'une phosphine tertiaire. Ils permettent la production sélective d'α-oléfines linéaires dans des conditions douces. Lorsqu'on ajoute à un tel complexe une substance capable de décoordiner et piéger la phosphine L, on obtient un nouveau catalyseur, qui cette fois est capable de polymériser l'éthylène. Plusieurs études ont démontré qu'un des processus de désactivation des catalyseurs SHOP est la conversion par transmétallation d'une espèce de type A en une espèce bis-chélatée inactive de type [Ni(P,O)2] (C), cette transformation se faisant via l'intermédiaire B (Eq. (2)) [21,22]. On comprend aisément que la présence d'un groupe R3 stériquement encombré est de nature à empêcher la formation d'une espèce de type B, ce qui devrait donc accroître la stabilité du catalyseur SHOP correspondant.
Afin de tester cette hypothèse, nous avons synthétisé les ylures de phosphore 10–13 dans lesquels le groupe céto est substitué par un fragment très encombré, à savoir une unité calixarénique. On notera que les calixarènes 10 et 12 ont une conformation conique, les composés 11 et 13 adoptant une conformation 1,3-alternée (Schéma 5) [23].
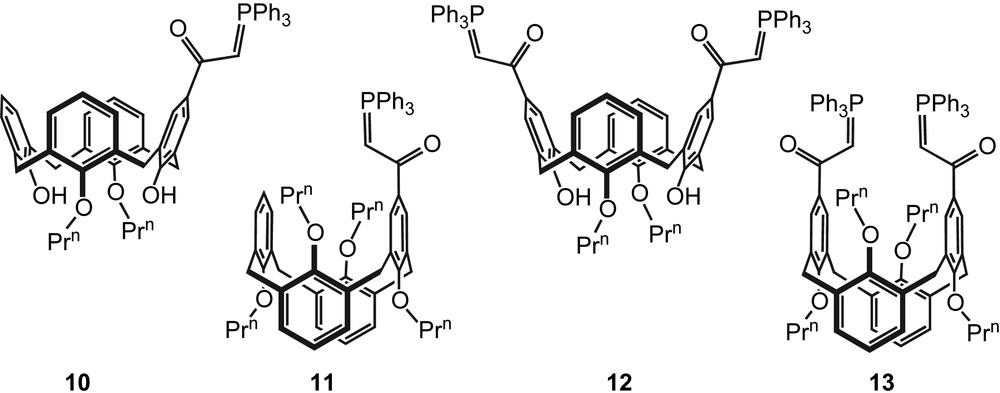
Calix-ylures de phosphore 10–13.
Les propriétés catalytiques de chacun de ces calix-ylures ont été évaluées en mélangeant l'ylure avec [Ni(COD)2], soit en présence de PPh3/éthylène pour obtenir un catalyseur d'oligomérisation, soit uniquement en présence d'éthylène pour disposer d'un catalyseur de polymérisation (Schéma 6).
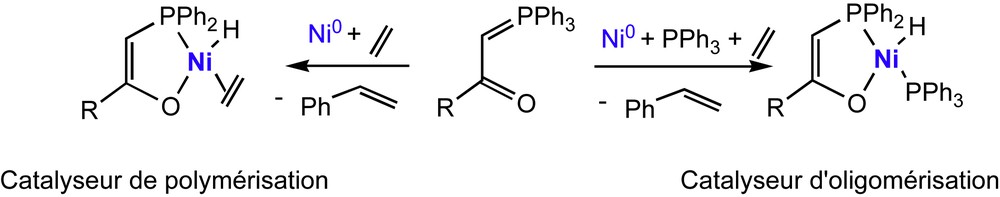
Synthèse in situ des catalyseurs.
La formation des complexes attendus a été confirmée par des études de RMN du 31P pour chacun des ligands 10–13 (observation d'un signal vers +20 ppm, caractéristique d'un groupement phosphinoénolate coordiné au nickel). La possibilité de former un complexe bimétallique à partir de 13 a été démontrée en isolant le complexe 14 obtenu en faisant réagir 13 avec [Ni(COD)2] en présence de PPh3 (Schéma 7). Ce complexe a été caractérisé par diffraction des rayons X (Fig. 3).

Synthèse du complexe 14.

Structure RX du complexe bis-nickel 14. Pour plus de clarté, seuls les atomes de carbones Cipso des unités PPh ont été représentés.
L'activité des précurseurs a été testée dans le cas de l'oligomérisation de l'éthylène en utilisant un rapport Ni/PPh3 de 1:1. L'ylure PPh3CHC(O)Ph (15) a été choisi comme référence. Les résultats des tests, effectués dans le toluène, sont résumés dans le Tableau 2. Tous les systèmes conduisent à des oligomères linéaires, la proportion en α-oléfines étant supérieure à 98% dans la plage des oligomères C4–C16. Les deux mono-ylures 10 et 11 présentent des activités significativement plus grandes que celle du complexe de référence 15 (Tableau 2, entrées 2 et 3), reflétant ainsi le rôle bénéfique d'un fragment calixarène capable de retarder la désactivation du catalyseur. Il est intéressant de constater que les deux bis-ylures 12 et 13 présentent des activités globalement inférieures à celles du catalyseur 15. Ainsi, alors que l'activité de 10 à 5 bar dépasse de 30% celle de 15, l'activité de 12 est d'environ 35% inférieure. Une étude approfondie réalisée avec le bis-ylure 13 montre que la proximité des deux centres catalytiques peut engendrer un processus de désactivation. En effet, lors de la phase d'activation du système, on observe la formation du complexe mononucléaire 16, comportant une entité phosphinoénolate chélatante ainsi qu'une entité P-monodentate (Fig. 4). Ce complexe présente une très grande stabilité et s'avère totalement inactif en oligomérisation.
Oligomérisation de l'éthylène avec les ylures 10–13 et Ph3PCHC(O)Ph 15a
| Entrée | Ylure | [Ni(COD)2] (μmol) | TOF (mol(C2H4) mol(ylure)−1 h−1) | αb | C2H4 consomméc (g) |
| 1 | 15 | 5 | 41 800 | 0,94 | 8,79 |
| 2 | 10 | 5 | 55 800 | 0,95 | 15,64 |
| 3 | 11 | 5 | 54 000 | 0,96 | 15,15 |
| 4 | 12 | 10 | 28 700 | 0,94 | 8,04 |
| 5 | 13 | 10 | 34 700 | 0,92 | 9,74 |
a Ylure 5 μmol, [Ni(COD)2]/PPh3 : 1/1, P(C2H4) = 5 bar, toluène 20 mL, 70 °C, 1 h.
b Paramètre de Schulz–Flory α = n(Cn+2)/n(Cn).
c Valeur calculée (standard interne: heptane).

Formule et structure RX du complexe chélate 16. Seuls les atomes de carbones Cipso des unités PPh ont été représentés.
Les réactions de polymérisation ont été effectuées sous pression constante d'éthylène. Des températures supérieures à 60 °C sont nécessaires pour initier la réaction. En polymérisation, les activités présentent les mêmes tendances qu'en oligomérisation, mais elles sont toutefois plus marquées. Ainsi, les composés mono-ylures 10 et 11 sont deux fois plus actifs que le complexe de référence, alors que les bis-ylures 12 et 13 donnent lieu à des activités plus faibles (Tableau 3). Une fois encore, l'encombrement du macrocycle semble jouer un rôle prépondérant.
Polymérisation de l'éthylène avec les ylures 10–13 et 15a
| Entrée | Ylure | quantité d'ylure (μmol) | t (h) | PEb (g) | TOF (mol(C2H4) mol(ylure)−1 h−1) | Mwc (g mol−1) | Mw/Mnc |
| 1 | 15 | 4 | 0,5 | 1,12 | 20 000 | 6600 | 2,5 |
| 2 | 10 | 2 | 0,25 | 0,30 | 21 400 | 7000 | 2,1 |
| 3 | 11 | 2 | 0,25 | 0,70 | 49 900 | 3300 | 1,6 |
| 4 | 12 | 2 | 0,25 | 0,18 | 6400 | 5700 | 1,9 |
| 5 | 13 | 2 | 0,25 | 0,47 | 16 800 | 4100 | 1,8 |
a [Ni(COD)2] / unité ylure = 10, toluène 20 mL, P(C2H4) = 5 bar, T = 70 °C.
b Après précipitation de la réaction avec du méthanol (200 mL) acidifié (HCl).
c Déterminé par SEC-HT vs. standard polystyrène, non corrigé.
Les polymères obtenus avec les ylures 10 et 12, caractérisés par la présence de OH phénoliques, conduisent à des masses moléculaires plus élevées (Mw = 6000 à 7000 g mol−1) que celles obtenues avec les ylures 11 et 13 (Mw = 3000 à 4000 g mol−1). Ce résultat est vraisemblablement lié au caractère inductif donneur des groupes –C6H4OH. Il est connu, en effet, que les catalyseurs SHOP ayant un centre métallique électroniquement enrichi favorisent les longues chaînes par suite d'une diminution de la β-élimination [24–28]. Dans tous les cas, les polyéthylènes formés sont hautement linéaires. Le taux de branchement restant très faible, on peut exclure tout effet bimétallique.
5 Formation de polyéthylène linéaire basse densité
Le polyéthylène linéaire basse densité (abréviation anglaise, LLDPE) consiste en une chaîne de polyéthylène comportant des chaînes latérales courtes, rendant ce polymère plus élastique et plus résistant à la rupture que le polyéthylène basse densité (LDPE). La présence de ramifications diminue la température de fusion et la cristallinité du polymère. Le matériau formé est moins brillant et plus facile à mettre en forme que le polyéthylène haute densité (HDPE).
Récemment, nous avons montré que le calixarène 4 pouvait avantageusement être utilisé, et ce, en association avec un autre complexe, ZrCp2Cl2 (17), pour la formation de LLDPE [29]. La synthèse de LLDPE a été effectuée, soit par catalyse tandem orthogonale (voie a, Schéma 8), soit par catalyse one-pot en deux étapes (voie b, Schéma 8) [30]. Ces procédés d'obtention du polymère se distinguent de la voie classique de synthèse de LLDPE, qui consiste à réaliser la copolymérisation d'éthylène avec des α-oléfines courtes préformées. S'affranchissant d'une étape de séparation, cette voie synthétique permet de réaliser une appréciable économie d'énergie.
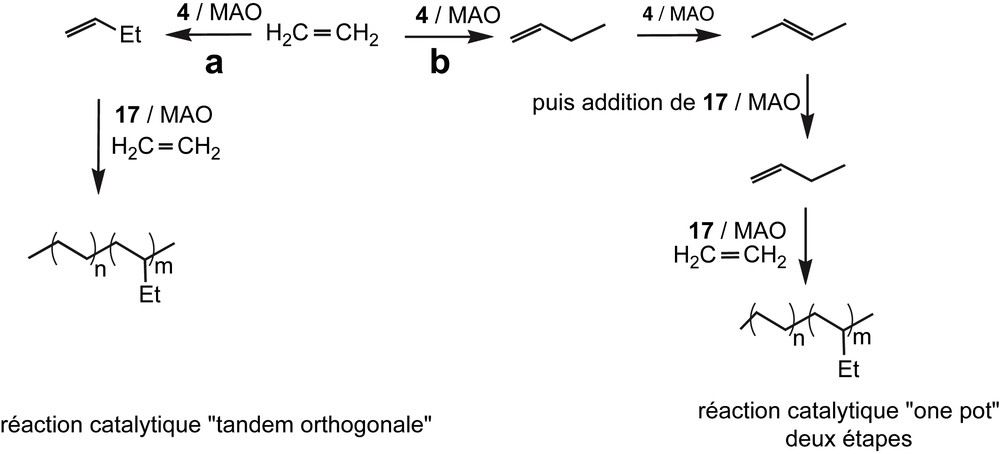
Synthèse de LLDPE via catalyse tandem orthogonale ou one-pot.
La différence entre les chemins (a) et (b) réside dans le fait que, dans le premier cas, le complexe 17 est présent dès le début de la réaction, alors que dans le second il est introduit lorsqu'une quantité suffisante d'oléfine C4 a été produite.
Deux séries de tests de polymérisation par catalyse tandem ont été réalisées, l'un sous une pression d'éthylène de 10 bar, l'autre de 20 bar. Les analyses des copolymères montrent qu'à l'intérieur de chaque série, plus le rapport 4/17 est important (ce qui augmente la quantité de but-1-ène formé), plus la quantité de butène incorporée dans le copolymère est grande (Tableau 4, entrées 2–5 et 6–9). Les polyéthylènes présentent alors des températures de fusion cristalline de plus en plus basses et un pourcentage de région amorphe de plus en plus important. Un accroissement sensible de la quantité de butène incorporé (2,7% de branches éthyles) peut être observé si le complexe de zirconium 17 n'est introduit que dans un second temps (Tableau 4, entrée 10). Cette façon de procéder permet de s'affranchir du contrôle de l'exothermie de la dimérisation, le trans-but-2-ène formé étant isomérisé en α-oléfine par le catalyseur de zirconium avant incorporation dans la chaîne de polyéthylène [31]. Dans tous les cas, les polymères ainsi synthétisés présentent uniquement des branches éthyle comme ramification (spectre RMN 13C, Fig. 5).
Copolymérisation de l'éthylène et du butène via catalyse tandem orthogonale/one-pota
| Entrée | P(C2H4) (bar) | Ni/Zr (4/17) | rendement (g) | branche éthyleb (%) | Mwc (g mol−1) | Mw/Mnc | Tmd (°C) | Xce (%) |
| 1f | 10 | 0 | 1,271 | 0,0 | 348 000 | 2,7 | 140,4 | 58,9 |
| 2 | 10 | 2 | 1,065 | traces | 307 400 | 2,9 | 139,1 | 61,0 |
| 3 | 10 | 20 | 0,330 | 0,4 | 346 700 | 2,1 | 131,8 | 48,4 |
| 4 | 10 | 50 | 0,237 | 0,6 | 265 000 | 2,0 | 131,3 | 39,5 |
| 5 | 10 | 100 | 0,138 | 1,0 | 212 800 | 2,0 | 128,0 | 39,6 |
| 6 | 20 | 2 | 1,126 | traces | 224 000 | 3,0 | 138,3 | 64,8 |
| 7 | 20 | 20 | 0,490 | traces | 323 400 | 2,0 | 132,4 | 45,2 |
| 8 | 20 | 50 | 0,402 | 0,5 | 316 600 | 2,0 | 131,6 | 45,2 |
| 9 | 20 | 100 | 0,252 | 0,9 | 285 800 | 2,0 | 128,4 | 43,7 |
| 10g | 4,5 | 0,616 | 2,7 | 139 400 | 6,1 | 120,1 | 48,8 |
a 4 4,5 μmol, toluène 22 mL, MAO 400 équiv./M, Tinitale = 20 °C, 30 min.
b Déterminé par RMN 13C [32].
c Déterminé par SEC-HT à 150 °C dans le 1,2,4-trichlorobenzène.
d Température de fusion cristalline déterminée par DSC.
e Cristallinité déterminée à partir de Xc (%) = (ΔHm/ΔHm∗) × 100 (ΔHm∗ = 293 J/g pour HDPE).
f 17 0,112 μmol.
g Catalyse one-pot en deux étapes (i) 4 4.5 μmol, toluène 22 mL, P(C2H4) = 30 bar, MAO 400 équiv/Ni, Tinitale = 20 °C, 1 h. (ii) 17 1 μmol, toluène 6,3 mL, P(C2H4) = 1 bar, MAO 1000 équiv/Zr, T = 40 °C, 22 h.

spectre RMN 13C de LLDPE (Tableau 4, entrée 10).
6 Formation de polyéthylène ultra haute masse moléculaire
La plupart des catalyseurs de Ziegler–Natta homogènes perdent de leur efficacité à haute température, ce qui en limite considérablement les possibilités d'application industrielle. Ce défaut résulte des réactions de désactivation qui conduisent à une diminution de l'activité et de la sélectivité du catalyseur. Par exemple, les masses moléculaires des polyéthylènes obtenus avec le complexe ZrCp2Cl2 chutent de 600 000 à 120 000 g mol−1 quand la température de polymérisation passe de 20 à 70 °C [33,34].
Nous avons étudié le complexe diphénolate–dichlorure de titane (IV) 19, obtenu par réaction entre le précurseur TiCl4 et le 25,27-dipropoxy-calix[4]arène 18 [35]. Comme le révèle une analyse structurale, les deux fonctions propyloxy permettent de stabiliser le complexe. À l'état solide, la distance Ti–OPr est de 2,11 Å (Fig. 6).

Formation du complexe titano-calixarène 19.
Après activation par le MAO, le complexe 19 conduit à la formation de polyéthylène d'ultrahaute masse moléculaire (abréviation anglaise, UHMWPE), les masses moléculaires observées étant de l'ordre de 3 à 4 × 106 g mol−1 pour des températures de polymérisation inférieures à 75 °C (Tableau 5, entrées 1–3). Le catalyseur est thermiquement très stable ; il reste en effet actif à 120 °C (Tableau 5, entrée 6). À 60 °C, avec une concentration d'éthylène de 1 mol L−1, l'activité atteint 2940 mol(C2H4) mol(Ti)−1 h−1 (Tableau 5, entrée 10). Les polydispersités des polyéthylènes sont inférieures à 1,5, démontrant le caractère vivant de cette polymérisation. L'analyse RMN 13C confirme l'absence de ramification des polyéthylènes formés (Fig. 7).
Formation d'UHMWPE catalysée par le complexe de titane 19a
| Entrée | [éthylène] (mol L−1) | P(C2H4) (bar) | T (°C) | rendement (g) | TOF mol(C2H4) mol(Ti)−1 h−1 | Tmb (°C) | Mnc/10−6 (g mol−1) |
| 1 | 0,50 | 5,3 | 45 | 0,089 | 380 | 135,8 | 3,98 |
| 2 | 0,50 | 6,1 | 60 | 0,252 | 1060 | 136,9 | 3,44 |
| 3 | 0,50 | 7,0 | 75 | 0,367 | 1560 | 137,0 | 2,56 |
| 4 | 0,50 | 7,9 | 90 | 0,504 | 2140 | 134,2 | 0,96 |
| 5 | 0,50 | 8,8 | 105 | 0,458 | 1950 | 137,0 | 0,73 |
| 6 | 0,50 | 9,6 | 120 | 0,227 | 960 | 135,5 | 0,60 |
| 7 | 0,25 | 3,0 | 60 | 0,082 | 350 | 138,3 | 3,24 |
| 9 | 0,75 | 9,1 | 60 | 0,452 | 1920 | 137,8 | 3,53 |
| 10 | 1,00 | 12,1 | 60 | 0,692 | 2940 | 138,3 | 4,22 |
a [Ti] = 2,1 × 10−5 M, MAO 1440 équiv/Ti, toluène 200 mL, T = 60 °C, 2 h.
b Température de fusion cristalline déterminée par DSC.
c Masse moléculaire moyenne déterminée par viscosimétrie.
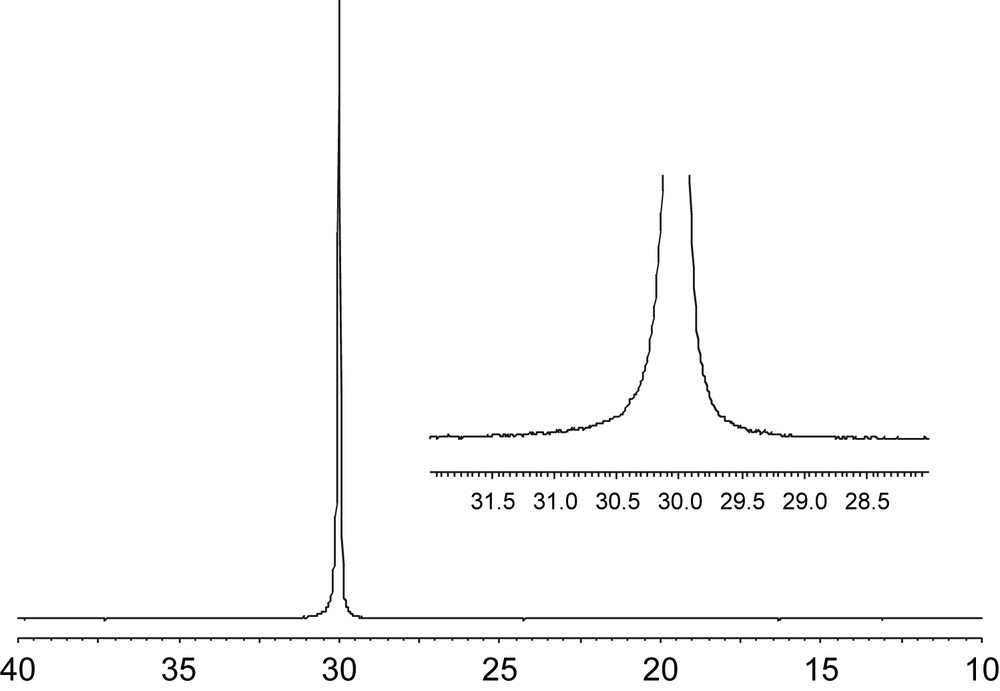
Spectre RMN 13C d'UHMWPE (Tableau 5, entrée 2).
Il est raisonnable d'admettre que ce sont les interactions entre les groupements propyloxy et le titane qui sont à l'origine de la grande stabilité de l'espèce active. Globalement, on peut donc considérer que le titane est fixé sur une surface tétra-oxo qui protège les deux tiers de la première sphère de coordination du centre métallique. Signalons qu'il a été possible de caractériser spectroscopiquement l'espèce cationique Ti–Me 20, formée lors de la réaction entre le complexe 19 et le MAO (Schéma 9). Ce dernier, mis sous pression d'éthylène, conduit bien à la formation d'UHMWPE.

Formation de l'espèce active 20.
7 Conclusions
Les calix[4]arènes sont des molécules macrocycliques aisément synthétisables et thermiquement robustes. Leur fonctionnalisation permet d'accéder à une grande diversité de ligands. Dans cette publication, nous avons présenté plusieurs complexes calixaréniques pouvant être employés en oligomérisation et/ou polymérisation de l'éthylène. Nous avons montré qu'un fragment calix[4]arène directement greffé sur le coordinat phosphinoénolate P,O d'un catalyseur de type SHOP pouvait, en raison de son fort encombrement, contribuer à la longévité du catalyseur. Un autre aspect intéressant des calix[4]arènes concerne leur dynamique intrinsèque. Celle-ci peut par exemple modifier la géométrie de la première sphère de coordination d'un métal auquel il est relié et, incidemment, comme dans le cas du complexe 4, augmenter la réactivité du catalyseur. Plus simplement, les calixarènes peuvent fonctionner comme de petites surfaces tétra-oxo servant à la fixation d'un centre métallique. Dans le cas du complexe Ti–O4 19, la protection stérique et électronique assurée par le calixarène est remarquable, permettant au catalyseur de polymérisation correspondant de rester actif à des températures élevées et ainsi de fournir du polyéthylène à ultra haute masse moléculaire.
Remerciements
Nous remercions MM. Marco Frediani, Pierre Kuhn et Manuel Lejeune pour leur contribution à ces résultats, ainsi que M. Pierre Lutz pour de fructueuses discussions. Nous tenons particulièrement à remercier le comité d'organisation du Gecom 2007 pour l'excellence du travail fourni à l'occasion de ces journées.