

1 Introduction
The use of small molecules to perturb biological processes has proven to be an attractive approach to investigate biological mechanisms and lies at the core of pharmacological intervention [1]. There is a tremendous demand for small molecules which can selectively inhibit pathways with temporal control. It can be argued that biologically active natural products are a privileged starting point in this endeavor based on the fact that they have been selected by evolution to bind to given protein motifs [2,3]. Given the prevalence of domain shuffling in protein evolution, it can be anticipated that the privileged scaffolds that have evolved to inhibit a given protein may be suitable for the inhibition of functionally distinct proteins that have a homologous domain. With these considerations, we have been particularly attracted to the resorcylic acid lactones (RALs – Fig. 1). Zearalenone was the first member of this family to attract the attention of both chemists and biologists for its potent agonism of the estrogen receptor which is a transcription factor. It was shown to adopt a conformation which mimics the steroid and competes with estradiol for the estrogen receptor. More recently, radicicol and the cis-enone resorcylides such as hypothemycin or LL-Z1640-2 have taken the center stage for their potent inhibition of HSP90 and MAP kinases, respectively. Despite the lack of structural similarity between radicicol or LL-Z1640-2 and ATP, a crystal structure of each compound in complex with their respective target has shown them to be bound in the ATP-binding pocket (Fig. 2) [4,5]. Pochonin C which is structurally closely related to radicicol was discovered from a screen for herpes simplex virus (HSV) replication, while aigialomycin D was reported to be an inhibitor of the malaria parasite. However, the precise targets of these compounds have not been reported. Thus, while zearalenone targets a hormone transcription factor, radicicol targets an ATPase and the cis-enone resorcylides target kinases. Based on the breadth of biological activity, we have been particularly interested in developing diversity-oriented syntheses which would enable us to broaden the chemical space surrounding this pluripotent natural product motif. We have recently reviewed the chemistry and biology of RALs [6]; herein we review our own synthetic efforts towards the resorcylic acid lactones and discuss their potential therapeutic application.
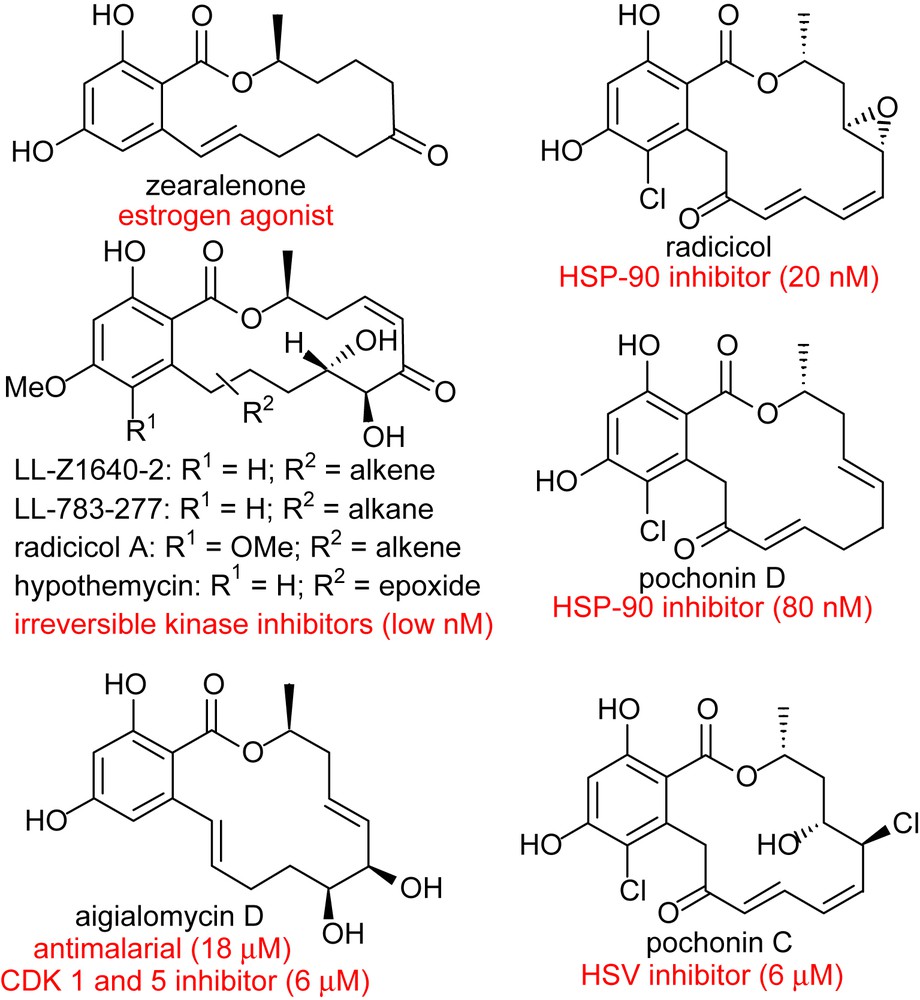
Selected members of the resorcylic acid lactone family of natural products.
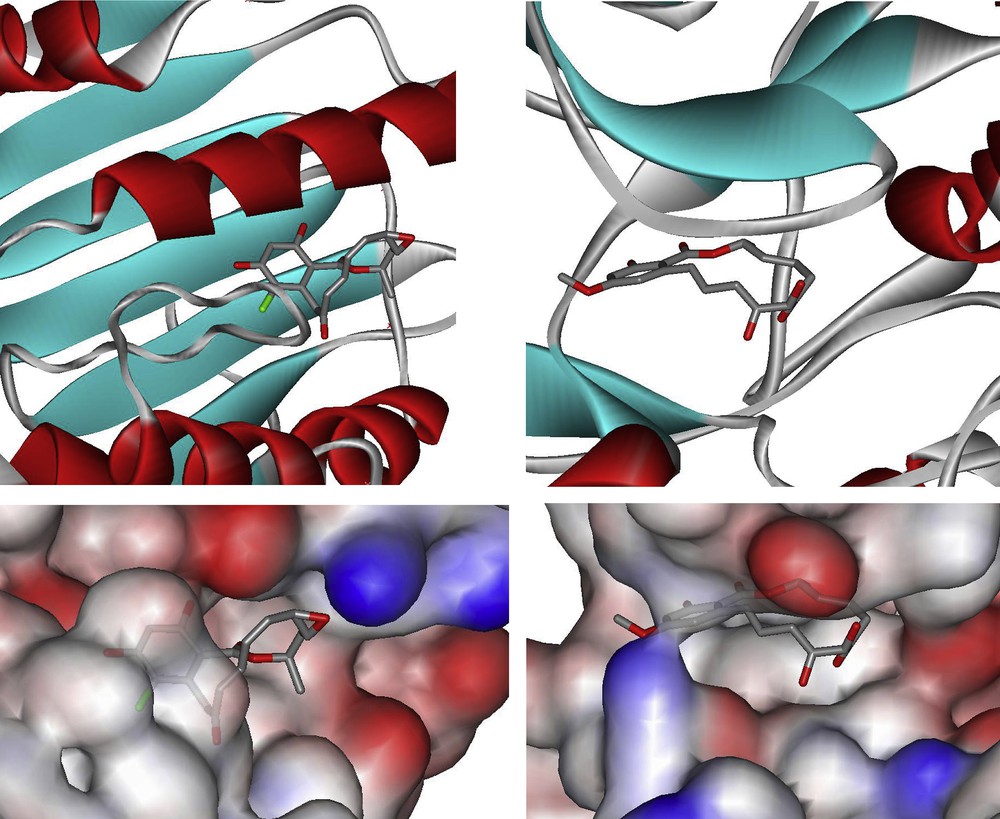
Co-crystal structure of radicicol bound to HSP90 (pdb ID: 1bjq) and LL-Z1640-2 bound to ERK2 (pdb ID: 2e14).
2 Radicicol – a potent HSP90 inhibitor
The heat shock protein 90 (HSP90) is a chaperone responsible for the folding and maturation of its client proteins and is one of the most abundant protein in cells (1–2% of proteomic mass). Despite the seemingly ubiquitous function of this constitutively expressed chaperone, its role in stabilizing conformationally labile proteins has implications in pathologies ranging from oncology to neurodegenerative diseases and has emerged as one of the most exciting therapeutic targets in recent years [7,8]. Most of HSP90's endogenous client proteins are key regulators of cell signaling which are destabilized and degraded in the absence of HSP90's chaperoning activity.1 The dependence of transformed cells on HSP90 is further accrued by the fact that many oncogenic mutations, while increasing the activity of pro-growth signaling pathways, are less stable than their wild-type counterpart and have an increased dependence on HSP90's chaperoning activity [9]. A clinically relevant example is the heightened dependence on HSP90 of drug resistant mutants of Bcr-Abl. Indeed, HSP90 inhibitors in combination with Abl inhibitors remain effective against cell lines bearing that mutation [10,11]. Accordingly, HSP90 inhibition might provide a broader, more effective target for anticancer treatment than more focused therapies targeting single oncogenic proteins, and can work synergistically with cytotoxics or focused inhibitors. Aside from its involvement in oncogenic processes and potentiating the evolution of drug resistance, HSP90 is also implicated in regulating the fate of conformationally unstable proteins leading to neurodegenerative diseases. Indeed, HSP90 inhibitors have been shown to reduce protein aggregates in cellular and animal models of Huntington [12], spinal and bulbar muscular atrophy [13], Parkinson [14], and other Tau protein related neurodegenerative diseases [15]. These facts have stimulated tremendous research to obtain inhibitors with good pharmacological properties for clinical application. The most advanced candidate is 17AAG, a semisynthetic analogue of geldanamycin, which is currently in multiple phase II studies, however its hepatotoxicity is limiting its therapeutic potential [16]. Rational design and high throughput screening have led to the discovery of several heterocyclic motifs [17–19] with good pharmacological profile.
Radicicol remains amongst the highest affinity ligand for HSP90 (20 nM) [5]. However, radicicol suffers from two pharmacological liabilities: a conjugated dienone which acts as a Michael acceptor and a strained allylic epoxide. Indeed, while radicicol is effective at the cellular level, it is not in animal models due to metabolic instabilities. It has been shown that thiols such as DTT readily react in a conjugate addition to yield a compound which is inactive [20]. This limitation was partly overcome by converting radicicol's ketone functionality to an oxime which attenuated its Michael acceptor properties and was shown to be active in mouse xenografts [21,22]; however clinical development for this compound series was interrupted due to ocular toxicity. Based on the fact that other HSP90 inhibitors do not engender this toxicity, it can be ascribed to an off-target activity which may be related to the epoxide serving as an alkylating agent. Mindful of the problematic issues surrounding the labile epoxide of radicicol, Danishefsky and coworkers reported an analogue wherein the epoxide was replaced by a cyclopropyl group and showed this compound to be nearly as active as radicicol [23].
Based on the therapeutic potential of HSP90 inhibition and the different biological activity of stucturally related pochonin C, we developed a synthesis to access both of the natural products [24,25]. An important criterion in our planning was the possiblity to use high throughput chemical synthesis techniques. It was clear at the onset of this program that the halohydrin of pochonin C could originate from a stereospecific opening of radicicol epoxide. As shown in Scheme 1, a key feature of the synthesis was to mask the conjugate alkene in the form of a thioether which could be converted to the α,β-conjugate system upon oxidation followed by elimination. The remaining disconnections involved a Mitusnobu reaction to form the ester and a ring-closing metathesis. Starting from the commercially available Weinreb amide 4, which was engaged in a chemoselective S-alkylation of 3-hydroxythiophenol followed by an O-alkylation to Merrifield resin, polymer-bound Weinreb amide 5 was obtained in one pot. While the selective oxidation to the sulfoxide proved difficult with traditional oxidants such as mCPBA and DMDO, it was found to proceed smoothly with H2O2 in the presence of HFIP [26] without any overoxidation to the sulfone. These conditions were compatible with solid-phase chemistry. Activation of the sulfoxide with trifluoroacetic anhydride in the presence of tributyl(allyl)tin afforded the Pummerer allylation product 6 in excellent yield. Intermediate 7 was obtained in two steps from 1 via Mitsunobu esterification/phenol protection. The use of a polymer-bound version of DIAD precluded the use of traditional purifications. Deprotonation of 7 with LDA and reaction with the polymer-bound Weinreb amide 6 afforded the cyclisation precursor 8 which was released from the resin prior to the metathesis. While the metathesis reaction could be achieved on the resin, carrying out the cyclisation on the triene rather than on the polymer-bound diene was preferable as it afforded exclusively compound 9 with the desired olefin geometry. Chlorination of the aromatic moiety with SO2Cl2 led to concomitant opening of the epoxide, thus affording pochonin C after deprotection of the two MOMs. Treatment of pochonin C with potassium carbonate led to radicicol in good yield.
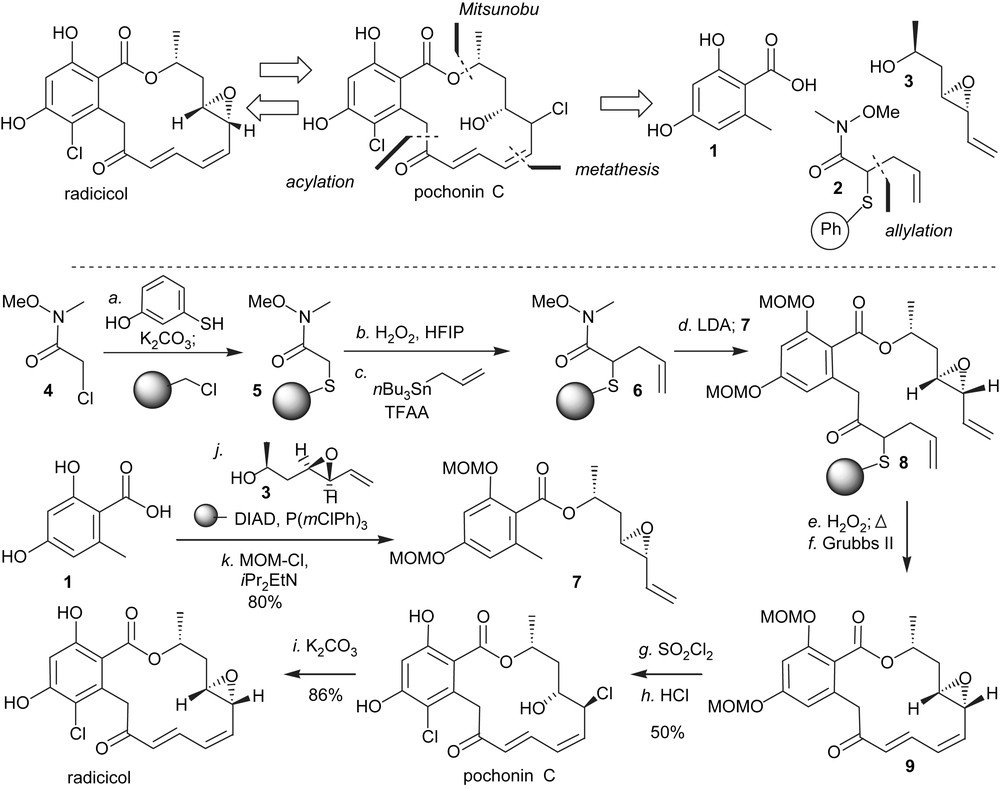
Polymer-assisted synthesis of pochonin C and radicicol.
Despite the structural similarity between pochonin C and radicicol, the NMR patterns of these two compounds were dramatically different. As shown in Fig. 3, radicicol exhibited strong NOE between the protons of methyl group (C1) and the protons of C8 and C11 in accordance with its conformation bound in the active site of HSP90 [5]. In comparison, pochonin C showed strong NOE between the protons of C5 and C9 while no NOE between the protons at C1 and C8/C11 suggesting a more planar and less organized conformation. The difference in biological activity reported by Hellwig and coworkers [27] for pochonin C and radicicol may be in part rationalized by their different conformational landscape rather than the difference in functionalities (epoxide vs halohydrin). In collaboration with Karplus and coworkers, the conformational landscape of radicicol and other resorcylides was calculated and it was found that indeed the lowest energy conformer of radicicol had a very similar conformation to the bioactive one while the corresponding compound having a diol instead of an epoxide had a minimum energy barrier of 2.7 kcal to adopt the bioactive conformation [28]. Further analysis of a series of resorcylides led to the identification of pochonin D as having a low energy barrier to the bioactive conformation, thus motivating us to pursue its synthesis. The synthesis of pochonin D followed the same logic as the one of pochonin C, namely a disconnection into three fragments (Scheme 2). The Weinreb amide fragment 13 was prepared in three steps using the polymer-bound thioether 5. The use of polymer-bound reagents [29] obviated the need for regular chromatography and afforded pochonin D in six steps from acid 1 and in 31% overall yield with only one traditional purification. Importantly, despite its simpler structure, pochonin D was indeed a good ligand for HSP90 with an affinity of 80 nM (compared to 20 nM for radicicol). The difference in affinity is consistent with the calculated 1.2 kcal change in internal energy of pochonin D upon binding. The C4–C5 olefin of pochonin D could be regioselectively epoxidized using dimethyldioxirane to obtain pochonin A which was also found to be an HSP90 ligand; however, it was not superior to pochonin D and presents a labile epoxide [30].
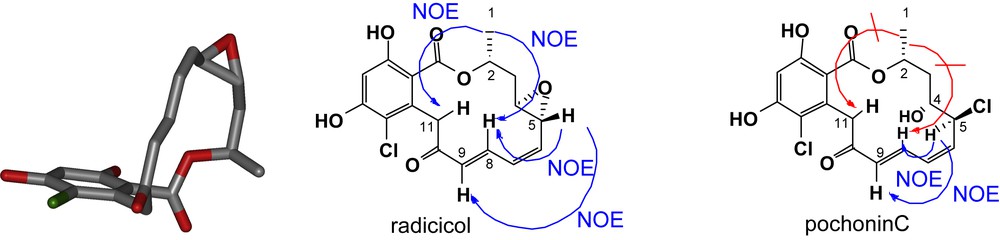
Conformational aspects of radicicol and pochonin C. Conformation of radicicol bound to HSP90 (left, pdb structure ID 1bgq), NOESY measured for radicicol (middle) and pochonin C (right) [numbering according to pochonin C definition].
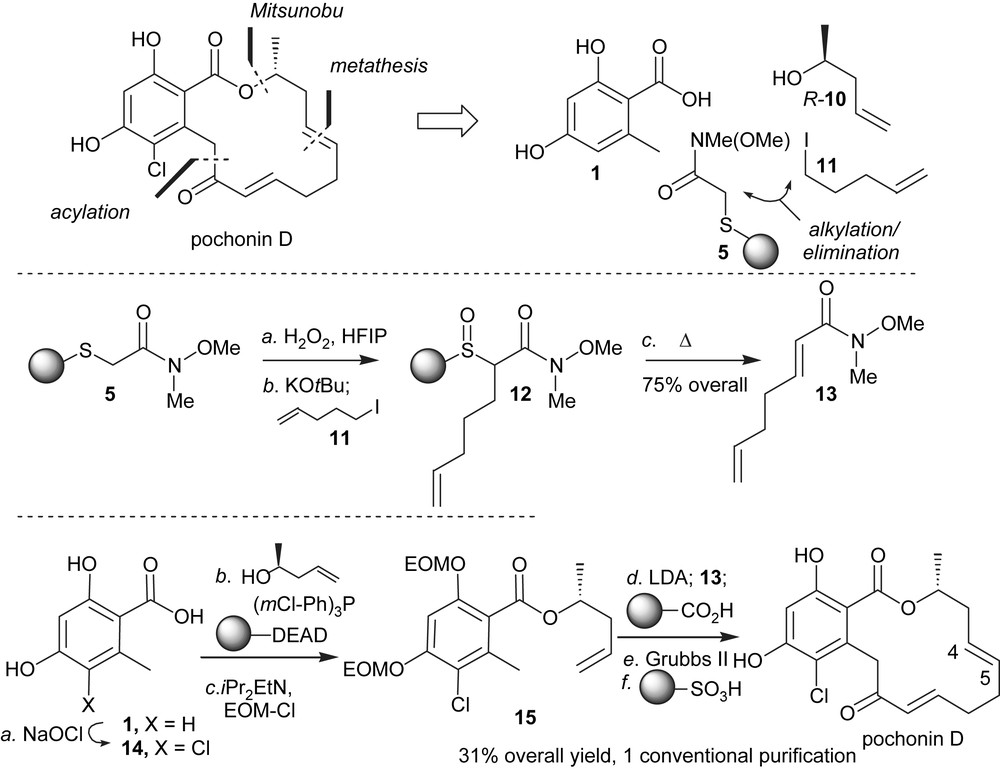
Polymer-assisted synthesis of pochonin D.
The fact that the synthesis of pochonin D required only six steps and one conventional purification rendered it amenable to combinatorial synthesis. As shown in Scheme 3, a library represented by the general structure 16 which bears five points of diversity was prepared [31] from intermediates 17, obtained through the aforementioned chemistry using different alcohols. The diversity of the library included the following modifications: the group on C2 (R1, both stereochemistries are present in natural resorcylides, however only with a methyl substituent); the meta position on the aryl ring (R2, a number of natural resorcylides bear a chlorine at that position); the substitution of the para phenol (R3, a number of natural resorcylides bear a methyl group at that position); the C4–C5 olefin (R4) which was converted to a diol or an epoxide; the C10 carbonyl (R5) which was reduced or converted to oximes and the olefin C8–C9 which was reduced. While not all permutations of the library were pursued, over 100 compounds were prepared using combinations of the chemistry shown in Scheme 3. Screening of this library against HSP90 revealed important structure–activity relationships which led us to the discovery of a compound with 10 0-fold higher activity than pochonin D in cellular assays and which was found to lead to tumor regression in a mouse xenograft bearing a breast tumor cell line [32]. With the goal of exploring the suitability of these compounds for kinase inhibition, a representative subset of the library (84 compounds) was tested for its inhibition in a panel of 24 kinases which led to the identification of 12 compounds with greater than 50% inhibition for one kinase at 10 μM. The high hit rate in this resorcylide library (14%) testifies to the potential of the RAL scaffold for kinase inhibition. Importantly, the promising kinase leads that were identified were not HSP90 inhibitors and showed diverse selectivity profiles of enzyme inhibition amongst the 24 tested kinases.
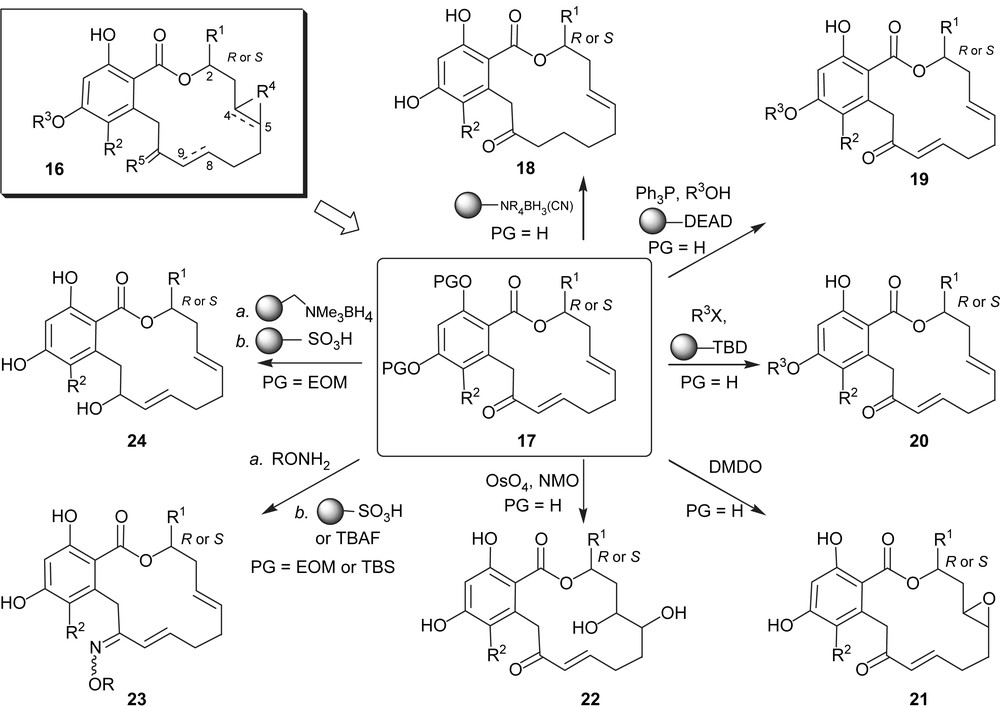
Diversity-oriented synthesis of a library of pochonins.
3 Aigialomycin D – an inhibitor of the malaria parasite (Plasmodium falciparum)
As part of our program to further the diversity of RALs beyond the naturally available analogues in the hope of finding new ATPase and kinase inhibitors, we became interested in the aigialomycins [33]. As for the pochonins, our goal was to develop synthetic protocols which were sufficiently flexible as to be carried out in a combinatorial fashion. As shown in Scheme 4, the molecule was disconnected in two fragments using a metathesis to close the macrocycle [34], and a seleno or thioether at the benzylic position to facilitate alkylation chemistry at that position and provide an attachment point to a resin for solid phase synthesis. The synthesis in solution started from benzoic acid 1 which was converted to the selenoether 28 in three steps. Deprotonation of the benzylic position with LDA followed by addition of bromide 26 afforded the alkylated product which was cyclized using Grubbs' second generation catalyst to obtain macrocycle 29 in excellent yield. Oxidation/elimination of the selenide followed by deprotection afforded aigialomycin D in seven steps from acid 1. The same chemistry was found equally productive with alkyl bromide 27 containing the allylic epoxide, however opening of the epoxide in macrocycle 30 under a variety of conditions failed to give the 1,2-cis diol and led in all cases to SN2′ reactions. While this route cannot be used to access aigialomycin D, this diversion proved quite general for different nucleophiles such as alcohols, cyanide or azide thus obtaining new RAL analogues 31. Importantly, as in the case of the pochonins, it was shown that the chemistry could be carried out on solid phase by replacing the phenyl selenide with a polymer-bound thioether. Thus benzyl chloride 32 was loaded on a thiol resin and both phenols were protected to obtain polymer-bound intermediate 33. Alkylation of the benzylic position with five different alkyl bromides (34) followed by RCM afforded macrocycles 35 which could be released from the resin either via oxidation/elimination or under reductive conditions to obtain aigialomycin analogues 36 and 37, respectively, after deprotection with sulfonic acid resin. It is interesting to note that the metathesis conditions that were successfully used with the selenoether in solution (toluene, 80 °C) proved to be ineffective on solid phase with the thioether. Excellent yields were nevertheless obtained by carrying out the RCM in CH2Cl2 at 120 °C using microwave irradiation. This pilot library was screened against a panel of kinases revealing that aigialomycin D is a moderate inhibitor of CDK1/cyclin B and CDK5/p25 at 5.7 and 5.8 μM, respectively, as well as GSK-3 at 14 μM but much less of PfGSK-3, the Plasmodium homologue of GSK-3. Closely related analogues were not inhibitors of these kinases suggesting that the particular functionalities present on aigialomycin D are important for its activity. It was further confirmed that aigialomycin D is not an inhibitor of HSP90 [23]. HSP90's ATP-binding pocket targeted by radicicol and pochonin D has a specific fold (Bergerat fold) [35] that is present in a superfamily which includes functionally diverse proteins such as DNA topoisomerase II, helicase, MultL and histidine kinase and is characterized by an L-shaped binding pocket [36]. The ATP-binding pocket of kinases on the other hand lies between the two lobes of the kinase fold which are connected by a so-called hinge region and is characterized by a more linear groove than the Bergerat fold. The selectivity of aigialomycin D for kinases rather than HSP90 or other ATPases bearing a Bergerat fold can be rationalized by the fact that this macrocycle's ground state conformation is planar and would need to overcome a minimum energy barrier of 14.7 kcal to adopt the L-shape conformation which fits in the Bergerat fold. Whether the antimalarial activity ascribed to aigialomycin D [33] can be attributed to kinase inhibition remains to be proven but its activity for selected kinases further supports the privileged nature of the RAL scaffold.
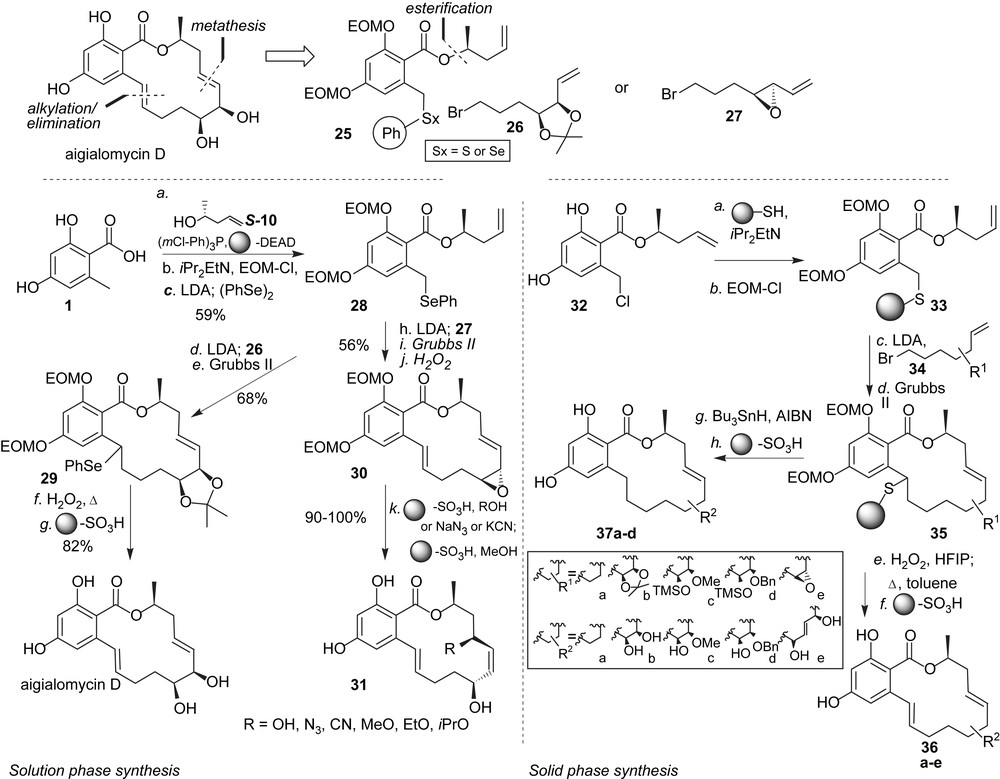
Solution- and solid-phase diverted syntheses of aigialomycin D and analogues.
4 Radicicol A: a potent MAP kinase inhibitor
Radicicol A (Fig. 1) belongs to the subset of RALs bearing a cis-enone and was first reported by researchers from Sandoz who identified this fungal metabolite from a screen for IL1β inhibition [37]. While it was observed that this compound accelerated the degradation of specific mRNA sequences including IL1β's and inhibited tyrosine phosphorylation, its precise molecular target was not identified [37,38]. Subsequently, two other related resorcylic acid lactones containing a cis-enone were reported to be potent irreversible yet selective kinase inhibitors [39,40]. LL-Z1640-2 (Fig. 1) was identified in a screen for TAK1 inhibition [39]. Importantly, it was shown that radicicol and zearalenone had no appreciable activity in this assay while LL-Z1640-2 had an IC50 of 8.1 nM. The authors further showed this compound to be competitive with ATP and to irreversibly inhibit TAK1. TAK1 is an MAPKKK involved in the p38 signaling cascade for proinflamation signals such as cytokines. In evaluating the selectivity of this compound for TAK1, the authors showed this compound to be 50-fold less active against MEK1 (411 nM), another MAP kinase and having no inhibitory effect on other MAP kinases such as MEKK1, ASKN and MKK4. The authors also demonstrated LL-Z1640-2 to effectively prevent inflammation in an animal model (topical application). LL-783-277 (Fig. 1) was found by researchers at Merck to be a potent and irreversible inhibitor of MEK1 (4 nM) [40]. The authors reported that hypothemycin (another cis-enone RAL, Fig. 1) was also an inhibitor of MEK (15 nM) and showed that the cis-enone funtionality was essential for their activities. It was later shown by Santi and coworkers that this irreversible inhibition observed can be attributed to a Michael addition onto the cis-enone of a cysteine residue present in the ATP-binding pocket of a subset of kinases [41]. A structure-bioinformatics analysis of the kinome revealed that 46 out of the 510 identified kinases contain a cysteine residue adequately positioned for reaction. Nevertheless, amongst 16 tested kinases containing this conserved cysteine, hypothemycin had a Ki in the low nanomolar range for five kinases (MEK1, MEK2, FLT1, FLT3 and KDR), a high nanomolar Ki for one kinase (TRKB), low micromolar Ki for seven of them (ERK1, ERK2, PDGFRα and β, PKD1, MAPKAP5, TRKA and SRC) while it inhibited the remaining two kinases only weakly (GSK2α and β). The difference in Ki clearly reflects the fact that, while the ATP-binding pockets are highly conserved and contain the required cysteine, hypothemycin is able to discriminate with some efficiency amongst the 16 tested kinases. These results provide a mechanism for earlier observations that hypothemycin is an inhibitor of the ras-signaling pathway [42] and inhibits the production of several cytokines (IL2, Il6, Il10, IFNγ and TNFα) [43]. Hypothemycin was found to inhibit several oncogenic cell lines dependent on kinase activation [41] as well as tumor growth in animal models [42]. As both MEK and TAK kinases are part of the 46 kinases containing the suitably positioned cysteine, the irreversible inhibition previously observed for LL-783-277 and LL-Z1640-2 can be rationalized by the same mechanism. More recently, Santi and coworkers reported semisynthetic derivatives of hypothemycin leading to some preliminary structure–activity relationship [44]. The co-crystal structure of LL-Z1640-2 with ERK2 (Fig. 2) provided the first structural information for this class of compounds and a rational for the importance of the alcohol stereochemistry [4].
The disconnections that were utilized for the synthesis of radicicol A and related cis-enone RALs are summarized in Scheme 5 [45]. A significant challenge is to control the cis-geometry of the enone as it can isomerize to the thermodynamically more favorable trans-isomer which is known to be significantly less active [40]. We thus planned to reveal the enone at the latest stage through a selective allylic oxidation and to form the cis-alkene using a vinyl lithium addition onto an aldehyde. The molecule was disconnected into three fragments of even complexity: 38, 39 and 40 where either the aldehyde or the iodide of fragment 40 would need to be masked depending on the order of assembly. While the order of coupling of these three fragments is conceptually possible in all permutations, we reasoned that starting the sequence with the coupling of 39 and 40 would allow the use of a fluorinated protecting group for the alcohol in 39 enabling the use of the fluorous isolation technology [46,47] until the penultimate cyclisation. Two features made this strategy particularly attractive, first and foremost, it has been shown that multiple components tagged with different lengths of fluorinated tags can be carried through a synthesis as a mixture and ultimately resolved using fluorous chromatography [48,49]. Second, this technology has now been automated making it amenable to high throughput synthesis [50]. The cis-vinyl bromide functionality was obtained from terminal alkene S-10 via cross metathesis with vinyl boronate and treatment with bromine under basic conditions thus affording 39 in good yield. Fragment 44 was prepared in three steps from the acetonide-protected 2-deoxy-d-ribose 43 and engaged directly in the coupling with 39. Protecting group manipulations afforded intermediate 46, which was then used to alkylate the different benzylic thioethers 38. Oxidation/elimination of the selenoether afforded compounds 47a–c. It is noteworthy that this reaction sequence starting from S-10 did not require a single traditional workup and that the products were isolated by “catch and release” extractions on fluorinated silica. The PMB group was removed under the action of DDQ and again, the byproducts of these reactions were easily sequestered using fluorous solid-phase extraction while the 2-(trimethylsilyl)ethyl ester was removed with TBAF. The hydroxyacids thus obtained were engaged in Mitsunobu macrocyclizations using fluorous-tagged triphenyl phosphine and diisopropyldiazodicarboxylate yielding the macrocycles 48a–c after a fluorous solid-phase extraction. All protecting groups were removed with BCl3 and the allylic alcohols were then selectively oxidized with the polymer-bound version of IBX affording radicicol A, LL-Z1640-2 and radicicol A analogue 49c in nearly quantitative yield following a simple filtration.
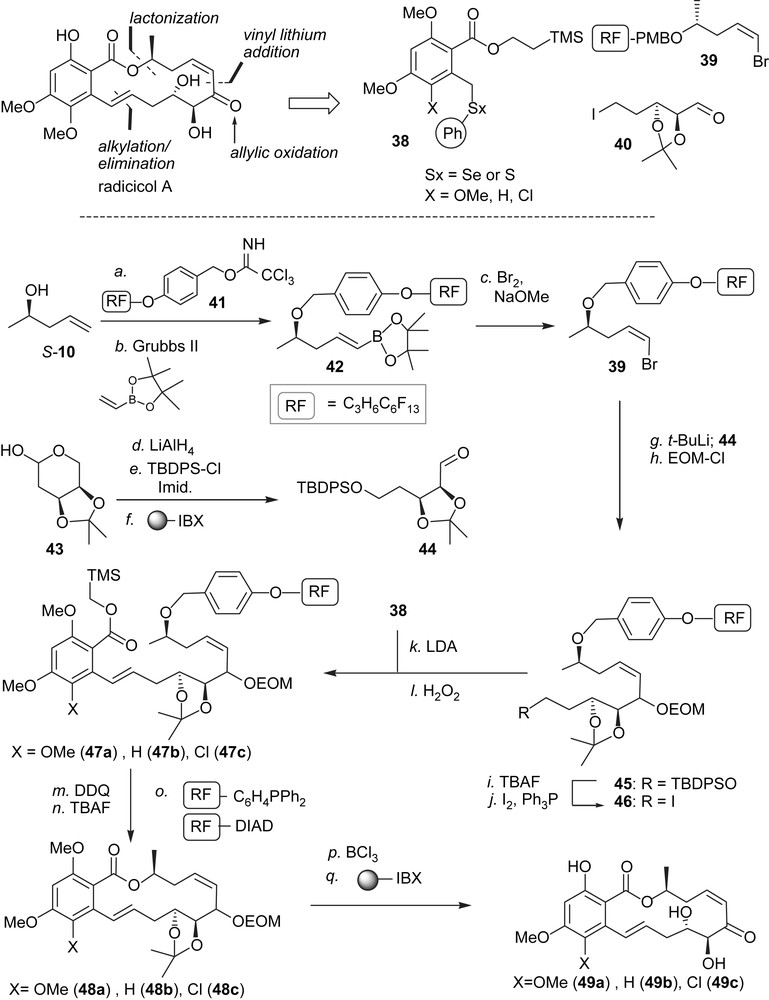
Fluorous synthesis of radicicol A, LL-Z1640-2 and chlorinated analogue.
Radicicol A was found to exhibit potent inhibitory activity (low nM) against VEGF-R2, and MEK and moderate activity against VEGF-R3, FLT3 and PDGFR-β when tested in a panel of 24 kinases. The demonstration that radicicol A inhibits key kinases involved in MAPK cascades can rationalize the original observation that it accelerates the degradation of Il1β's RNA as there is mounting evidence that MAP kinases are involved in the regulation of protein translation by controlling the stability of specific mRNAs [51,52]. However, from a therapeutic perspective, radicicol A may not be suitable as we found it quite prone to form quinone under mild oxidative conditions. On the other hand, analogue 49c was not prone to quinone formation due to the chlorine substitution on the aryl ring. We found this compound to be nearly as potent as radicicol A. Evaluation of this inhibitor in a larger panel of kinases (127) showed it to be quite selective for therapeutically important kinases (MEK1, VGEFRs, and to a lesser extent PDGFR-α and β, Fig. 4). All inhibited kinases bear the suitably positioned cysteine residue, yet the discrimination among the subset of potential kinases indicates that unique interactions with its target are necessary for the Michael addition to proceed and that its selectivity could be honed or tuned to other kinases. The potential efficacy of this compound was also demonstrated in cells by measuring the level of VEGF-R2 autophosphorylation in the presence of its ligand (VEGF165). From a chemotherapeutic perspective, the fact that the multiple kinases inhibited by the cis-enone containing RAL are involved in pro-growth signaling pathways and angiogenesis endow these compounds with potential to have a broader therapeutic application than selective kinase inhibitors. The first broad spectrum kinase inhibitor (sunitinib – SU11248) targeting VGFR1-3, PDGFRs, KIT, FLT3, RET and CSF1R has been approved in 2006 for the treatment of renal and gastrointestinal cancers [53]. An irreversible kinase inhibitor targeting the EGFRs is currently in phase II clinical trials. This inhibitor targets the same kinase as two approved reversible inhibitors (Iressa and Tarceva). Interestingly, it has been shown that this inhibitor remains effective against mutants that have evolved a resistance to the reversible inhibitor [54]. In addition to the therapeutic potential of this family of natural products, the irreversible nature of these inhibitors also offers a new lead structure for the design of selective inhibitors towards engineered kinases [55,56] as well as for chemical proteomics [57].
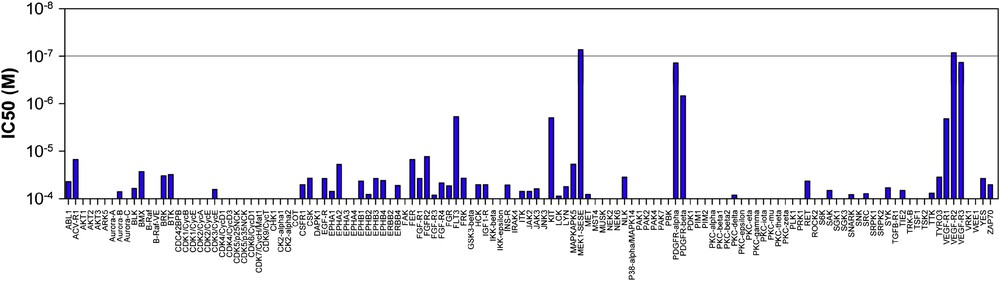
Profile of compound 49c against a panel of 127 kinases. In this assay, only VEGF-R2 and MEK are inhibited below 100 nM.
5 Chemical synthesis methodology to access all the functionalities present in the RALs
Beyond the aforementioned syntheses, part of our goal was to develop a general methodology suitable to access the diversity of functionalities present in the RALs which is compatible with high throughput synthesis techniques. In that regard, the thioether functionality is particularly attractive as it offers interesting reactivity but also provides an immobilization strategy. This was exploited in the synthesis of radicicol and aigialomycin D. We have further explored the potential of this linker as shown in Scheme 6. Thus thioether 38 can be oxidized to the sulfoxide 50 which participates in alkylation chemistry under milder conditions than the thioether 38. Conversely, sulfoxide 50 can also participate in allylation chemistry under palladium catalyzed π-allyl transfer conditions thus affording 55. Product 51 or 55 can be converted to the styrenyl functionality (52) present in a number of RALs. Alternatively, they can also be converted to alkane 53 or ketone 54 under Pummerer condition in the presence of a reducing agent or an acid, respectively. Last but not least, S-alkylation of thioether 38 affords the sulfur ylide 56 which rapidly reacts with aldehydes to afford the benzylic epoxide functionality (57) present in hypothemycin and aigialomycins.
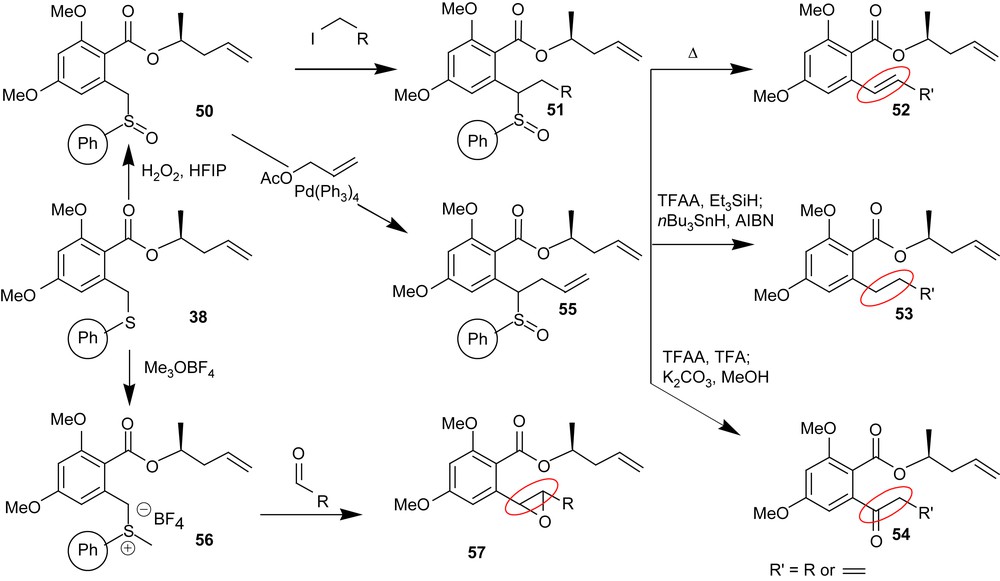
Divergent chemistry of the thioether linker.
6 Conclusion and future perspective
It is clear from the diverse biological activity of natural and synthetic resorcylides that this scaffold is a privileged starting point for the discovery of new inhibitors. The thioether methodology developed in our laboratory provides access to all the functionalities present in natural RALs and is compatible with high throughput synthesis. From a therapeutic perspective, HSP90 and kinase inhibition are on the center stage of chemotherapeutic development and the demonstration that several RALs are effective in animal model raises the hope that a natural member, semisynthetic derivative or synthetic analogue may enter clinical development.
Acknowledgments
The work reviewed in this manuscript was partly supported by grants from the Agence National de la Recherche (ANR), ARC, COST (network D28), HFSP. MENRT and BDI fellowships are also gratefully acknowledged.
1 For a complete list of HSP90's clients, see: <http://www.picard.ch/downloads/Hsp90interactors.pdf>.