1 General introduction
Polyketide natural products are a structurally diverse family of compounds showing a wide range of biological activities. Some examples include antibiotic erythromycin A [1], immunosuppressant rapamycin [2], and antitumor antibiotic doxorubicin [3] (Fig. 1). Specifically, ansamycin antibiotics have become an important class of naturally occurring bioactive compounds with the discovery of the antibacterial properties of rifamycin B in 1959 [4]. Specifically, rifamycins have been used to treat tuberculosis, leprosy and AIDS-associated mycobacterial infections [5]. These macrolactams are named because of their “basket-like” structure consisting of an aromatic chromophore connected at nonadjacent positions by an aliphatic chain, like a handle or ansa [6]. The structure of the aromatic moiety distinguishes the two types of these natural products (Fig. 2).
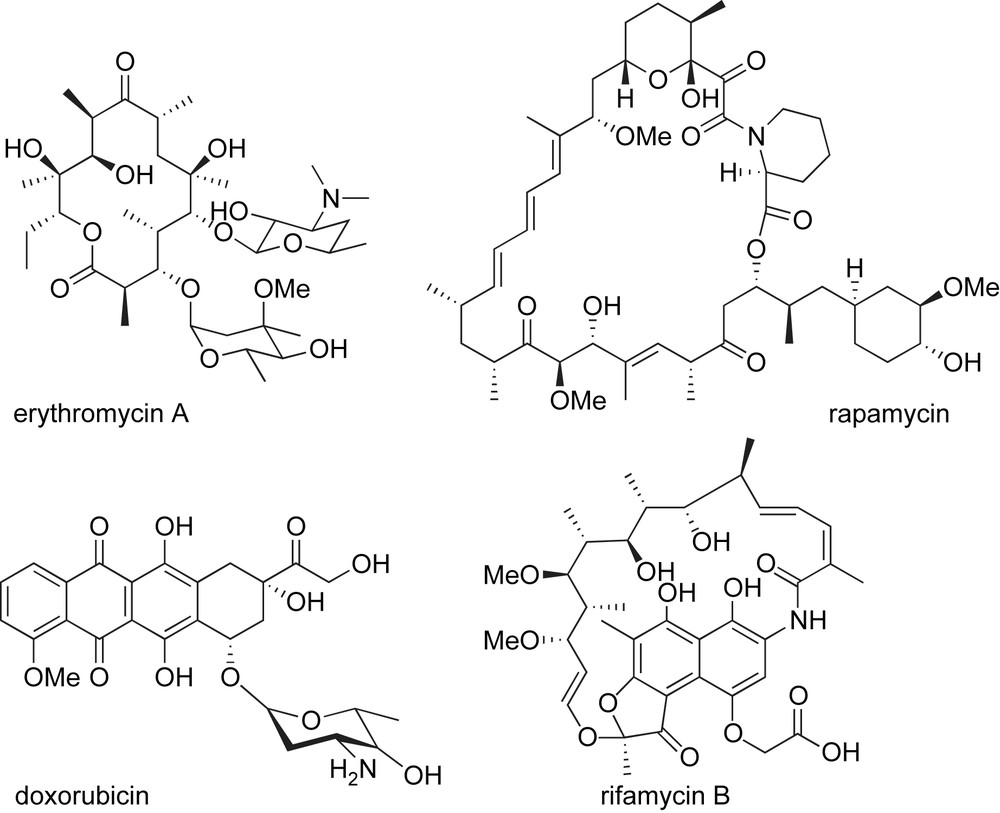
Representative structures of erythromycin A, rapamycin, doxorubicin, and rifamycin B.
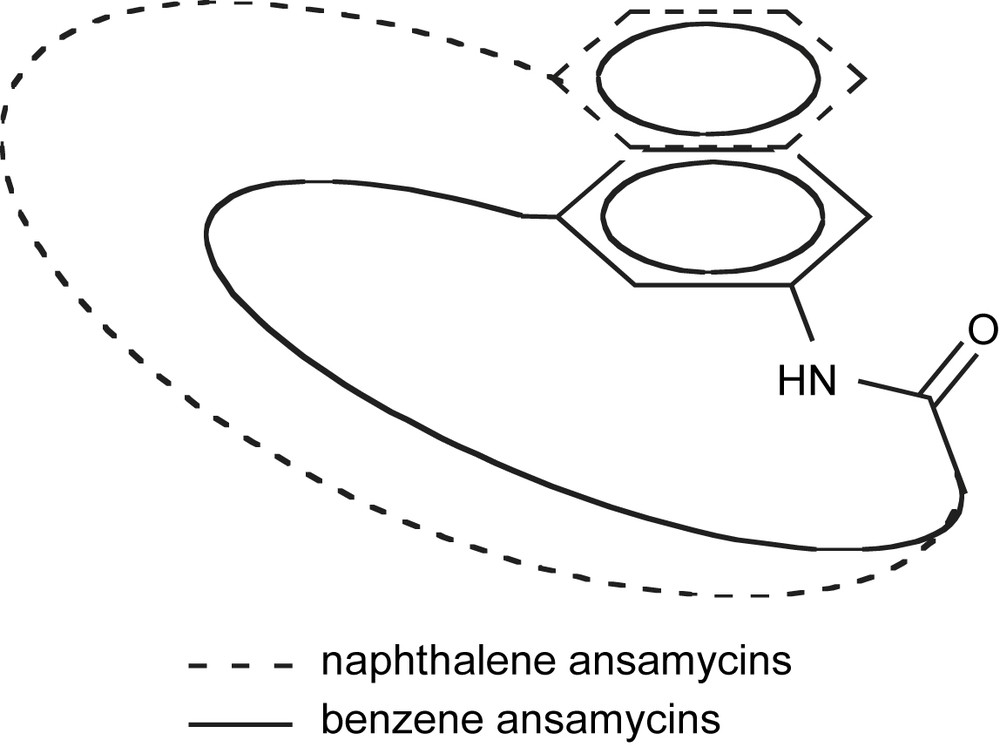
“Basket-like” structure of ansamycins.
Naphthalenic ansamycins (rifamycin, streptovaricin, naphthomycin) exhibit strong antimicrobial activities [7], whereas benzenic ansamycins (geldanamycin, herbimycin, trienomycins) have demonstrated their potential as antineoplastic agents [8]. An additional mode of differentiation can be made within the ansa chain (C15- and C17-) of the benzene ansamycins (Fig. 3). The geldanamycin type ansamycins IIa are comprised of a 19-membered lactam that contains an isolated trisubstituted olefin. Alternatively, the 21-membered macrolactam of mycotrienin type ansamycins IIb consists of an all (E)-triene system; this subclass of ansamycins will be subsequently referred to as ansatrienins.
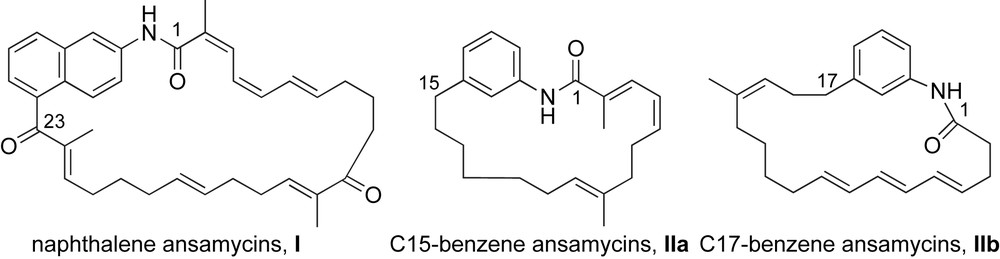
Representative structures of naphthalene ansamycins I and C15- and C17-benzene ansamycins II.
1.1 Biosynthesis of benzenic ansamycins
The biosynthesis of ansamycin natural products occurs via the common precursor 3-amino-5-hydroxybenzoic acid (AHBA), Fig. 4 [9]. The ansa chain of geldanamycin is assembled from AHBA by sequential addition of 4 propionate, 2 glycerate/glycolate and 1 acetate groups [10]. These seven chain-elongation steps utilize substrate condensation and various reductions or dehydrations to produce the nascent polyketide framework. The methoxy carbons are derived from methionine while the carbamate carbon comes from the ureido group of citrulline. In a similar fashion, the aliphatic chain of ansatrienins is derived from 6 acetate and 2 propionate subunits [11]. Subsequent formation of a macrolactam between the terminal C1-carboxyl and 3-amino group of AHBA results in characteristic “basket-like” molecular conformation of the C15- and C17-benzene ansamycins.
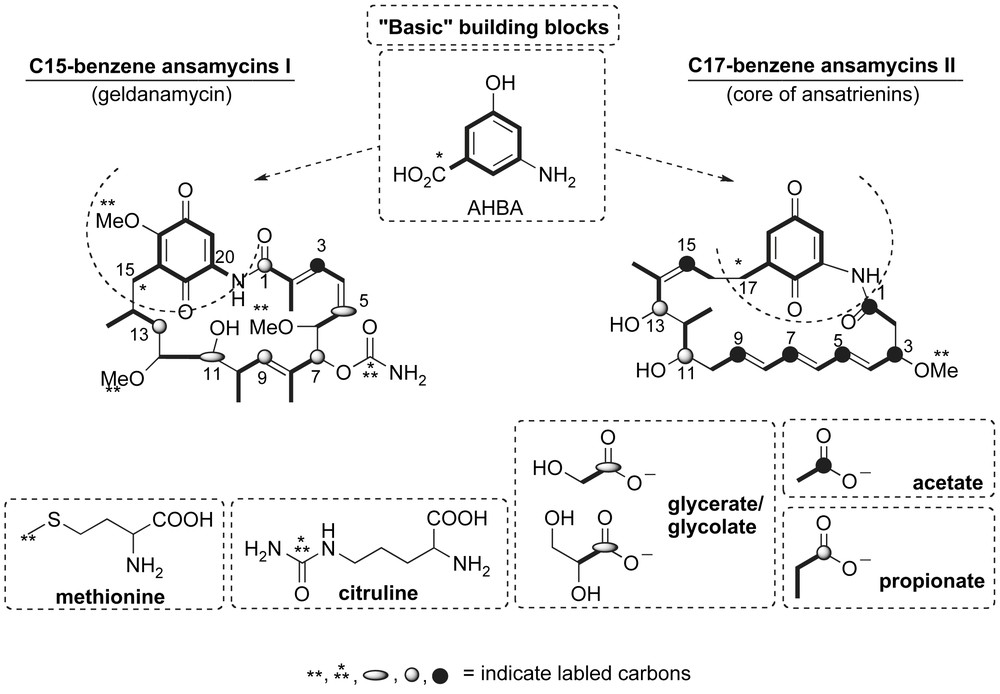
Biosynthesis of geldanamycin and ansatrienins.
In recent years, the production of novel structural variants of these macrolides has been achieved by genetic manipulation of these biosynthetic pathways [12]. For example, Kosan Biosciences reported that alteration of a gene cluster responsible for the biosynthesis of geldanamycin led to discovery of KOSN1559 with 8-fold greater binding affinity to Hsp90 than natural geldanamycin [13]. Some of these geldanamycin derivatives are currently being evaluated in clinical trials as antitumor agents [14]. Although the biological activity of C15- and C17-benzenic ansamycins have been reviewed [15], there are few reports on their chemical syntheses. Accordingly, the next two sections will provide a detailed overview of the chemical approaches and associated bioactivities of these 19- and 21-membered macrolactams.
2 C15-Benzoquinone ansamycin antibiotics
2.1 Isolation, structure and biological activity
The C15-benzoquinone ansamycins were discovered between 1970 and 2000 from several strains of Streptomyces and Nocardia in screens for antibacterial, antifungal, and antiviral compounds (Fig. 5). Structurally, these macrolactams contain six or seven stereogenic centers, a C7-carbamate, a trisubstituted double bond, and a C1–C5 dienoate. The different substitution patterns within the carbon skeleton at C6, C11, C12, C15, and C17 as well as the oxidation state of the aromatic chromophore define this class of polyketide antibiotics.
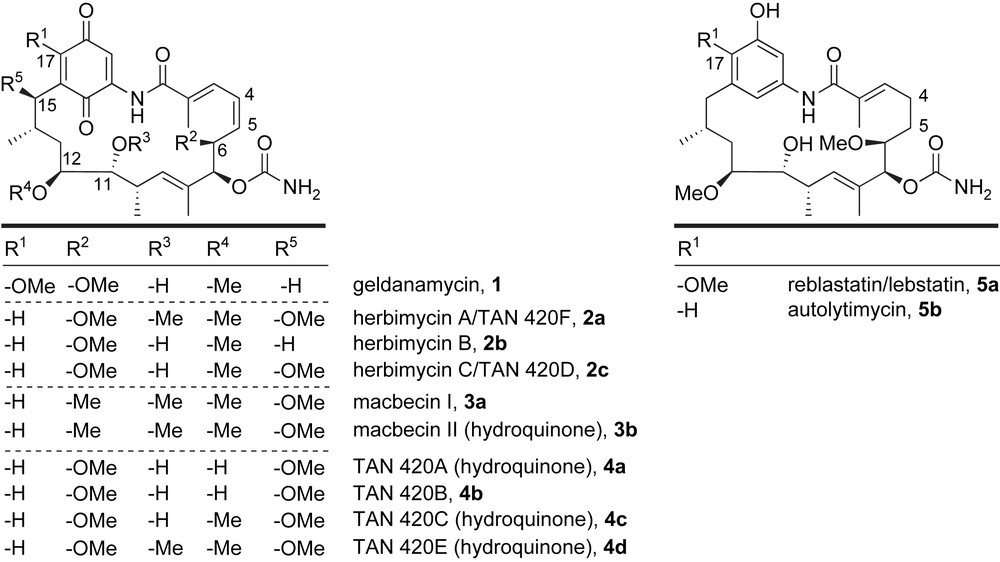
Molecular architectures of C15-benzene ansamycins.
Geldanamycin 1 was the first member of this family to be isolated from a strain of Streptomyces hygroscopicus var geldanus [16–18]. Initial screenings showed modest biological activity against Gram-positive bacteria, fungi, and parasites [17]. However, a report by Neckers [19] in 1994 found that geldanamycin was a potent inhibitor of heat shock protein 90 (Hsp90). The molecular chaperone Hsp90 regulates proteins in cell growth and survival as well as assisting the folding and maturation of client proteins such as signaling kinases and transcription factors. The Hsp90 client proteins seem to be involved in all hallmarks of cancer, thus their inhibition is supposed to disrupt multiple oncogenic signaling pathways. This finding initiated considerable interest in this class of natural products and resulted in synthesis of various analogues as potential anticancer agents. Most of these semi-synthetic derivatives are generated from functionalization at the C17-position [20] of the natural product and some have advanced to phase II clinical trials [14].
Herbimycin A 2a was isolated from the fermentation broth of S. hygroscopicus var geldanus strain AM-3672 in 1979 [21]. The structure and absolute configuration of herbimycin A were based on 1H and 13C NMR spectroscopic analyses and X-ray crystallographic studies [22]. Not surprisingly, this macrolide is quite similar to geldanamycin with retention of configuration at all shared stereocenters, differing only in the substituents at C11, C15, and C17 (Fig. 5). Along with herbimycin A, two other isomers were resolved, herbimycin B 2b [23] and herbimycin C 2c. [24]. These natural products are identical to the parent compound except for functionality at C11 and C15. Similar to geldanamycin, the herbimycins were shown to have antibacterial, antifungal, herbicidal, antiangiogenic and antitumor properties, including inhibition of Hsp90 [21,22,25].
Macbecin I 3a, along with its hydroquinone derivative macbecin II 3b, was isolated in 1980 from the strain of Nocardia sp. [26]. Their structures and absolute configurations were determined from partial degradation studies and X-ray crystal structure analysis [27]. The macrolide ring system and stereochemistry of macbecin I once again resemble the architecture of geldanamycin except for C6, C11, C15, and C17 positions. Interestingly, macbecin I is the only member of the C15-benzoquinone ansamycin family to contain a methyl group instead of a methyl ether at C6. Macbecin I and II were shown to have moderate activity against Gram-positive bacteria and fungi in vitro and good antitumor activity in vivo [26,27]. Only recently, macbecin I 3a has been evaluated as an Hsp90 inhibitor [28].
In 1986, TAN 420 series of ansamycins 4a–d were isolated from a strain of S. hygroscopicus [24,25b,29]. Three hydroquinone (A, C and E) and three related quinone (B, D and F) isomers were elucidated; however, only TAN 420A and TAN 420B were structurally different from other ansamycins with the presence of C12-hydroxy group. These natural products displayed biological activity against Gram-positive bacteria and fungi [29].
The most recent additions to the C15-ansamycin class of natural products, isolated from a culture of Streptomyces, are reblastatin 5a [30,31] and autolytimycin 5b [31,32]. In 2000, Takatsu and co-workers reported a benzenoid-type antibiotic that was very similar to geldanamycin except for the aromatic moiety (phenol versus quinone) and saturation of C4–C5 olefin within the C1–C5 dienoate. In the original report, reblastatin was shown to exhibit antitumor activity [30]. The related natural product autolytimycin 5b was isolated in 2001 from a strain of Streptomyces autolyticus JX-47 [32]. Autolytimycin differs structurally from reblastatin in the aromatic region, specifically at C17, and was shown to possess activity in a cell based oncostatin M signaling assay [31]. The architectural similarities of reblastatin and autolytimycin to geldanamycin and other members of the ansamycin family have led to exploration of these molecules as potential Hsp90 inhibitors [13].
2.2 Introduction: synthetic approaches toward the C15-ansamycins
This section provides a comprehensive, chronological overview of synthetic approaches developed toward the benzenic ansamycins. Specifically, previous syntheses of geldanamycin 1, herbimycin A 2a, (+)-macbecin I 3a, and reblastatin 5a are presented. In all, there are 12 reports from nine different groups detailing total and formal syntheses. Reports on advanced fragment syntheses [33] and model studies [34] of these ansamycins will not be discussed in this review.
The first common disconnection of the C15-ansamycins occurred at the C1–N20 amide bond. Macrocyclization of these 19-membered lactams IIa was performed on seco-amino acid IIIa using BOP–Cl as the coupling reagent (Scheme 1). Baker reported the first use of this approach in 1990 and will be discussed in more detail in the following section (Section 2.3.1) [35]. For redundancy purposes, conversion of these different acyclic frameworks to ansamycins will be omitted after the first total synthesis, with one exception. Panek utilized intramolecular copper-mediated amidation of bromo-amide IIIb to form the 19-membered macrolactam of reblastatin (and later geldanamycin), see Section 2.3.6 for discussion [36]. This is the first example of a Buchwald aryl amidation reaction used for macrocyclization. The following discussion is divided into two distinct parts and is based on a second common disconnection at the C15–C16 benzylic bond or at the C14–C15 aliphatic bond of the ansamycin framework. Of the 12 syntheses, there are seven reports that utilize aromatic aldehyde IVa as the starting point for ansa chain construction (i.e. aromatic elongation). The remaining five employ aryl lithium IVb to install the C15-alcohol of the ansamycin framework (i.e. late-stage assembly). These two different synthetic pathways allow for direct comparison of methodologies utilized to arrive at similar complex fragments and will be reviewed as such in the following section.
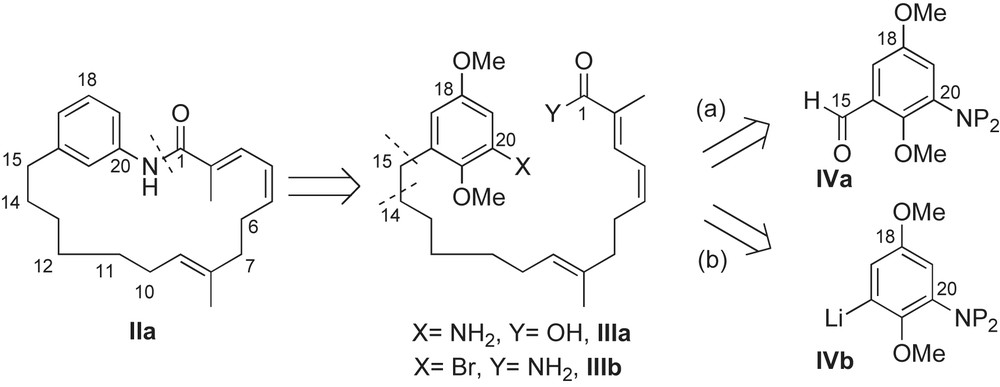
2.3 Syntheses of ansamycins utilizing “aromatic elongation”
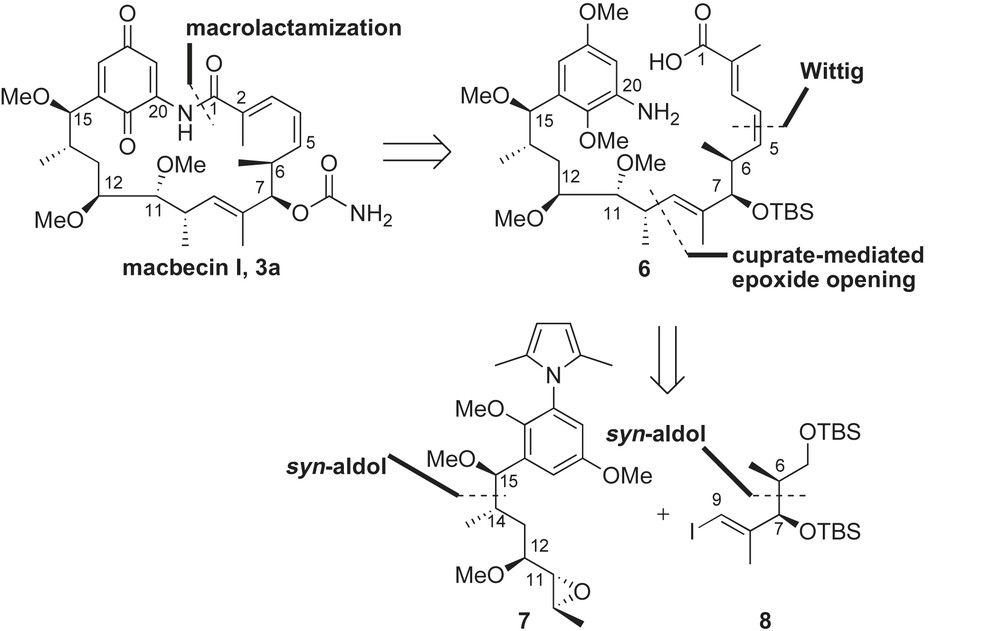
2.3.1 First total synthesis of (+)-macbecin I by Baker (1989)
Baker and co-workers reported the first approach to the ansamycin class of natural products in 1989 with their enantioselective synthesis of macbecin I 3a [35]. Retrosynthetic disconnection at the C1–N20 amide bond reveals the prevalent acyclic framework 6 (Scheme 2). The (E,Z)-conjugated diene of the seco-amino acid 6 was envisaged to be formed from successive Wittig olefinations, while cuprate-mediated coupling of epoxide 7 with vinyl iodide 8 would afford the crucial C9–C10 bond construction. The syn-relationship at C6–C7 and C14–C15 stereocenters was installed using Evans' metal enolate-based aldol strategy [37], while Sharpless asymmetric epoxidation (SAE) was used to install the C10–C11 stereocenters.
Installation of the C6–C7 stereocenters of the vinyl iodide fragment 8 was achieved using Evans' chiral auxiliary [38] (Scheme 3). The vinyl iodide 9 was prepared in four steps (40% yield) from commercially available diethyl methyl malonate [35d]. Reaction of aldehyde 9 with the Z(O)-boron enolate of chiral propionyl oxazolidinone 10 under soft enolization conditions with 9-BBN-OTf provided the syn-C7-allylic alcohol 11 in 65% yield. Removal of the auxiliary and protection of the resulting alcohols as TBS ethers completed the preparation of vinyl iodide 8 in 93% yield.
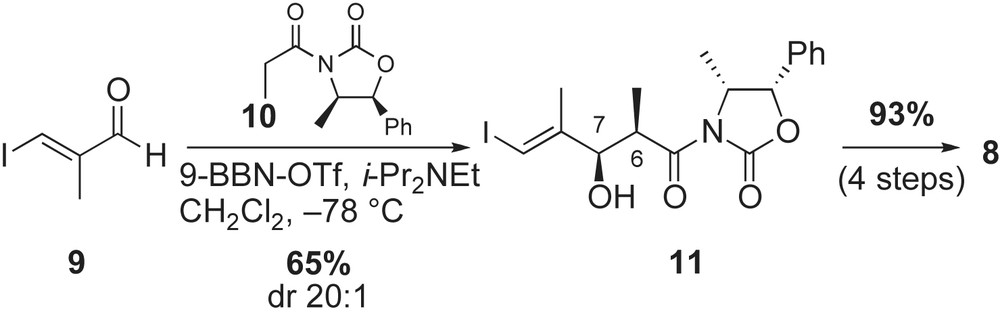
Synthesis of the advanced intermediate 7 featured asymmetric aldol, enolate hydroxylation, and Sharpless epoxidation reactions (Scheme 4). The benzaldehyde 12 was prepared in three steps starting from p-methoxyphenol (33% yield) [35a]. Treatment of 2,5-dimethoxy-3-nitrobenzaldehyde 12 with the Z(O)-boron enolate [37] of 10 afforded the syn-aldol adduct 13 in 88% yield and 20:1 diastereoselectivity. The benzylic alcohol 13 was converted to the acyl oxazolidinone 14 in 11 steps (62% yield). The desired C12-alcohol was installed using, at the time, recently developed asymmetric hydroxylation of enolates. Thus, reaction of sodium enolate of 14 with Davis' reagent 15 [39] gave the required alcohol 16 in 83% yield and excellent diastereoselectivity. The oxazolidinone 16 was converted to the C12-allylic alcohol intermediate 17 in five steps (73% yield). Sharpless asymmetric epoxidation of (E)-allylic olefin with stoichiometric amounts of (+)-DIPT and Ti(OiPr)4 afforded the required fragment 7 after protection of the alcohol as its methyl ether (88% over two steps).
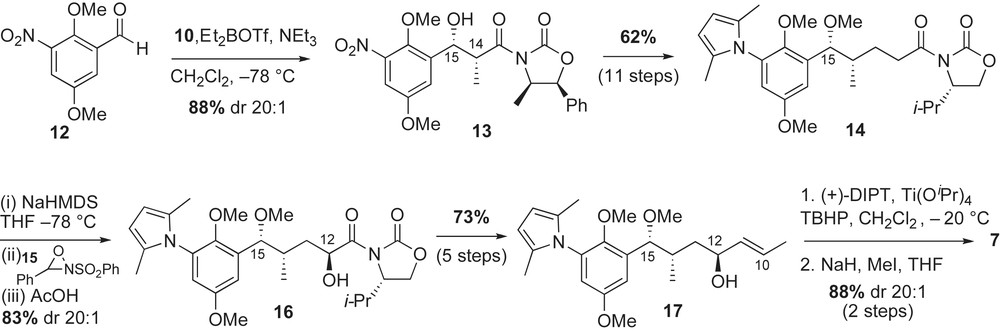
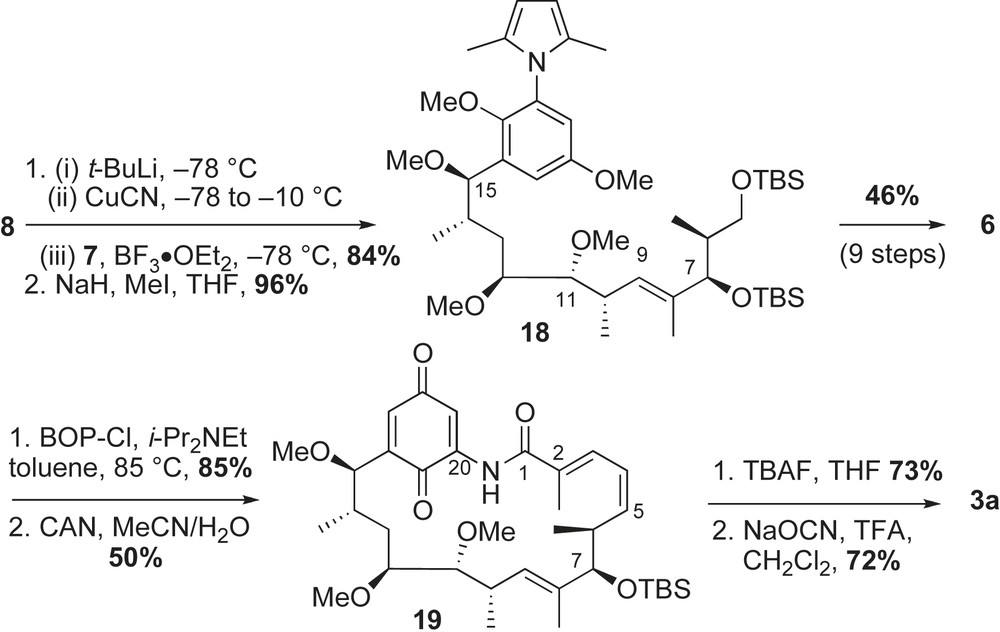
Fragment coupling, diene insertion and macrocyclization were required for completion of the synthesis. The crucial coupling of cyanocuprate 8 with activated epoxide 7 (BF3·OEt2) yielded the desired homoallylic alcohol (Scheme 5), which was subsequently protected as its methyl ether 18 (81% over two steps). The advanced intermediate 18 was converted to the (Z,E)-conjugated acid 6 via sequential Horner–Emmons and Wittig reactions with good selectivities. Macrocyclization of the seco-amino acid 6 with BOP–Cl, followed by CAN oxidation provided macrolactam 19 in 43% yield (two steps). Removal of the C7-TBS ether with TBAF and installation of the carbamate functionality with sodium cyanate completed the total synthesis of macbecin 3a. The authors also reported that urethane installation can precede the quinone oxidation with a similar yield.
The first synthesis of (+)-macbecin 3a was achieved in 38 steps with 0.79% overall yield beginning with p-methoxyphenol (longest linear sequence). As such, Baker's synthesis of macbecin I generated enormous interest from the synthetic community from this important class of natural products.
2.3.2 Total synthesis of (+)-macbecin I by Evans (1992)
Evans' synthesis of macbecin featured late-stage macrocyclization, Still–Gennari homologation and asymmetric oxazolidinone-based aldol reactions [40]. The retrosynthesis in Scheme 6 began with a disconnection at the C12–C13 bond of the seco-amino acid 6 to reveal two fragments of similar size and complexity. In a forward sense, the union of aromatic 20 and aldehyde 21 fragments was accomplished using a syn-aldol reaction. All three syn methyl-methoxy group relationships of macbecin (C6–C7, C10–C11 and C14–C15) were introduced using sequential asymmetric boron-mediated aldol processes [37]. The C12-stereocenter was introduced using chelate-controlled reduction of ketone.
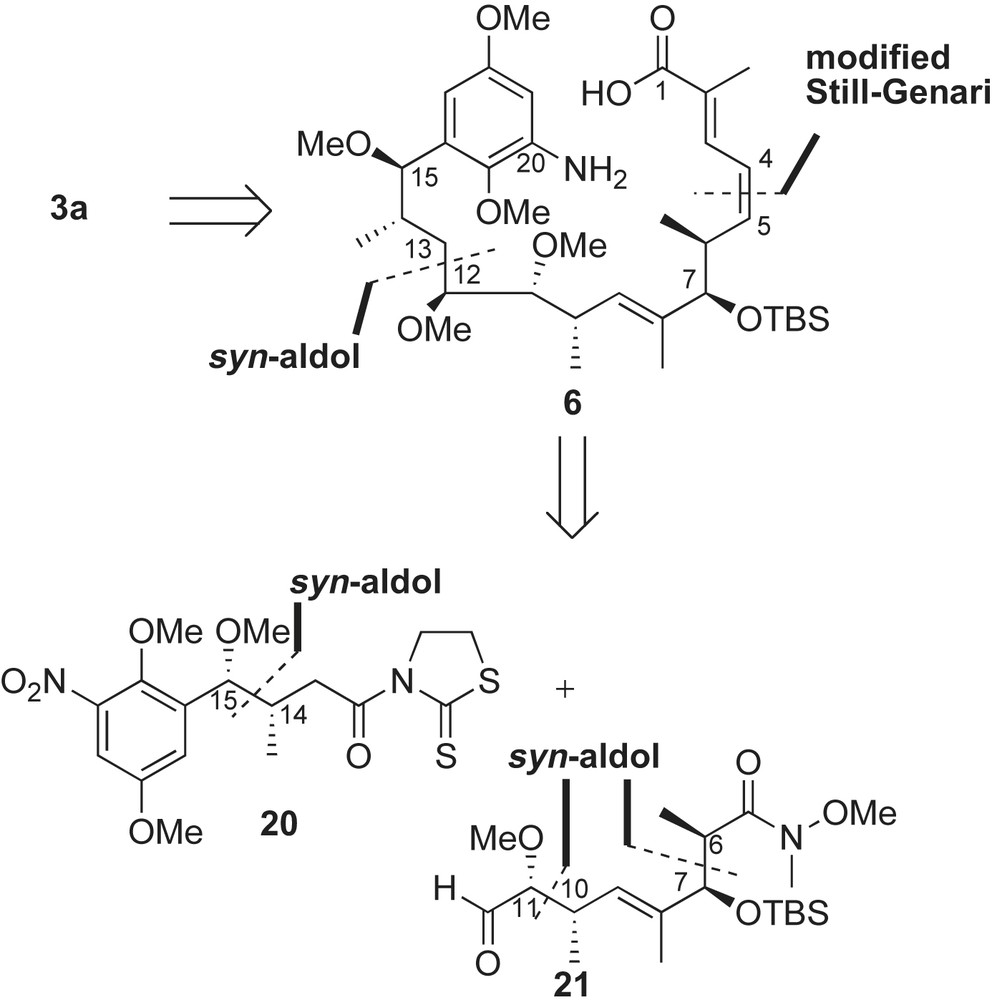
The syn-stereocenters (C14–C15) of the 2-mercaptothiazoline fragment 20 were generated using previously described sequence (refer to Baker's preparation of aromatic fragment 13 in Scheme 4).
The syn methyl-oxygen stereogenic centers (C6–C7 and C11–C10) of complex fragment 21 were installed using sequential Evans' aldol reactions [37] (Scheme 7). Treatment of trans-cinnamaldehyde 22 with the boron enolate of imide 10 afforded the aldol adduct 23 in 70% yield (dr 20:1). This allylic syn-alcohol 23 was converted to the (E)-α,β-unsaturated aldehyde 24 in six steps (60% overall yield). Second iteration of the aldol reaction with imide 10 furnished the allylic alcohol 25 in 78% yield with 20:1 diastereoselectivity. Protection of C7-alcohol, transamidation, and ozonolysis were necessary to convert adduct 25 to the chiral aldehyde intermediate 21.

Coupling of thioimide 20 with aldehyde 21 in a double stereodifferentiating aldol reaction was initially problematic (Scheme 8). Complex mixtures of products and low selectivity were obtained using boron and tin enolates. In a clever modification, the authors were able to obtain the desired aldol adduct 26 as a single diastereomer using the titanium enolate of 20. Intended radical decarboxylation of the carboxylic acid derivative, obtained via hydrolysis of thiamide moiety of 26, resulted in competitive β-lactone formation or reduction of the aromatic nitro group. This required an alternate decarboxylation method at C13 to complete the synthesis. Oxidation of C12-alcohol with Dess–Martin periodinane (90%), followed by lithium hydroxide hydrolysis of the thiazolidinethione and in situ formal decarboxylation, afforded the desired ketone 27 in 73% yield. Chelate-controlled reduction of ketone 27 with Zn(BH4)2 provided the (S)-C12-alcohol as a single diastereomer in 85% yield. Methylation of the resulting secondary alcohol with Meerwein's reagent, followed by DibalH reduction produced aldehyde 28 in 79% yield over two steps. The C2–C5 diene of the seco-amino acid 6 was incorporated using Horner–Emmons olefination of aldehyde 28 with phosphonate reagent 29. The resulting (E,Z)-conjugated ester was obtained in 70% yield, as a 3.7:1 mixture of olefinic isomers. Reduction of the nitro group with Lindlar's catalyst (94% yield) gave the anilinic ester that was subsequently hydrolyzed with LiOH to give the desired carboxylic acid 6. The remaining steps in Evans' synthesis of macbecin I 3a are similar to the reported sequence developed by Baker. This convergent synthetic route toward macbecin was completed in just 25 steps from trans-cinnamaldehyde (longest linear sequence) and 1.2% overall yield.
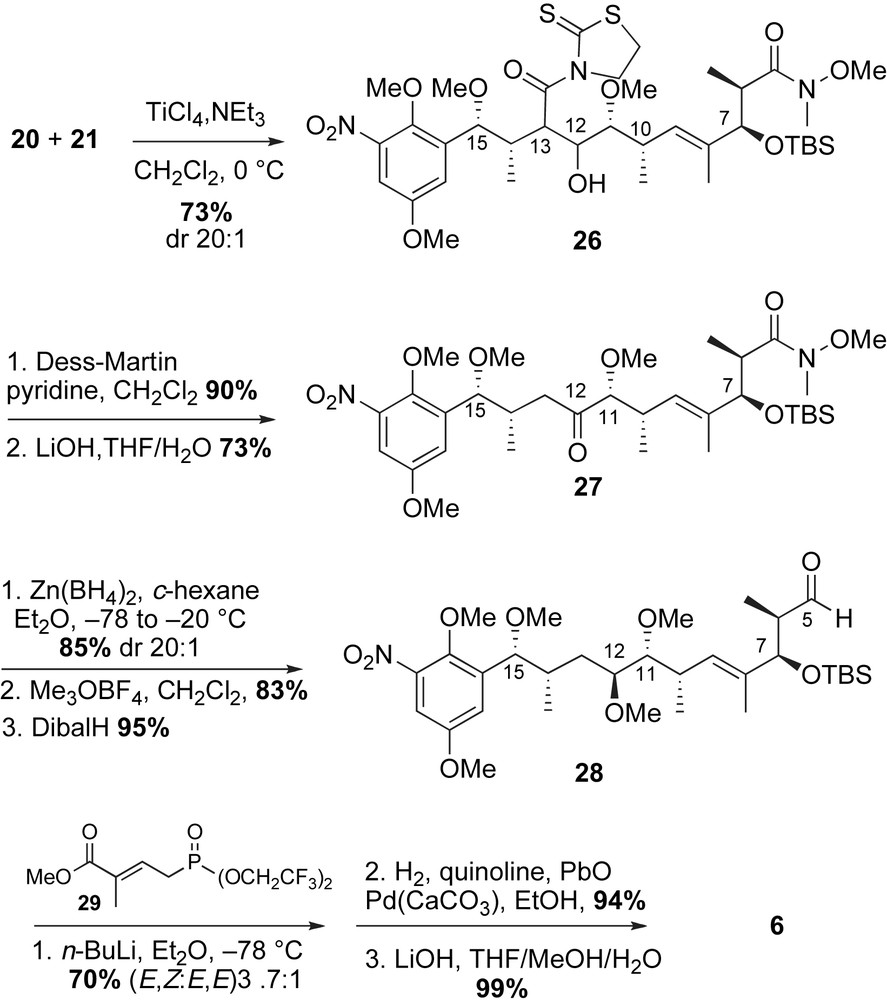
2.3.3 Panek's total synthesis of (+)-macbecin I (1995)
While the first two enantioselective syntheses of (+)-macbecin I utilized Evans' chiral metal enolate-based bond construction to introduce stereogenic centers of the ansa chain, the approach by Panek and co-workers featured use of chiral crotylsilane methodology (mechanistically, fundamentally different) (Scheme 9) [41,42]. Homologation of the aromatic acetal 30 to the (E,Z)-dienoate 6 was achieved using crotylsilane reagents 31a–c. This crotylation approach was used to install six out of seven stereogenic centers of the molecule (C6–C7, C10–C11, and C14–C15). The remaining C12-stereocenter was introduced using an alkoxy-directed hydroboration reaction developed in the Panek laboratory [43].
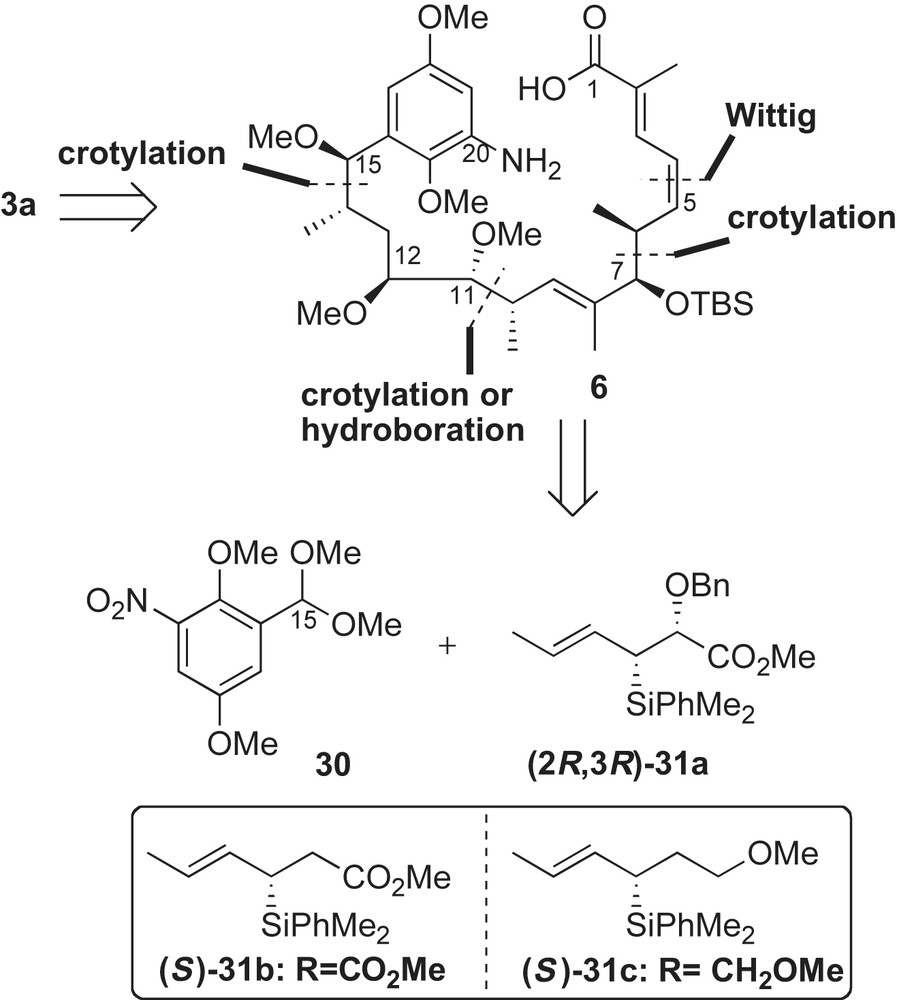
The synthesis of macbecin I 3a was initiated with a TMSOTf-catalyzed condensation reaction between 2,5-dimethoxy-3-nitrobenzaldehyde dimethyl acetal 30 and (E)-crotylsilane reagent 31a (Scheme 10). The homoallylic C15-methyl ether 32 was obtained in 89% yield and with high diastereoselectivity via in situ-generated oxocarbenium ion, which is illustrated in the antiperiplanar transition state in Scheme 10. The resulting α-alkoxy-β,γ-unsaturated methyl ester 32 was subjected to a borane-dimethyl sulfide hydroboration reaction [43]. In this intramolecular process, selective reduction of the methyl ester afforded good levels of regio- and diastereoselection in the olefin oxidation (dr 8.5:1) and the desired 1,3-diol 33 was obtained in 85% yield.
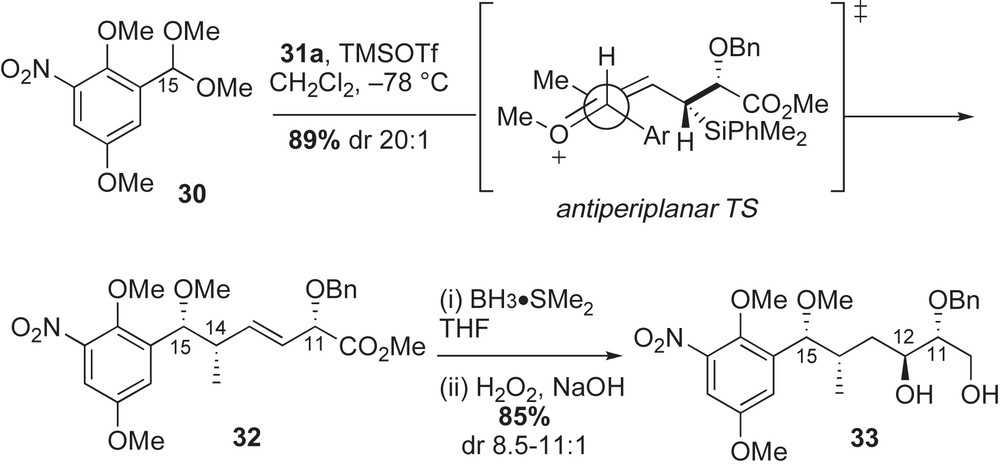
The 1,3-diol 33 was initially converted to α-methoxy aldehyde 34 in five steps and 64% yield (Scheme 11, Eq. (1)). Crotylation of aldehyde 34 with silane reagent 31b installed C10–C11 stereocenters in 80% yield and moderate diastereoselectivity (dr 12:1). This syn bond construction reinstalled the C11-alcohol that was removed during preparation of 34. In this double stereodifferentiating reaction, C11–C12 stereocenters emerged via an anti-Felkin relationship, with crotylsilane 31b overriding the chirality of aldehyde 34. Ozonolysis of the aforementioned product gave the desired α-methyl aldehyde 35 in 65% yield.
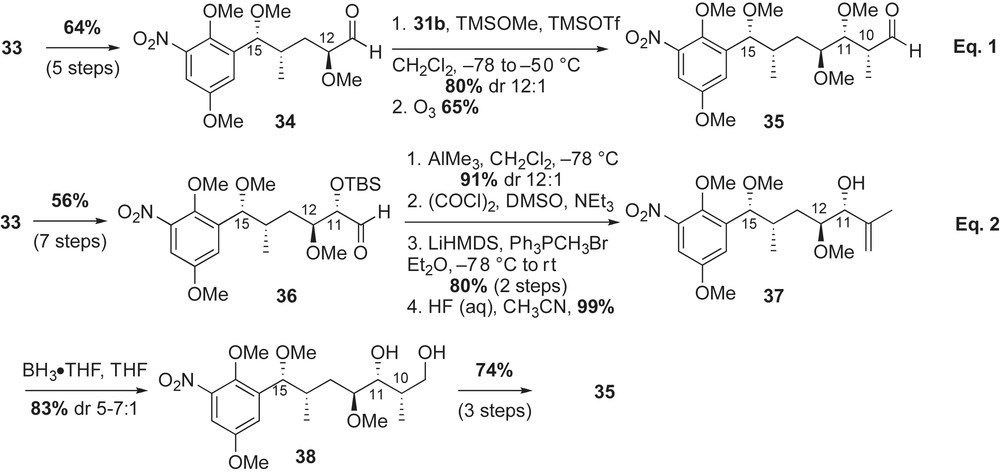
An alternate route to α-methyl aldehyde 35 is illustrated in Scheme 11, Eq. (2). Conversion of 1,3-diol 33 to aldehyde 36 preserved the previously established C11 stereocenter (Scheme 10). Chiral aldehyde 36 was converted to the 1,1-disubstituted olefin intermediate 37 via a four-step sequence (alkylation with AlMe3, oxidation, Wittig homologation, silyl deprotection) in 72% yield. Hydroboration of terminal allylic alcohol 37 with BH3·THF complex afforded the desired 1,3-diol 38 in excellent yield (83%) and moderate diastereoselectivity (dr 5:1) [44]. Protection of the secondary C11-hydroxyl group in 38 and subsequent oxidation of the primary alcohol completed the preparation of the aldehyde 35. Both reaction pathways produced the desired aldehyde 35 in ∼30% yield from 1,3-diol 33 (seven steps for Eq. (1) versus 15 steps for Eq. (2), Scheme 11).
The final two stereocenters (C6–C7) of macbecin I were also introduced using a crotylsilane-based bond construction methodology (Scheme 12). Homologation of α-methyl aldehyde 35 to the α,β-unsaturated aldehyde 39 was accomplished via Wittig olefination, subsequent DibalH reduction of the ester and Swern oxidation of the primary alcohol. Condensation of aldehyde 39 with crotylsilane reagent 31c in the presence of 4-acetoxybenzyl trimethylsilyl ether afforded the protected syn-alcohol 40 in 92% yield (dr 20:1). Intermediate 40 was converted to the (E,Z)-conjugated acid 6 via sequential phosphorus-based olefinations (Horner–Emmons and Wittig). Similar known methods for macrocyclic ring closure and functional group interconversion completed the synthesis.
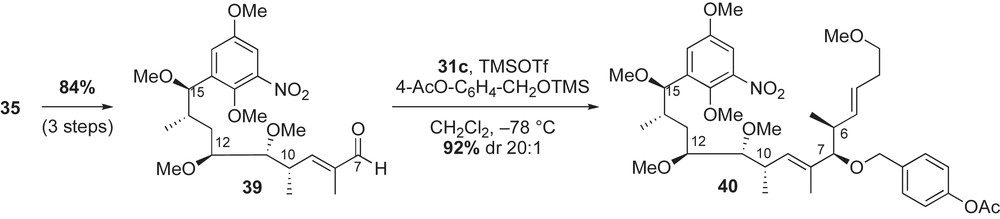
Panek's synthesis of (+)-macbecin I 3a relied on use of chiral (E)-crotylsilane methodology to construct six of seven stereogenic centers of the molecule. Macbecin was completed in 29 steps and 0.78% overall yield starting from p-methoxyphenol (longest linear sequence). This convergent approach differed from Baker's and Evans' use of metal enolate-based methodology to prepare the ansa chain.
2.3.4 First total synthesis of geldanamycin by Andrus (2002)
Andrus and co-workers accomplished the total synthesis of geldanamycin 1 in 2002 [45]. The long delay between isolation (1970) and synthesis may be attributed to challenges accompanied with the molecular architecture of geldanamycin. Unlike herbimycin A and macbecin I, geldanamycin possesses a C17-methyl ether on the quinone, but lacks the methoxy group at C15. The latter prevents use of aldol reaction to generate the C14–C15 stereochemistry and represented different synthetic challenges not directly amendable to an alcohol based bond construction (Scheme 13). In addition, the C11 stereocenter is a hydroxyl group as opposed to the methyl ether present in other benzoquinone ansamycins.
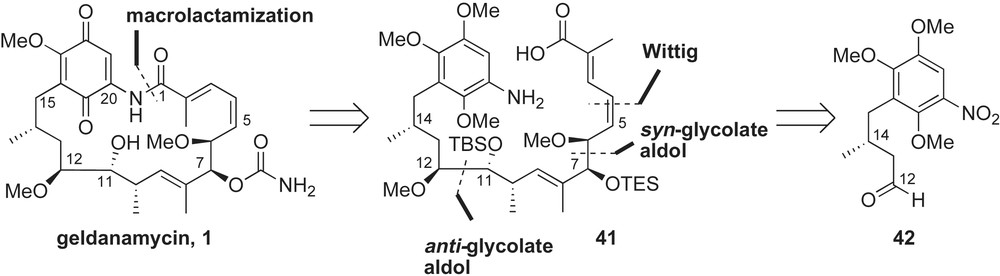
A linear approach was undertaken by Andrus to accommodate structural changes present in the ansa chain of geldanamycin (Scheme 13). Assembly of the seco-amino acid 41 was envisioned to come from sequential aldol and olefination reactions starting from aldehyde 42. Specifically, anti-glycolate aldol with an oxapyrone boron enolate was utilized to set the stereochemical relationship between the C11–C12 functionalities [46].
Synthesis of acyclic geldanamycin 41 utilized auxiliary-directed addition reactions to install the stereogenic centers (Scheme 14). The necessary benzyl bromide 43 was prepared from 1,2,4-trimethoxybenzene in four steps (56% yield). Asymmetric Evans' alkylation [47] was used to introduce the methyl group at C14. Treatment of alkyl bromide 43 with the sodium enolate of (S)-44 afforded the desired adduct 45 in good yield and selectivity. The auxiliary product 45 was converted to the aldehyde 42 in just three steps. To install the C12-stereocenter, the authors applied an anti-glycolate aldol reaction using chiral dioxanone 46 [46]. Condensation of aldehyde 42 with “locked” E(O)-boron enolate of oxapyrone 46 yielded the anti-aldol product 47 with 10:1 diastereoselectivity (70% yield). This methodology complements the predominantly syn-glycolate adducts obtained from acyclic auxiliaries (Z(O)-enolates) or addition of allylmetal reagents containing (Z)-γ-alkoxy functionality [48]. Removal of the auxiliary and installation of the C10-methyl and trisubstituted alkene were accomplished using known methods [41,44]. The C6–C7 diol was introduced using the syn-glycolate aldol reagent 49, derived from (−)-norephedrine as previously developed by Andrus [49]. Reaction of aldehyde 48 with the Z(O)-enolate of glycolate 49 afforded allylic alcohol 50 in 90% yield and high selectivity. Homologation of alcohol 50 to conjugated acid 41 proceeded smoothly using previously described olefination procedures.
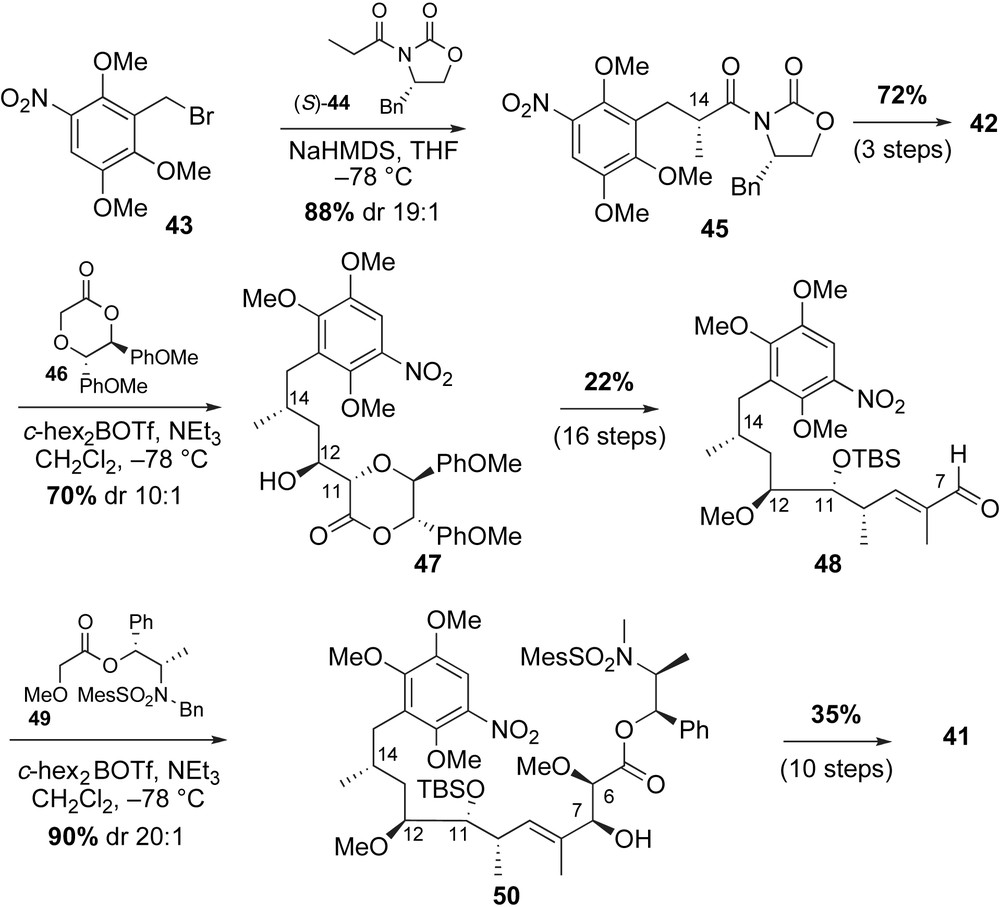
The key steps for completion of the synthesis were macrocyclization, carbamate installation and quinone oxidation. Cyclization of 41 in the presence of BOP–Cl provided the 19-membered core 51 in 76% yield (Scheme 15). Selective removal of TES ether with TBAF, followed by urethane formation using trichloroacetylisocyanate [50] proceeded in 80% yield over two steps. Removal of C11-TBS ether was achieved using aqueous hydrogen fluoride to furnish geldanamycin precursor 52 in 95% yield. Formation of the p-quinone from the trimethoxy precursor 52 proved challenging due to unforeseen conformational and electronic issues of the molecule. Various oxidants such as CAN or AgO/HNO3 were examined, but the dominant product from these reactions was the aza-quinone 53. Ultimately, the use of nitric acid permitted formation of the desired p-quinone product 1, albeit as the minor product (1:10, p-quinone/o-quinone).
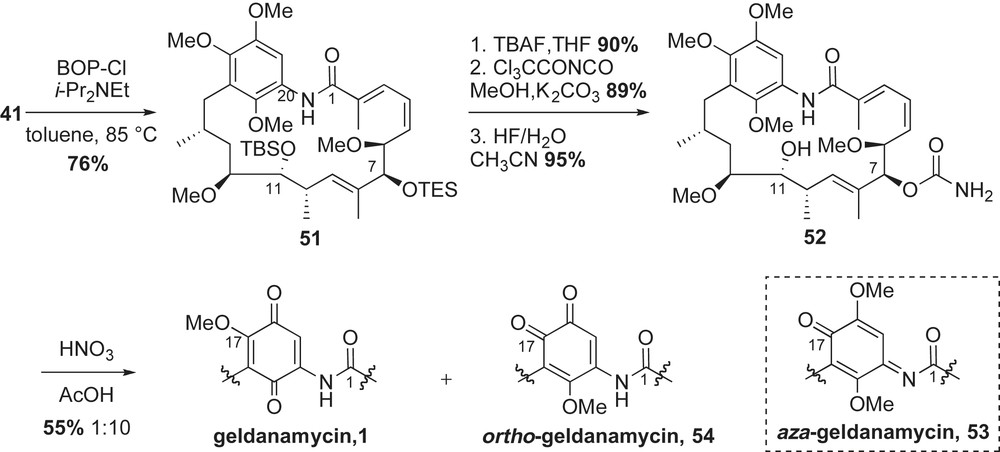
The structurally distinct ansa chain of geldanamycin proved to be a challenging molecule for total synthesis. Andrus and co-workers developed two glycolate aldol reactions to introduce the key stereogenic relationships at C11–C12 and C6–C7. The synthetic product 1 was obtained in low overall yield (0.05%, 41 steps, starting from 1,2,4-trimethoxybenzene) due to competitive o-quinone formation 54. Since their initial publication, Andrus and co-workers reported a model study on the p-quinone formation, which can be selectively produced from mild oxidation of the 1,4-dihydroquinone precursor [51].
2.3.5 Panek's approach to the synthesis of herbimycin A (2004)
Due to their complex structural architecture and biological properties, the Panek laboratory has maintained an interest in the synthesis of ansamycins for over 15 years. The stereochemistry of the ansa chain inspired development of the crotylsilane-based reagents to construct key fragments of this class of natural products. Prior to this publication [52], the only previous synthesis of herbimycin A 2a was reported by Tatsuta in 1991 (see Section 2.4.1) [44,53]. This natural product differs from macbecin I 3a at the C6 position by having a methyl ether instead of a methyl group. The retrosynthesis of herbimycin A begins with the disconnection of the C1–N20 amide bond (Scheme 16). The two syn-methoxy-methyl stereocenters at C10–C11 and C14–C15 were introduced using sequential crotylations with chiral silane reagents 31a–b. The C6–C7 syn-diol of acyclic framework 55 was envisioned to come from Brown's asymmetric allylboration methodology [54].
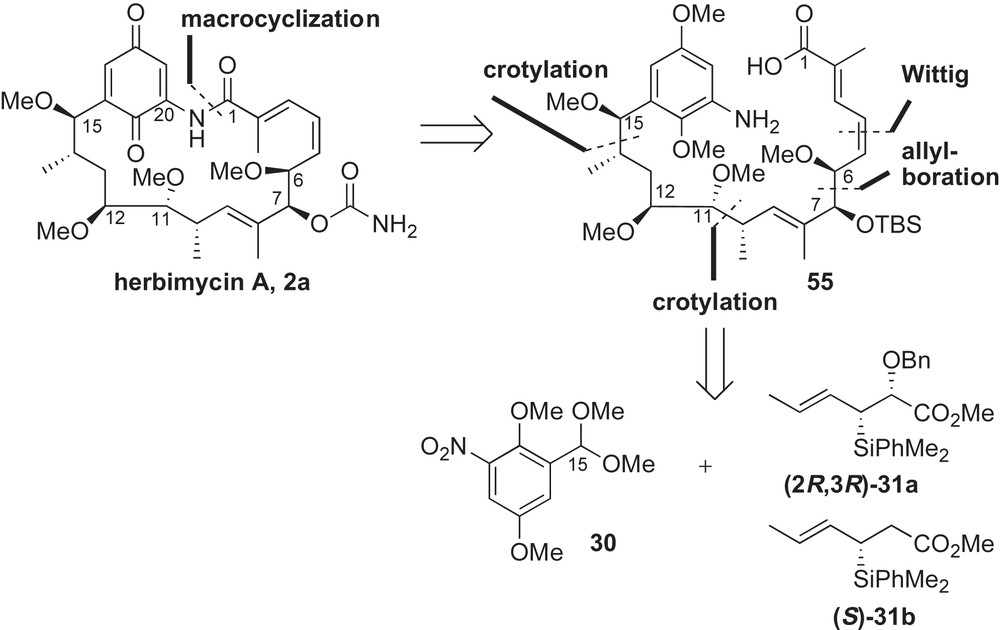
The common left-hand motif of macbecin and herbimycin, aromatic fragment 39, was prepared as described previously (Section 2.3.3) [41]. Asymmetric allylboration of aldehyde 39 with γ-methoxyallyl organoborane reagent 56 introduced the desired syn-relationship between the C6–C7 stereocenters (Scheme 17). The corresponding allylation adduct 57 was obtained as one diastereoisomer in 77% yield. Following previously published methodology for the synthesis of macbecin, Panek and co-workers completed the synthesis of herbimycin A 2a.
This report documents reliability and high levels of stereoselectivity that can be attained using crotylsilane methodology. With that said, synthesis of herbimycin A was realized quickly (31 steps with 0.57% yield beginning with p-methoxyphenol) following reported protocols due to structural similarities of these ansamycin natural products.
2.3.6 First total synthesis of reblastatin by Panek (2005)
In 2000, Takatsu isolated the benzenoid ansamycin, reblastatin 5a, possessing structural similarity to geldanamycin [30]. The presence of a phenol and C17-methoxy group, as well as saturation at C4–C5 olefin of the C1–C5 dienoate, presented a new synthetic challenge. Mindful of these subtle architectural differences within the ansa chain the Panek group set out to design a convergent synthesis of the natural product.
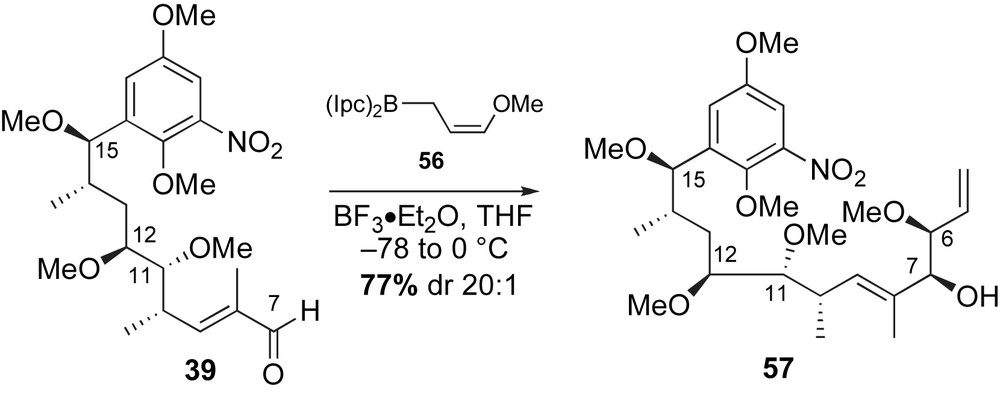
Strategy for the synthesis of reblastatin 5a focused on a late-stage copper-mediated aryl amidation for macrolide formation (Scheme 18). Intramolecular copper-mediated amidation of acyclic skeleton 58 was anticipated to form the 19-membered macrolactam. A second disconnection was envisaged at the C7-allylic alcohol to give alkyne 59 and aldehyde 60. Addition of an (E)-vinyl zirconium species, derived from 59, to aldehyde 60 would allow for convergent assembly of the acyclic framework 58. Sequential crotylation reactions installed the four stereogenic centers of the aromatic intermediate 59, while the α-methoxy aldehyde 60 was obtained from a readily available compound present in the chiral pool.
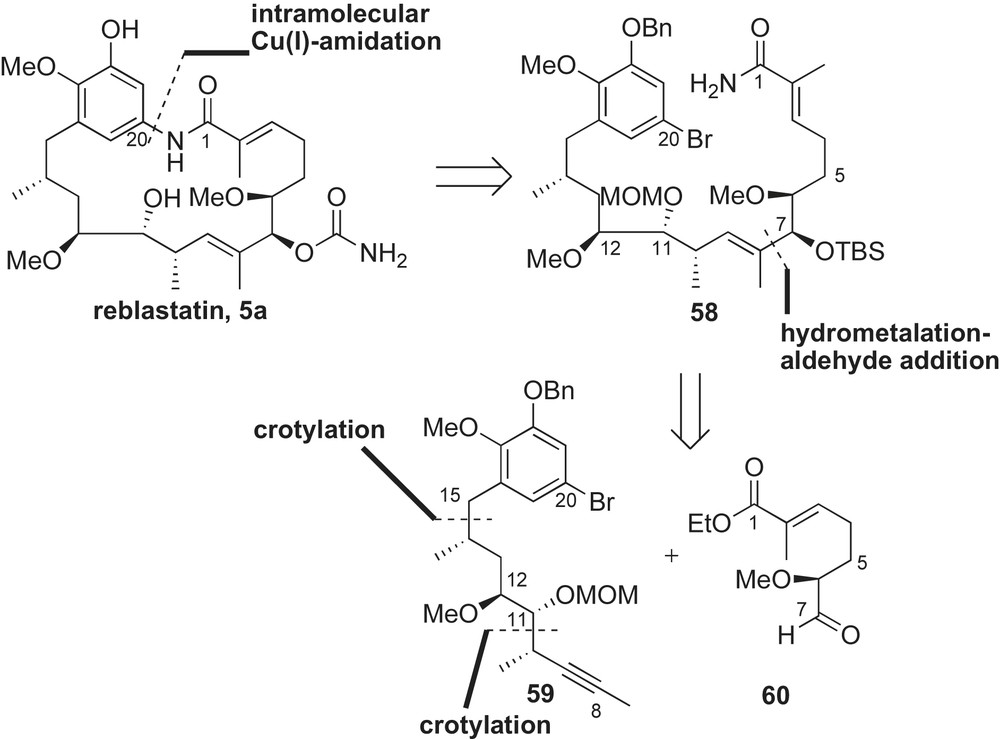
The preparation of aldehyde 60 was possible from chiral furanone 61 (Scheme 19). Lactone 61 was prepared from l-glutamic acid in three steps (64% yield) [55]. Protection of the alcohol in 61 as a TBS ether, followed by DibalH reduction of lactone provided the intermediate lactol. Treatment of the resulting lactol with (carbethoxyethylidene)triphenyl phosphorane furnished the desired unsaturated ethyl ester 62 in 73% yield (three steps). The C6-alcohol was converted to the desired aldehyde partner 60 in three steps (93%).

The preparation of the methyl alkyne fragment 59 is shown in Scheme 20. Commercially available 2,3-dihydroxybenzaldehyde was functionalized to 5-bromobenzaldehyde 63 in three steps (57% yield). Crotylation of aldehyde 63 with (E)-crotylsilane reagent 31a provided the desired C15-syn-homoallylic alcohol (dr 20:1) that was subsequently removed under ionic conditions (Et3SiH, BF3·OEt2) to afford methyl ester 64 in 60% yield. Conversion of ester 64 to the α-methoxy aldehyde 65 was achieved utilizing a similar six-step sequence reported in Panek's synthesis of macbecin [41] and herbimycin [52] (Sections 2.3.3 and 2.3.5, respectively). A matched crotylation between aldehyde 65 and (S,S)-crotylboronate 66 [56] yielded syn-homoallylic alcohol as a single diastereomer. The resulting C11-alcohol was protected as a MOM-ether 67 (71% yield, two steps) and converted to the methyl alkyne 59 in a four-step sequence (Scheme 21).
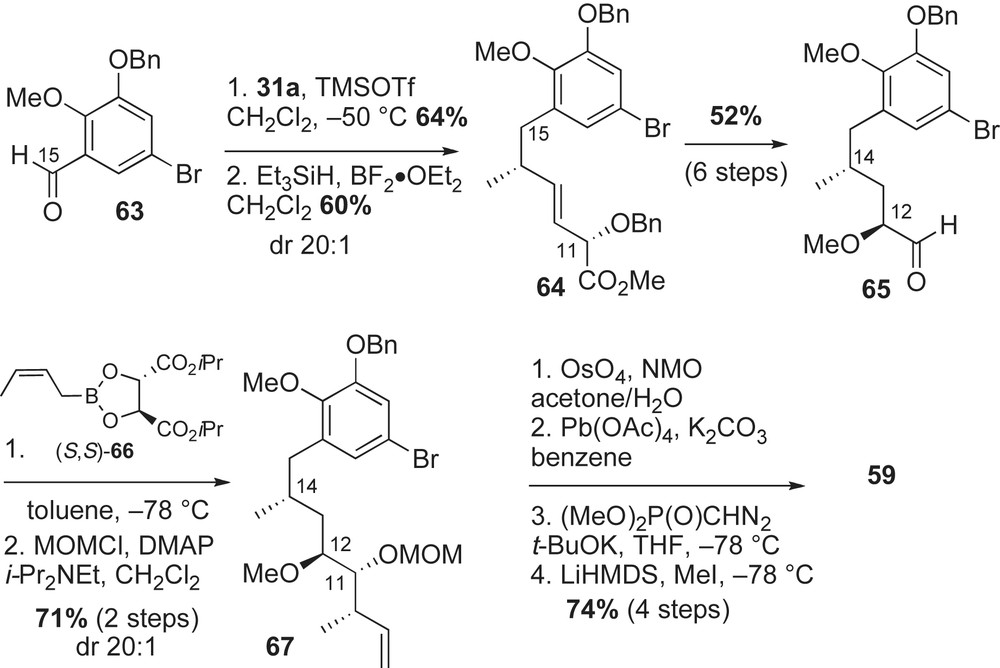

Completion of the synthesis featured an asymmetric coupling of alkyne and aldehyde and an intramolecular aryl amidation reaction. To achieve crucial fragment assembly of advanced intermediates 59 and 60, hydrometalation-aldehyde addition sequence was explored [57]. Hydrozirconation of 59 followed by transmetalation and addition to aldehyde 60 afforded the desired allylic alcohol 68 as one diastereomer in 55% yield. In order to obtain the desired regioselectivity (dr 20:1) in the hydrozirconation process, elevated temperatures and greater than 2 equiv of Schwartz's reagent were necessary. After protection of the C7-alcohol as a TBS ether, the ethyl ester was converted to the unsaturated amide 58. The crucial macrocyclization step was attempted using Buchwald's amidation conditions [58]. Although Buchwald demonstrated that his Cu(I) system can generate five-membered heterocycles [58,59], Panek and co-workers wanted to determine if the reaction conditions would transfer to macrocyclic lactams. Accordingly, subjection of the bromo-amide 58 to amidation conditions (CuI, K2CO3, diamine ligand) provided the desired 19-membered macrolactam 69 in 83% yield. Following macrolactamization, completion of the first total synthesis of reblastatin 5a was accomplished in three steps (37% yield). Since the initial publication [36], several examples of intramolecular amidation to form macrolides of 10-membered rings or greater have been reported [60].
The first synthesis of phenol containing ansamycin, reblastatin 5a, was achieved in just 25 steps and 0.79% overall yield beginning with 2,3-dihydroxybenzaldehyde.
2.3.7 Panek's total synthesis of geldanamycin (2008)
In 2008, Panek and co-workers reported the second synthesis of the known Hsp90 inhibitor, geldanamycin 1 [61]. The convergent synthesis was highlighted by a scandium triflate pyran-ring opening reaction [62] and an enantioselective crotylation reaction to introduce the (E)-trisubstituted double bond and set the syn-relationship between the C10–C11 stereocenters of the molecule (Scheme 22). Macrocyclization was effected with an intramolecular copper-mediated amidation reaction of bromo-amide 70, which was first utilized in the total synthesis of reblastatin 5a [36]. The geldanamycin aromatic moiety was protected as bis-isoproxy ethers to facilitate late-stage deprotection followed by oxidation to the dihydroquinone, a problem encountered by Andrus and co-workers in their synthesis of geldanamycin [45].
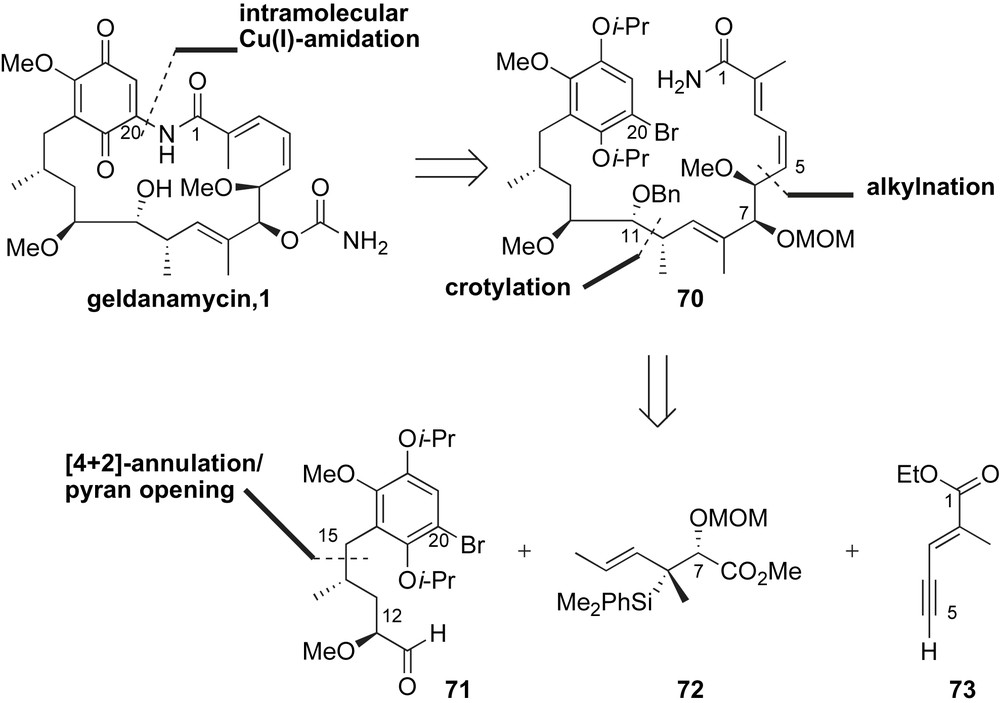
The synthetic strategy for C13–C21 fragment 71 of the ansamycin framework featured a [4 + 2]-annulation followed by ScOTf3/Et3SiH mediated opening of an aryl C-glycoside (Scheme 23) [62]. Accordingly, annulation of 3-bromo-2,5-diisopropoxy-6-methoxybenzaldehyde 74 with (E)-anti-crotylsilane 75 was mediated with trifluoromethanesulfonic acid. The desired 2,6-trans-dihydropyran 76 was obtained in 84% yield with moderate diastereoselectivity (dr 4:1). Regio- and diastereoselective hydroboration of the endo cyclic olefin followed by methylation of the resulting secondary alcohol yielded the pyran opening substrate 77 in excellent yield. Reduction of the benzylic C–O σ-bond with scandium triflate in the presence of Et3SiH [62] furnished the stereochemically well-defined acyclic fragment 78 in 72% yield and the de-isopropoxy byproduct 79. Fortunately, phenol 79 was recycled back to the desired product 78 and two-step functionalization procedure afforded α-methoxy aldehyde 71.
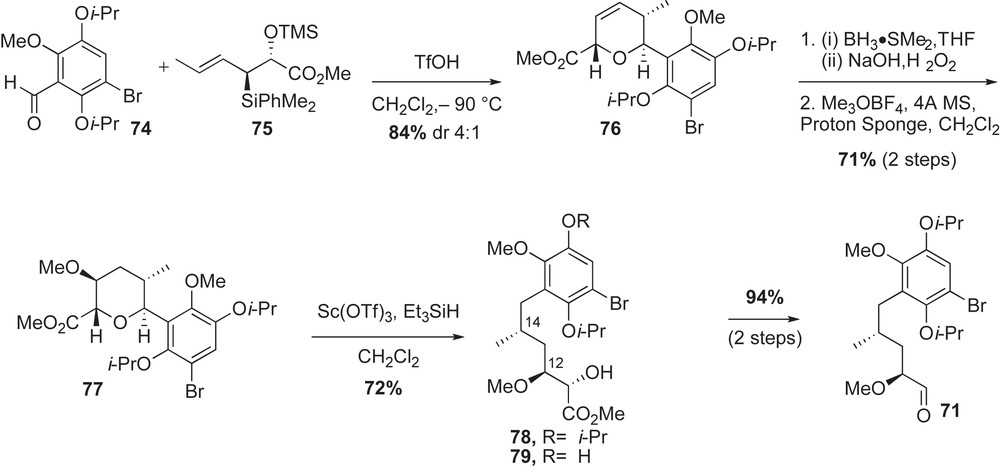
Having the desired aldehyde 71 in hand, Panek group applied the newly developed crotylsilane reagent 72 [63] for installation of the remaining stereocenters of the C6–C21 fragment of geldanamycin (Scheme 24). Utility of methyl-substituted crotylsilanes was previously exploited in the [4 + 2]-annulation reaction during synthesis of a tetrahydropyran fragment of kendomycin by Panek and co-workers [63]. As anticipated, crotylation of silane 72 with aldehyde 71 in the presence of BF3·OEt2 proceeded to give benzyl protected homoallylic ether 80 in 70% yield with moderate selectivity (dr 9:1). In a one step fashion, crotylation reaction installed the C10,C11-syn stereocenters, the C7-ether and the (E)-trisubstituted double bond.
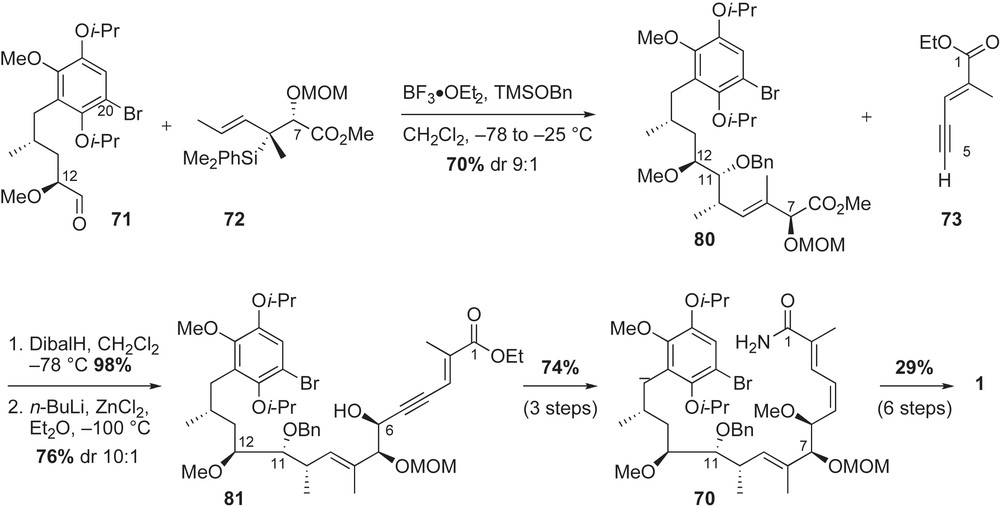
Completion of the fully functionalized acyclic framework of geldanamycin featured a chelation-controlled addition of acetylene, Lindlar's reduction and intramolecular copper-mediated amidation reaction (Scheme 24). Sequential treatment of ester 80 with DibalH and metallated acetylide 73 furnished the propargylic alcohol 81 in 76% yield and good diastereoselectivity (dr 10:1). The Cram-chelate product 81 was further modified with the (E,Z)-conjugated amide 70 in three steps. Following a reported protocol for copper-mediated ring macrocyclization [36], the 19-membered core of geldanamycin was observed in 81% yield. Installation of the C7-carbamate moiety as well as bis-isopropoxyether deprotections [61] occurred without complication to provide the synthetic geldanamycin 1 in 2% overall yield (20 linear steps starting from commercially available 2-methoxy hydroquinone).
Panek's synthetic strategy of geldanamycin 1 differed vastly from the linear approach reported by Andrus. Specifically, reductive opening of pyran substrate and subsequent crotylation reaction yielded the functionalized C7–C21 fragment in high yield [61]. Further application of the intramolecular Buchwald amidation to stereochemically similar substrates (19-membered macrolactams) expanded the scope of the useful reaction. Having a short and high-yielding synthetic route toward geldanamycin allows for useful generation of structural derivatives of the natural product which could act as potential Hsp90 inhibitors.
2.4 Syntheses of ansamycins via late-stage union of aromatic moiety and C15-ansa chain
The second strategy employed in the synthesis of ansamycins involved coupling of the aryl lithium fragment with a functionalized C15-aldehyde ansa chain. Tatsuta first described this approach in 1991 in his synthesis of herbimycin A 2a. A more recent account of this sequence was reported by Cossy in 2007. The two formal syntheses of macbecin by Martin and Kallmerten utilized masked aldehyde (lactol) ansa frameworks for union with the aromatic chromophore.
2.4.1 First total synthesis of herbimycin A by Tatsuta (1991)
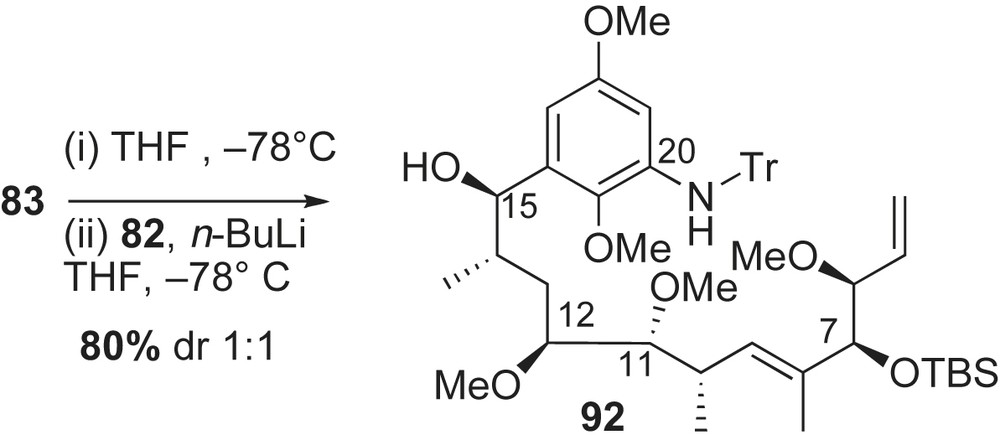
The first total synthesis of herbimycin A 2a by Tatsuta and co-workers featured fragment coupling of an aryl lithium derivative with a polyketide unit [44,53]. Disconnection at the C15–C16 bond of the seco-acid 55 reveals bromobenzene 82 and chiral aldehyde 83 (Scheme 25). In a forward sense, halogen–lithium exchange of 82 and addition to aldehyde 83 would introduce the required C15-hydroxyl group of the natural product. The assembly of fragment 83 was envisioned to come from homologation of methyl α-d-mannopyranoside (C11–C12, C14) and use of a metal-based asymmetric allylation reaction [54] to set the syn stereochemistry at C6–C7.
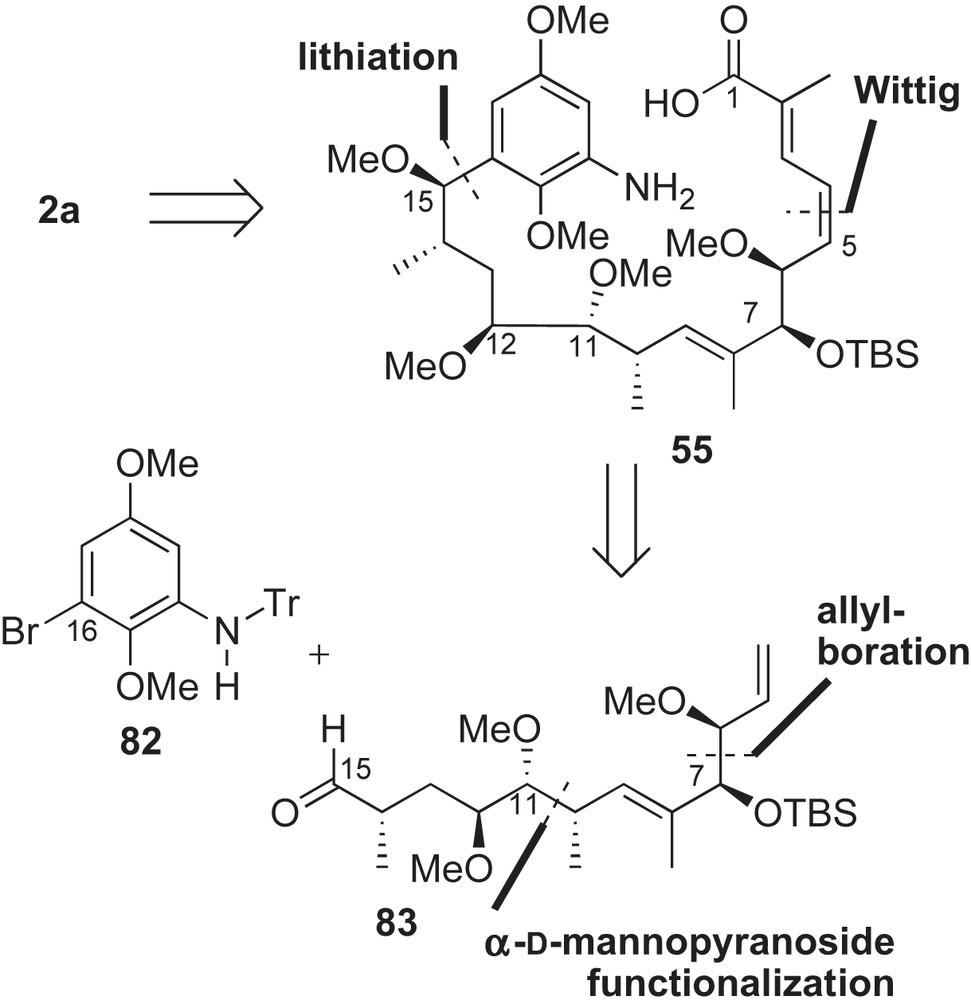
Synthesis of C5–C15 aldehyde fragment 83 proceeded in a linear fashion from a natural sugar (Scheme 26). Commercially available methyl-α-d-mannopyranoside 84 was converted to 1,1-disubstituted olefin intermediate 85 [64]. Hydrolysis of 85 was followed by NaBH4 reduction to afford diol 86 with the required C11, C12, and C14 configurations. Asymmetric hydroboration of terminal olefin 86 provided triol 87 with the newly installed stereogenic center at C10 (dr 5:1). The triol 87 was functionalized to α-methyl aldehyde 88 and subjected to Corey–Schlessinger's olefination [65] conditions with imine 89 to provide the α,β-unsaturated aldehyde 90 in 85% yield (E:Z 20:1). Condensation of intermediate 90 with Brown's allylboration reagent 56 [54] afforded the syn-homoallylic alcohol 91 in 76% yield with moderate selectivity (dr 6:1). Protection of the C7-alcohol, MPM deprotection, and subsequent oxidation of the resulting primary alcohol produced aldehyde intermediate 83.

Completion of herbimycin commenced with the assembly of the acyclic framework 92 (Scheme 27). Subjection of aldehyde 83 to the lithiated aromatic chromophore 82 resulted in formation of the secondary benzylic alcohol 92 in a combined yield of 80% (dr 1:1). The undesired C15-epimer was conveniently recycled into the natural C15-epimer via a two-step process (56% yield). Following reported protocol for macbecin I 3a [35], advanced intermediate 92 was converted to herbimycin A 2a and thus concluded the first enantioselective synthesis of this ansamycin in 41 steps with 1.9% overall yield starting with methyl-α-d-mannopyranoside 84 (longest linear sequence).
2.4.2 Formal synthesis of (±)-macbecin I by Kallmerten (1990)
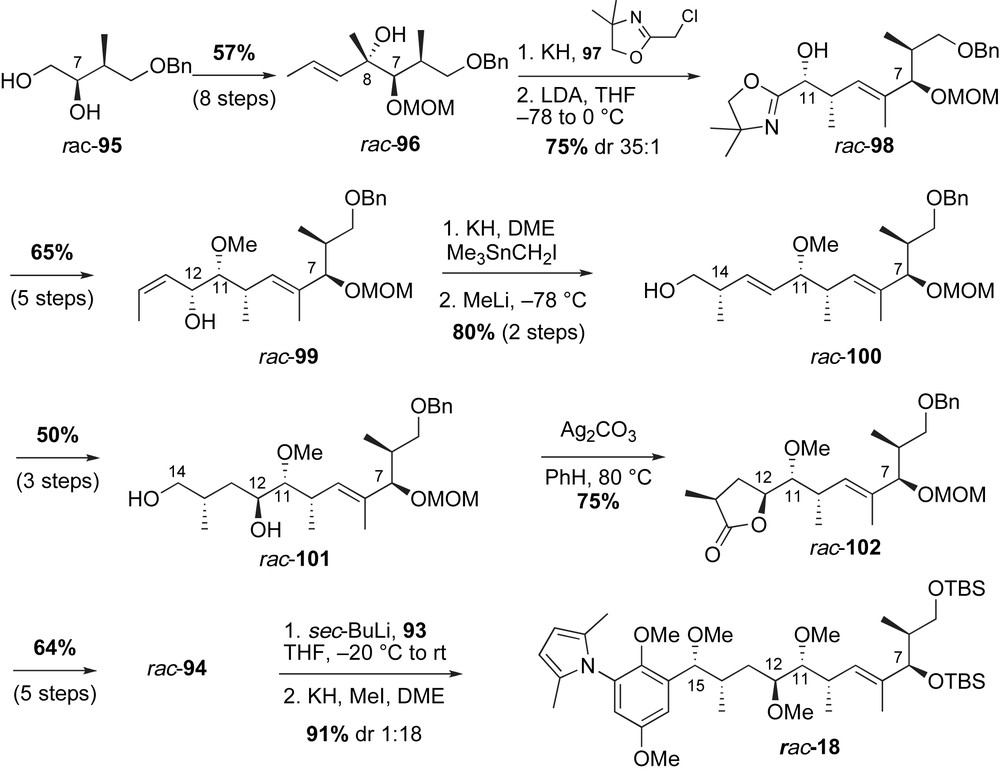
In 1990, Kallmerten reported a racemic synthesis of the carbon backbone of macbecin I with elegant use of sequential sigmatropic rearrangements (Scheme 28) [66]. Disconnection at the C15–C16 bond of rac-18 reveals the aromatic bromide 93 and the lactol subunit rac-94. The aniline functionality was masked with a 2,5-dimethylpyrrole group. Racemic lactol 94 was envisioned to arise from successive [2,3]-Wittig rearrangements.

The preparation of the required C5–C21 fragment of macbecin I is illustrated in Scheme 29. Racemic diol 95 was transformed to tertiary allylic alcohol rac-96 in eight steps and 57% overall yield. The alcohol was alkylated with chloromethyl oxazoline 97 and the obtained ether was treated with LDA to trigger [2,3]-Wittig rearrangement to produce rac-98 in 75% yield with high diastereoselectivity (35:1). The Wittig rearrangement established the syn methyl-oxygen relationship at C10–C11 and simultaneously generated the desired (E)-trisubstituted olefin. A second sigmatropic rearrangement was exploited to establish the remaining stereocenters of the ansa chain. The desired sigmatropic precursor, (Z)-allylic alcohol rac-99, was prepared from the Wittig adduct rac-98 in 5 steps. Alkylation of the C12-alcohol rac-99 with iodomethyl trimethylstannane and subsequent treatment with methyl lithium initiated the rearrangement. The resulting [2,3]-Wittig product rac-100 installed the C14-methyl group in 80% yield and as a single diastereomer. The disubstituted olefin in rac-100 was chemoselectively oxidized to introduce the C12-hydroxyl rac-101 via epoxidation and oxirane reduction sequence. Selective oxidation of diol rac-101 with Fetizon's reagent afforded the desired lactone rac-102 in 75% yield. The masked aldehyde rac-94 was prepared from DibalH reduction of lactone rac-102. Exposing lithiated aryl chromophore 93 to lactol rac-94 gave a mixture of diols that were subsequently alkylated with methyl iodide. Unfortunately, it was the minor product, rac-18, that contained the C15-methyl ether in the correct configuration. The authors suggested that the addition of anion 93 to lactol rac-94 occurred via internal chelation with the C12-alcohol to give the Cram-chelate product as the major isomer.
It is important to note that aryl lithium additions to other ansamycin skeletal frameworks have proceeded in Felkin–Anh additions, resulting in the correct C15-stereochemistry. These experiments were first reported by Tatsuta and will be further discussed in the following two syntheses by Martin and Cossy.
2.4.3 Formal synthesis of (+)-macbecin I by Martin (1992)
In 1992, Martin and co-workers reported an asymmetric synthesis of macbecin I (Scheme 30) [67]. The assembly of macbecin I precursor 103 would arise from an aryl addition of 93 to lactol 104. The construction of skeletal framework 104 was expected to come from a furan-hydropyranone transformation adduct via functional group manipulation. The tetrahydropyran-ring system in 104 proved to be an excellent template for stereoselective introduction of C14-methyl and C12-methoxy substituents. The syn methyl-oxygen relationships at C6–C7 and C10–C11 were incorporated using Evans' aldol reactions [37].
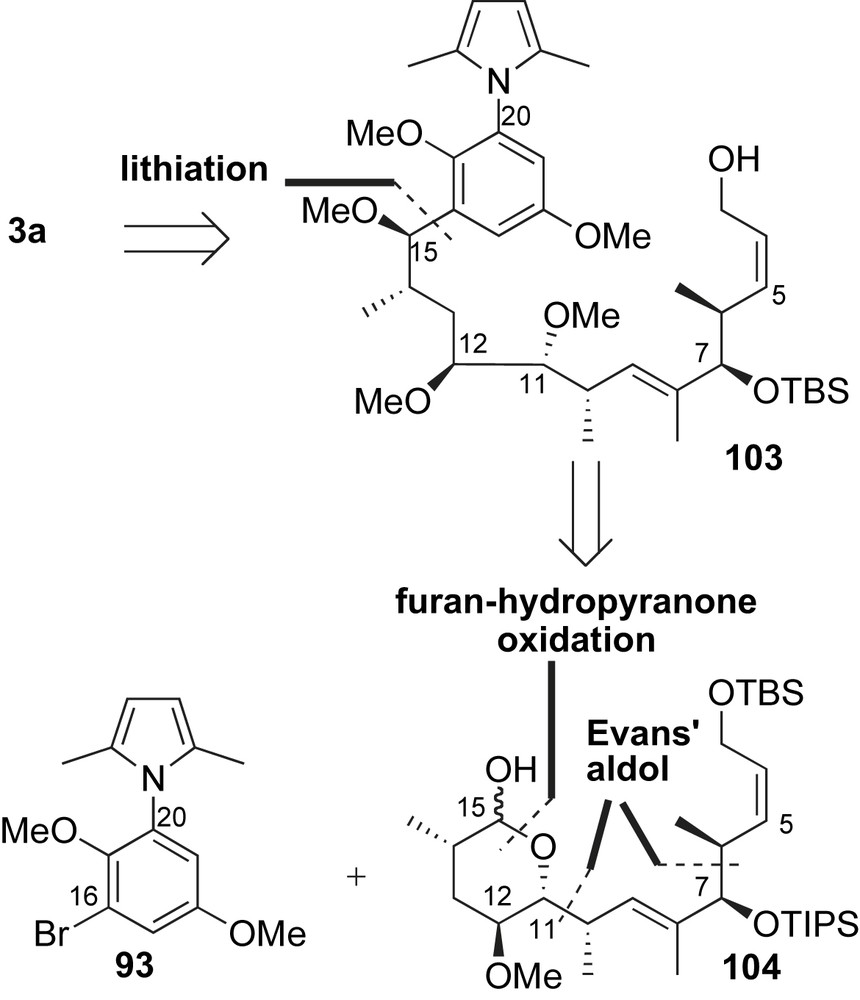
Martin's synthesis of the ansa chain utilized furan to pyranone oxidation, Evans' aldol and aryl alkylation reactions (Scheme 31). Condensation of 2-furaldehyde 105 with the Z(O)-boron enolate of 10 afforded the aldol adduct 106 in 92% yield. Oxidation of the furan 106 to dihydropyranone, followed by TBS protection of the resulting anomeric alcohol furnished a separable mixture of C15-isomers 107 (dr 3:1). Conjugate addition of lithium dimethyl cuprate to enone 107 in the presence of trimethylsilyl chloride yielded C14-methyl pyranone in 97% yield. Reduction of the pyranone with NaBH4 proceeded stereoselectively with concomitant cyclization to give the γ-lactone 108 in 89% yield. Unfortunately, the various hydride donors examined all produced the undesired C12 axial alcohol. Inversion of the stereocenter to the correct C12-epimer was attained with a Mitsunobu reaction. Accordingly, lactone 108 was reduced to a diol and the primary alcohol was selectively protected as a TBS ether 109 in 58% yield (two steps). Exposure of the secondary alcohol 109 to p-nitrobenzoic acid and DEAD in benzene afforded the desired product 110 after hydrolysis (80% yield over two steps). The inverted C-12 alcohol was methylated and subsequently homologated to α,β-unsaturated aldehyde 111. Treatment of this aldehyde with the Z(O)-boron enolate of 10 gave the syn allylic alcohol 112 in 91% yield with high diastereoselectivity. Imide 112 was further derivatized to compound 104 over a 7-step sequence. Subjection of advanced intermediate 104 to lithium reagent 93 produced benzylic alcohol 113 as the major product (C15, dr 3.5:1). In contrast, addition of five-membered lactol to anion 93 provides the undesired epimer at C15 as the major product (see Section 2.4.2). The major isomer 113 was converted to alcohol 103, an intermediate in Baker's synthesis of macbecin I (Section 2.3.1) which completed the formal synthesis of this ansamycin.

2.4.4 Cossy's synthesis of herbimycin A (2007)
Cossy and co-workers reported the most recent synthesis of herbimycin A. In their publication [68], the authors utilized aryl alkylation reaction of 115 and 83 to provide the seco-amino acid 114 (Scheme 32). The (Z)-olefin of the diene system 114 was envisioned to arise from a ring closing metathesis (RCM) [69]. Assembly of the aldehyde fragment 83 would arise from elongation of Roche ester 116 (16 steps and 25% overall yield) using sequential allylmetal additions to install five of six stereogenic centers (C6–C7 and C10–C12). In comparison, Tatsuta homologated the readily available methyl α-d-mannopyranoside 84 (28 steps and 3.6 overall yield) to establish three of six stereocenters of intermediate 83 (Scheme 26) [64].
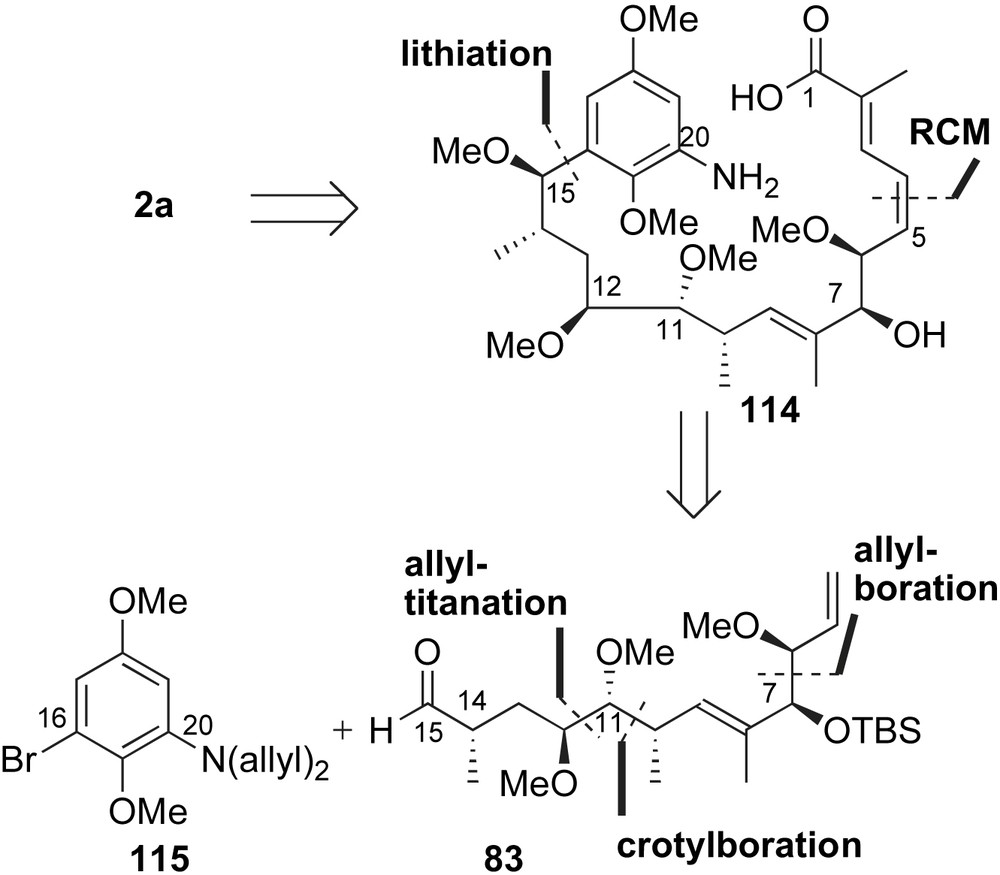
The acyclic skeleton of herbimycin A was prepared using sequential allylmetal addition reactions (Scheme 33). Commercially available hydroxy ester 116 was converted to the homo-methyl aldehyde 117 in four steps and 80% yield. Exposing aldehyde 117 to allyltitanium reagent (S,S)-118 provided the desired C12-homoallylic alcohol 119 with high yield and diastereoselectivity. Methylation of alcohol 119, followed by olefin isomerization and subsequent oxidative cleavage, afforded α-methoxy aldehyde 120. The syn stereochemical relationship at C10–C11 was constructed using a matched crotylation reaction of aldehyde 120 and (Z)-crotylboronate 66 [56]. The resulting homoallylic alcohol 121 was modified to α,β-unsaturated aldehyde 122 in four steps, including a Corey–Schlessinger olefination [65]. Addition of (Z)-γ-methoxy allylborane 56 [54] to aldehyde 122 afforded the syn product 123 in 69% yield after TBS protection of the nascent C-7 alcohol. The low diastereoselectivity (dr 4:1) observed in this oxyallylboration reaction is consistent with Tatsuta's report on a similar system (Scheme 26). Interestingly, the presence of an aromatic chromophore in the aldehyde electrophile yields higher reaction selectivity (Scheme 17) [52]. Selective deprotection of TBDPS group with ammonium fluoride and oxidation of the resulting primary alcohol afforded the desired C5–C15 fragment 83.
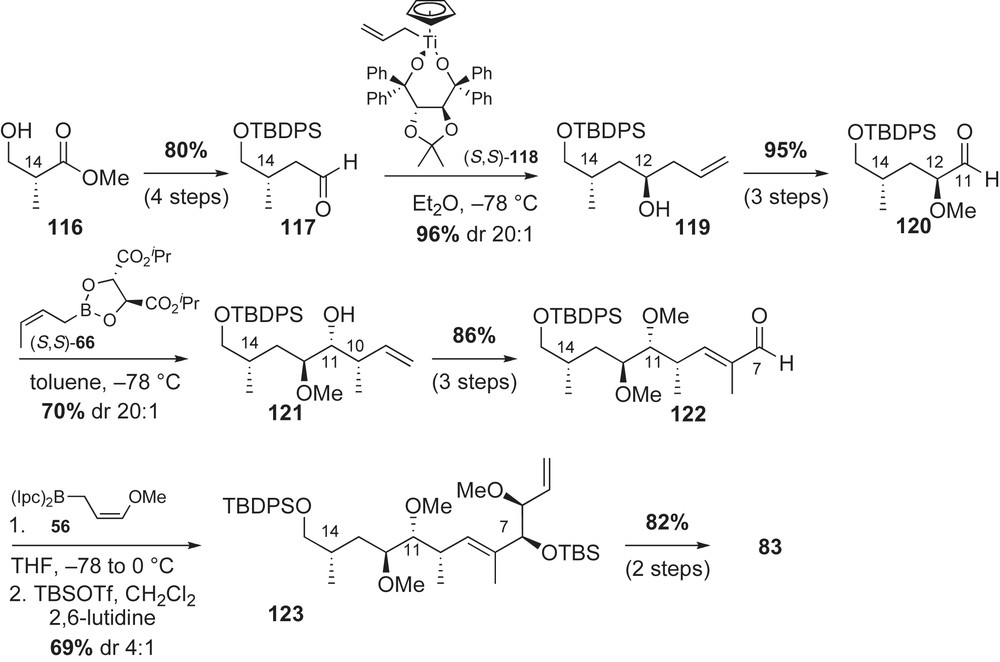
Completion of the ansa framework of herbimycin A utilized late-stage aromatic alkylation and RCM [69] reactions (Scheme 34). Addition of organolithium reagent 115 to aldehyde 83 gave a separable mixture of benzylic alcohol epimers. The desired Felkin–Anh product 124 was obtained in 54% yield from a 1.6:1 ratio of adducts. Advanced intermediate 124 was further functionalized to provide the RCM precursor 125 in five steps. The C7-acrylate was subjected to metathesis conditions with Grubbs II catalyst 126 to afford unsaturated lactone 127 in 72% yield. Reduction of the lactone 127 with DibalH and treatment of the resulting lactol with a stabilized phosphonate reagent completed the preparation of the (E,Z)-dienoate system 128. Following reported protocols for macrocyclization and urethane formation, Cossy and co-workers completed the total synthesis of herbimycin A 2a.
The synthesis of herbimycin A by Cossy is very comparable to the other reports in overall yield (1.5%, longest linear sequence) and number of steps (30, beginning with Roche ester 116). The authors utilized selective allylmetal additions to install stereochemical relationships within the ansa chain of the natural product. Use of RCM to produce α,β-unsaturated lactone and thus setting the (Z)-olefin of the dienoate is a complimentary approach to the standard olefination procedures.
2.4.5 Micalizio's synthesis of (+)-macbecin I (2008)
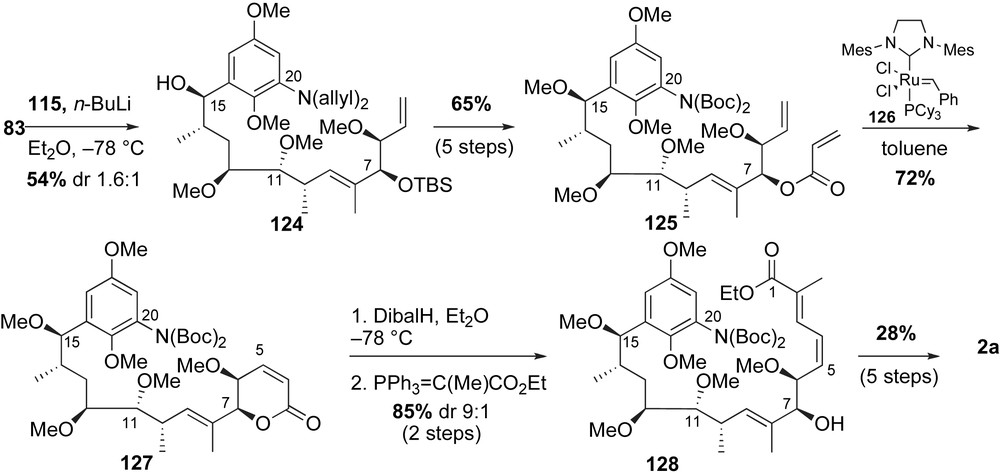
The most recent total synthesis of (+)-macbecin I 3a was reported by Micalizio and co-workers (Scheme 35) [70]. The preparation of the ansamycin antibiotic precursor 129 was envisioned through a metal-mediated reductive coupling of aniline methyl alkyne fragment 130 with β,σ-unsaturated aldehyde 131. Nucleophilic addition and substitution as well as propargylation installed the required stereochemical relationships of the alkyne fragment 130. Conversely, the (E,Z)-diene of the aldehyde fragment 131 was obtained via a palladium cross-coupling strategy.
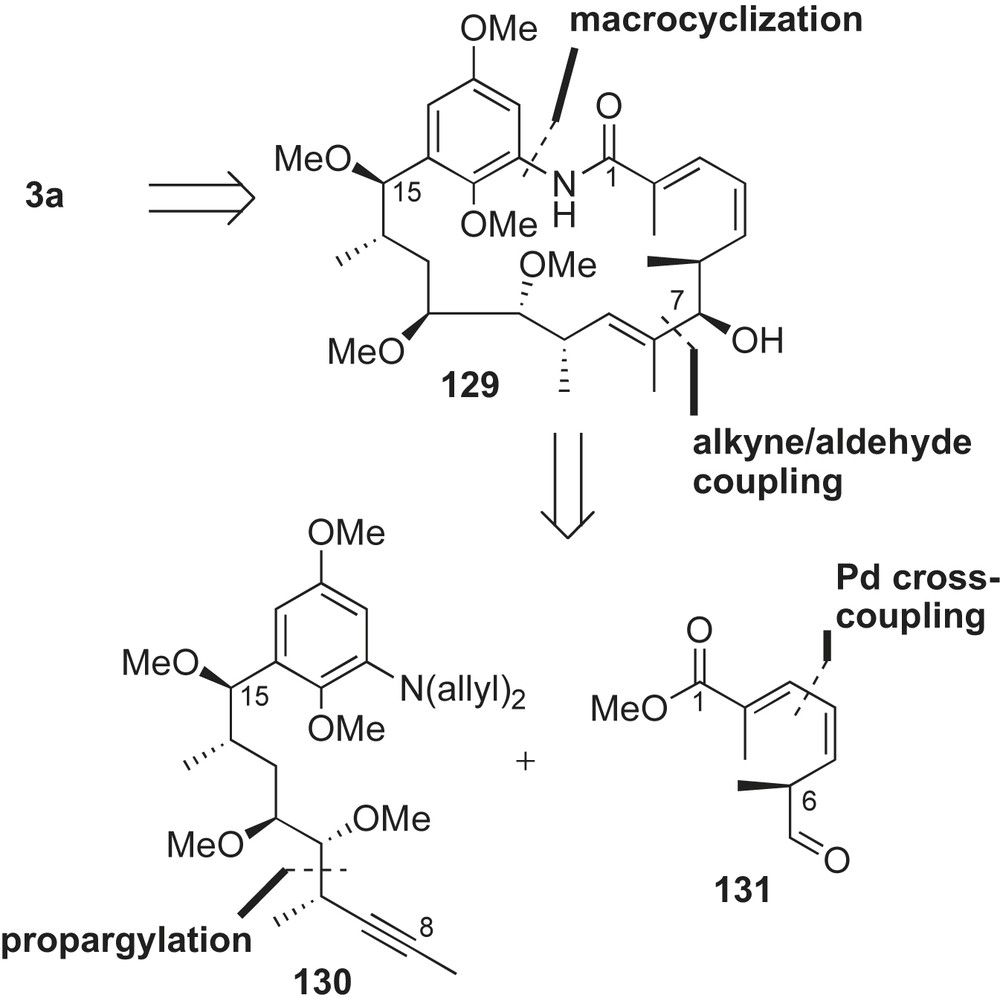
Preparation of the C8–21 coupling partner 130 of macbecin is depicted in Scheme 36. Subjection of Roche ester derived α-methyl iodide 132 (three steps from Roche ester 116) to pseudoephedrine based auxiliary 133 triggered formation of the alkylated adduct in moderate selectivity (dr 9:1). Reductive cleavage of the auxiliary and Dess–Martin oxidation of the resulting primary alcohol yielded α-methoxy aldehyde 134 in 70% yield over two steps.
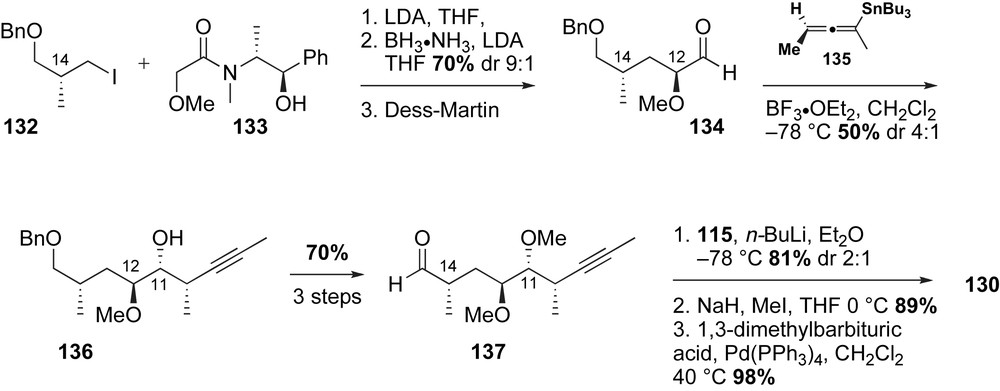
Installation of the C11,C12-anti-stereochemistry of alkyne fragment 130 was triggered with a diastereoselective propargylation utilizing chiral allenyl stannane 135. The newly emerged C11-homopropargylic alcohol 136 was obtained in modest yield and selectivity (50% yield, dr 4:1); partial epimerization of stannane 135 was responsible for the low diastereoselection. The C11-alcohol was methylated and subsequently converted to aldehyde 137 in two steps. In analogy to Cossy's method, nucleophilic addition of the aryl lithium reagent derived from 115 to aldehyde 137 proceeded to give C15-benzylic alcohol in 81% yield, albeit low stereoselection (dr 2:1). Fortunately, the minor C15-isomer was recycled to the desired alcohol via a two step oxidation/reduction sequence. Methylation of C15-alcohol followed by deallylation furnished the required C8–C21 alkyne fragment 130 of macbecin.
The synthesis of the homoconjugated aldehyde 131 featured two successive Pd(PPh3)4 catalyzed reactions (Scheme 37). Accordingly, exposure of α-silyloxy aldehyde 138 with alkyne 139 in the presence of palladium(0) provided the anti-homopropargylic alcohol 140 in 82% yield and modest diastereoselectivity (dr 5:1). Sonogashira coupling of 140 with readily available vinyl iodide 141 furnished enyne 142 in high yield. Finally, a three-step derivatization of 142 effected formation of (E,Z)-diene containing aldehyde 131 in 90% yield.
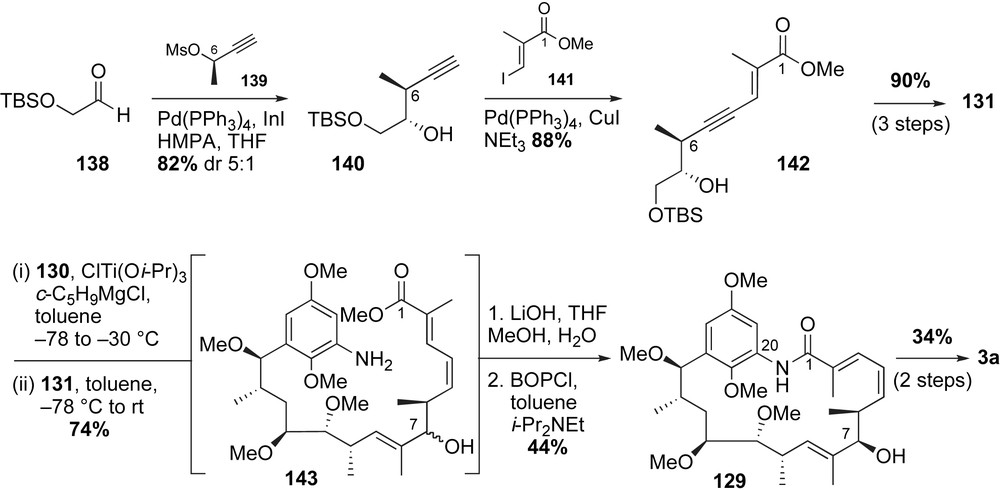
Completion of the total synthesis of macbecin 3a was highlighted by the titanium-mediated coupling reaction developed in Micalizio's laboratories (Scheme 37) [70]. Reductive titanium alkoxide mediated cross-coupling of a highly functionalized methyl alkyne 130 with the sensitive polyunsaturated aldehyde 131 provided a mixture of regio- and diastereomers 143 in 74% yield. Sequential exposure of alcohol 143 to LiOH and BOP–Cl afforded the desired macrocycle of macbecin 129 in 44% yield. Gratifyingly, the minor regio- and stereoisomers obtained from the titanium-mediated coupling reaction were effectively removed. Urethane installation at C7-alcohol and oxidation of the aromatic moiety to quinone with CAN completed the preparation of (+)-macbecin 3a.
Micalizio's synthesis of macbecin incorporates a more convergent route to the 19-membered macrolactam. The synthesis featured key reaction methodologies which afforded products in moderate yields and selectivities. Overall, the synthesis of macbecin was accomplished in 17 steps (1.3% yield, longest linear sequence) starting from commercially available Roche ester 116.
2.5 Conclusions
In this account, different approaches toward the synthesis of C15-ansamycin antibiotics were surveyed. Macrocyclization of the ansa chain framework was completed utilizing either BOP–Cl coupling of the seco-amino acid or intramolecular copper-mediated amidation of a bromo-amide. The assembly of these acyclic skeletons relied on the biomimetic elongation of the aromatic moiety through sequential aldol, allylmetal, and/or alkylation reactions. An alternate strategy exploited late-stage union of the aromatic chromophore to the preformed C15-ansa chain. In this approach, various methodologies were successfully applied to install the required stereocenters of the molecule. Among these, [2,3]-Wittig rearrangement and a furan–pyran oxidation were developed. The C15-ansamycin class of natural products has served to further develop novel macrocyclization and asymmetric C–C bond forming reactions.
3 C17-Benzene ansamycin antibiotics
3.1 Isolation, structure and bioactivity
Similar to the C15-ansamycins, the ansatrienins (or triene-ansamycins) constitute more than 30 members of the benzene ansamycin antibiotics (Figs. 6 and 7). Structurally, the C17-aliphatic ansa chain encompasses a C3-methyl ether and a C11–C13 stereotriad flanked by a (Z)-trisubstituted olefin and a conjugated (E,E,E)-triene. Notably, the ansatrienin biological diversity arises from the structural variations of the aromatic chromophore and the amino acid side chain at C11. The various oxidation states and substitution patterns define the aromatic moiety, whereas the C11 side chain is one of many N-acylated amino acids that are predominately derived from d-alanine or aminocyclopropane. These subtle structural changes originate in wide range of biological activities for ansatrienin ansamycins.

Triene-ansamycin antibiotics.
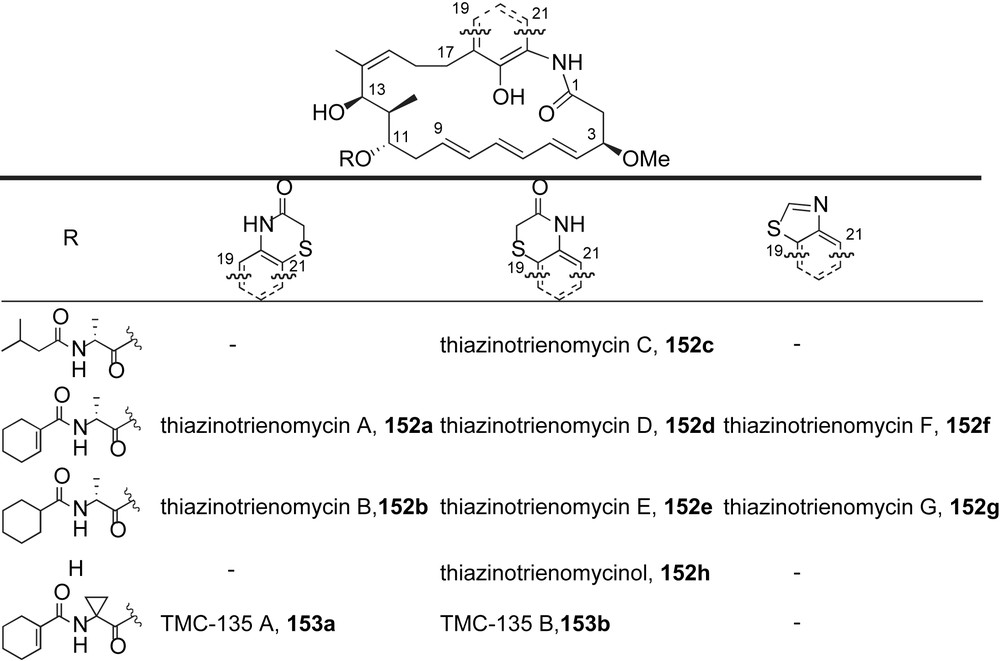
Triene-ansamycin antibiotics.
The parent compound of the C17-ansamycins, mycotrienin (renamed mycotrienin II), was first isolated from a strain of Streptomyces in 1967 by Coronelli [71]. Between 1981 and 1983, mycotrienins I and II (ansatrienins A and B) 144a–b were isolated from fermentation broths of Streptomyces collinus [72] and Streptomyces rishiriensis T-23 [11a,73]. Additional three congeners, ansatrienins A2–A4 144c–e [11b,74], were elucidated as minor components of these cultures. They differ from the parent at the C11-acyl residue. The mycotrienins were shown to have potent antifungal activity, yet weak antitumor activity [11b,73b]. (+)-Mycotrienol I 145a and its hydroquinone derivative (+)-mycotrienol II 145b [75] are the least active natural products of this family, presumably due to lack of alanine-derived side chain at C11.
In 1985, Umezawa and co-workers isolated five new ansamycins, trienomycins A–E 146a–e, from the culture broth of Streptomyces sp. No. 83-16 [76]. Not surprisingly, the trienomycins are very similar to ansatrienins except for the aromatic moiety (phenol versus quinone). In 1991, Smith and co-workers discovered and characterized another congener of the family, trienomycin F 146f [77]. Although structurally similar to mycotrienins, trienomycins showed very little antimycobacterial properties, but rather exhibited promising in vitro cytotoxicity against HeLa-S3, PLC hepatoma and L5178Y murine leukemia cell lines [76a,78]. The difference in the bioactivity (antibacterial versus antitumor) of the triene-ansamycins was later ascribed to the nature of the benzenoid moiety.
Between 1995 and 2007, distinct members of the C17-ansamycins were isolated possessing various structural modifications. Hydroxymycotrienins A–B 148a–b, produced by Bacillus strains [79], contain hydroxyl groups at C23 of the benzoquinone. On the other hand, cytotrienins A–D 149a–d [80] possess an unusual aminocyclopropane carboxylic acid side chain. Among these, cytotrienin A notably demonstrated an ability to induce apoptosis in human acute promyelotic leukemia HL-60 cells with an ED50 value of 7.7 nM [81]. More recent additions to the ansatrienin family, trierixin 150 [82], UCF-116 A–C 151a–c [83], and trienomycin G 146g [84] were obtained from diverse culture broths of Actinomycete strains. These natural products were found to inhibit various enzymes and transcription factors involved in carcinogenesis or cellular stress.
A novel class of cytotoxic ansamycin antibiotics, thiazinotrienomycins A–E 152a–e [85,86], was isolated in 1995 by Hosokawa and co-workers (Fig. 7). Thiazinotrienomycins are distinct from other ansatrienins with the presence of a benzothiazine ring system. These heterocyclic ansamycins differ in the connectivity of the benzothiazine moiety and the structure of the C11-acyl appendage. Related compounds, thiazinotrienomycins 152f–g [87], incorporate a benzothiazole ring as the aromatic chromophore. Other in vitro cytotoxic members of this family include TMC-135 A–B 153a–b [88].
Although triene-ansamycin antibiotics displayed promising antitumor properties, they were overshadowed by the C15-ansamycins upon discovery of their effectiveness in disrupting the Hsp90 complex.
3.2 Structure elucidation: determination of absolute and relative stereochemistry of triene-ansamycins
In 1982, Zeeck and co-workers elucidated the molecular structure of triene-ansamycins using extensive NMR analysis [11b,74]. However, it took nearly 10 years before the relative and absolute configurations of the C17-ansamycins were assigned. Toward this end, Smith and co-workers conducted degradative and analytical studies on trienomycins A–C 146a–c [89]. Deacylation of trienomycin A provided trienomycinol 147 (Fig. 6) that was subsequently converted to acetonide 154 (Scheme 38, path a). Ozonolysis followed by reductive workup furnished ketoaldehyde 155. Analysis of the 1H coupling constants within the dioxane ring of ketoaldehyde 155 revealed the C11–C12-anti and C12–C13-syn relative stereochemistry. To elucidate the absolute configuration of the C11–C13 fragment, Smith undertook and reported the enantioselective synthesis of the stereotriad's enantiomer, ent-155. The C9–C14 fragment synthesis of trienomycins by Fürstner [90] and Yadav [91] later confirmed these findings.
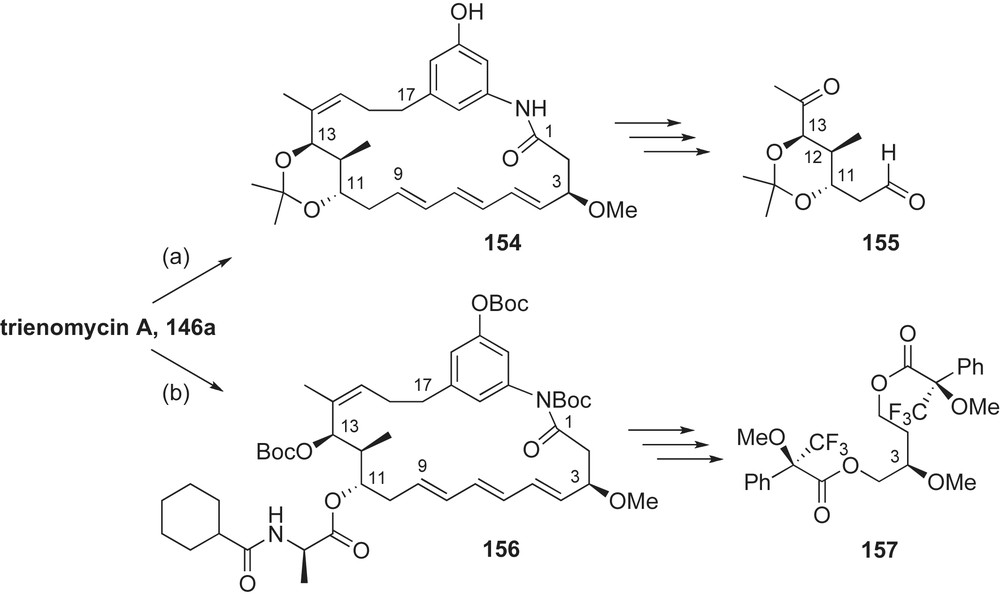
In order to assign the C3-methyl ether configuration of trienomycin A 146a, the natural product was protected as the tris-Boc derivative 156 (Scheme 38, path b). Reductive ozonolysis furnished a 1,4-diol that was subsequently derivatized as the bis-Mosher ester 157. Comparison of spectroscopic data of 157 with authentic samples unequivocally established the (R)-configuration at C3. In a similar study, Smith and co-workers established the relative and absolute stereochemistries of mycotrienins I and II via chemical correlation of (+)-trienomycin A 146a [92]. This investigative approach was also applied to the related heterocyclic thiazinotrienomycins [93]. These structure determination–stereochemical correlation studies have allowed for extension of stereochemical assignments (by analogy) to other members of the C17-ansamycin family.
3.3 Introduction: total syntheses of the triene-ansamycin antibiotics
This part of the account will provide a comprehensive overview of the synthetic approaches toward the ansatrienins. Specifically, previous total syntheses of (+)-trienomycins A 146a and F 146f, (+)-mycotrienin I 144a/(+)-mycotrienol I 145a and thiazinotrienomycin E 152e are presented. In addition, two approaches for preparation of the macrocyclic cores of cytotrienins 149 and ansatrienins 144 are also depicted. In the course of these contributions, two distinct strategies emerged with regard to the retrosynthetic disconnections. The first approach utilizes a late-stage installation of the (E,E,E)-triene system with concomitant macrocyclization (Scheme 39, path a). The macrolactam formation V → IIb was achieved through tandem inter/intramolecular reactions (double stitching) or olefin metathesis. An alternative sequence relies on late-stage assembly of the aromatic core VI with the fully functionalized ansa chain VII (Scheme 39, route b). This second strategy offers more flexibility in the choice of the aromatic moiety and thus allows for rapid assembly of the different ansatrienins.
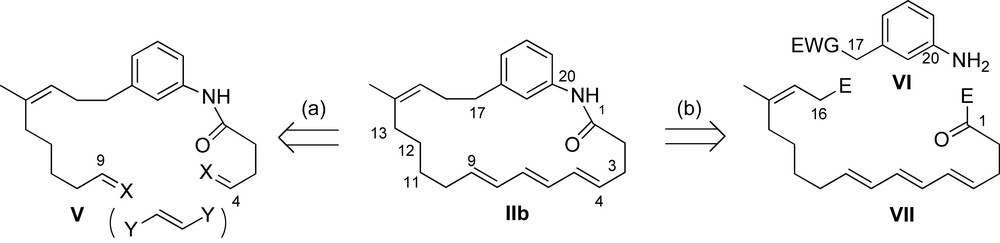
The next two sections will describe the different synthetic pathways, with particular emphasis on the installation of the C11–C13 stereotriad and the all (E)-triene system.
3.4 Approaches involving concomitant installation of the (E,E,E)-triene and macrocyclization
3.4.1 First total syntheses of trienomycins A and F by Smith (1995)
Smith and co-workers were the first to open up the field of the synthesis of triene-ansamycin antibiotics in 1995 [94]. From a retrosynthetic perspective, the trienomycins A 146a and F 146f were envisioned to come from a common advanced intermediate, (+)-trienomycinol 147 (Scheme 40). Construction of the (E,E,E)-triene moiety and thus macrocyclization was anticipated from a bis-Wittig olefination reaction of aldehyde 158 and phosphonium salt 159. The C4–C23 fragment 158 would arise from alkylation of sulfone 160 with allylic iodide 161. Acylation of aniline provided the aromatic fragment 160.
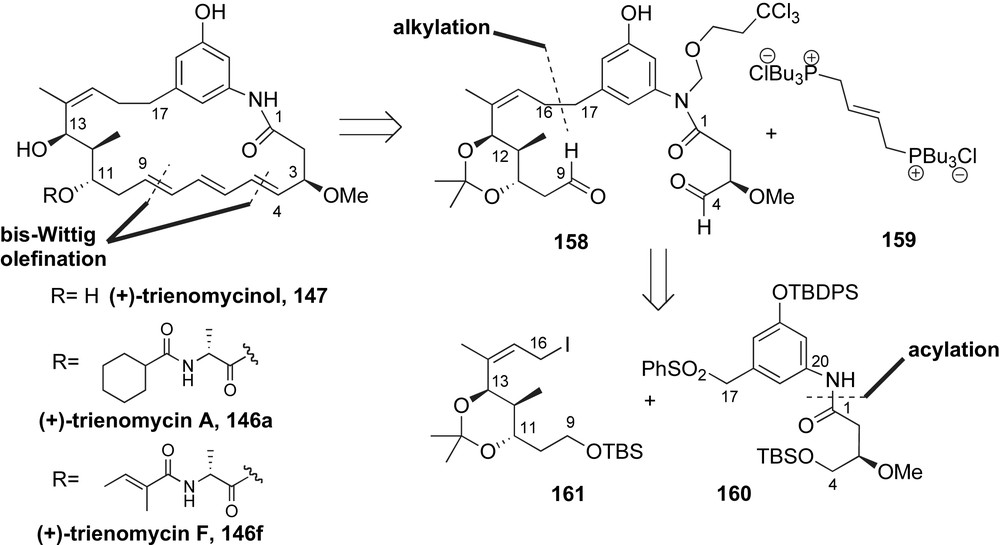
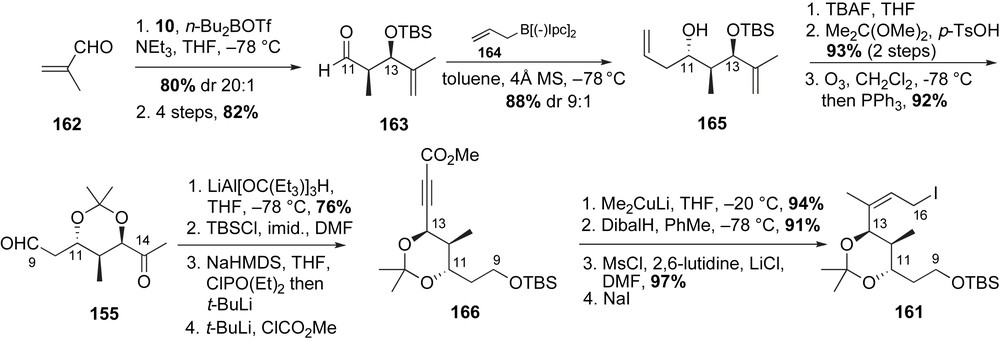
The C11–C13 stereotriad of the allylic iodide fragment 161 was installed using Evans' aldol and Brown's allylboration reactions. Treatment of methacrolein 162 with the Z(O)-boron enolate of oxazolidinone 10 [37] afforded the desired syn-C13-alcohol in 80% and high diastereoselectivity (Scheme 41). The aldol adduct was then converted to the C11-aldehyde 163 in 82% yield over four steps. The anti-stereochemical relationship at C10–C11 was installed using Brown's allylboration reagent 164 [95]. The resulting homoallylic alcohol 165 was produced in 88% yield and with good selectivity (dr > 9:1). Removal of the C13-TBS ether with TBAF, followed by acetonide formation and ozonolytic cleavage afforded the ketoaldehyde 155 in 86% yield over three steps. The C9-aldehyde was chemoselectively reduced with lithium tris[(3-ethyl-3-pentyl)oxy]aluminum hydride to primary alcohol and protected as TBS ether. Following Negishi's protocol [96], the C14-ketone was converted to the α,β-acetylenic ester 166 in 78% yield over two steps. Conjugate addition of lithium dimethyl cuprate to alkyne 166 furnished the required (Z)-trisubstituted olefin as one isomer (94% yield). A three-step transformation of methyl ester to allylic iodide completed the preparation of the advanced fragment 161.
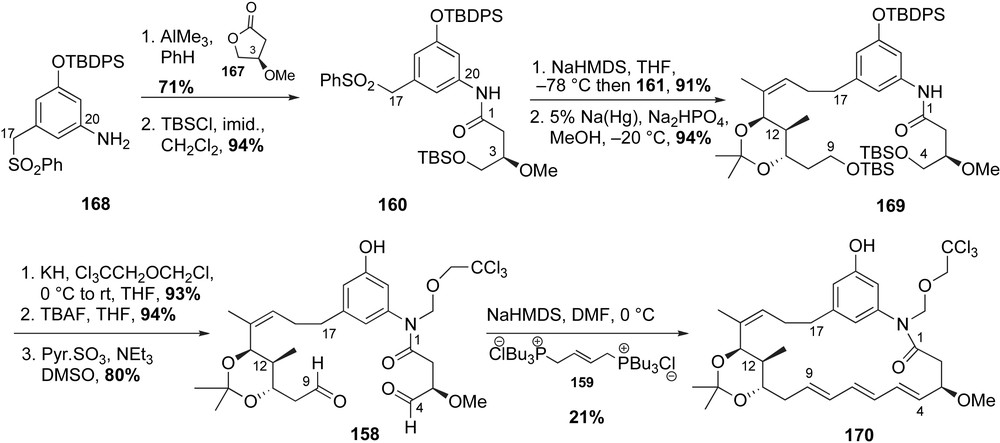
The key steps for formation of the macrocyclic core of trienomycins were aniline acylation, sulfone alkylation and Wittig olefination. The required acylation coupling partners, γ-lactone 167 [97] and aromatic sulfone 168,1 were obtained from d-malic and readily available 3-hydroxy-5-nitrobenzoic acids, respectively. Acylation of aniline 168 via ring opening of chiral lactone 167 and subsequent protection of the primary alcohol afforded the desired amide fragment 160 in 73% yield (Scheme 42). Sulfone 160 was deprotonated with NaHMDS and treated with allylic iodide 161. Mercury-mediated desulfonylation furnished the desired Wittig precursor 169. At this stage, the free amide was protected to prevent competitive formation of an N-acylhemiaminal 171, which did not react under Wittig conditions (Scheme 43). Subjection of amide 169 to chloromethyl 2,2,2-trichloroethyl ether [98] followed by silyl ethers deprotection and Parikh–Doering oxidation delivered the bis-aldehyde 158 (70% yield, three steps). The critical Wittig olefination of 158 with phosphonate salt 159 effected macrocyclization with formation of the (E,E,E)-triene 170 (21% yield). A significant amount of other triene isomers (34%) was also detected.
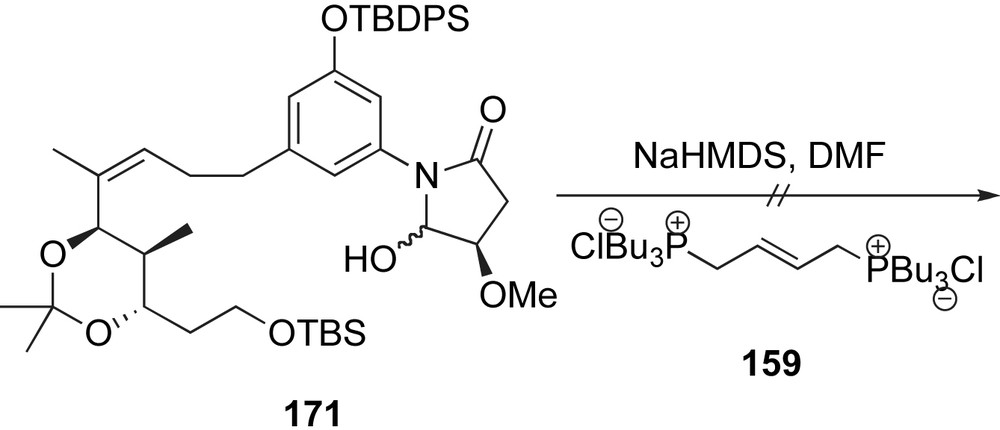
Completion of the synthesis required selective installation of the amino side chains (Scheme 44). As such, phenol 172 was protected as a TBS ether and the amide was deprotected with (Na)Hg in MeOH to reveal intermediate 173. Exposure of acetonide 173 to CSA unveiled the 1,3-diol at C11–C13. This intermediate was regioselectively esterified at C11 by a symmetrical anhydride of FMOC-d-alanine [89]. Liberation of the primary amine group with diethylamine followed by BOP-mediated coupling with either cyclohexenecarboxylic or tiglic acid provided the respective natural product precursors. Removal of the final protecting group ensued synthesis of trienomycin A 146a and F 146f in 38% and 23%, respectively, over the last three steps.

Smith and co-workers completed the first total synthesis of trienomycins and provided a viable route toward the formal synthesis of mycotrienins. Trienomycin A was completed in 29 steps with 0.77% overall yield starting from methacrolein (longest linear sequence), while trienomycin F was obtained in 0.47% overall yield. The benzenoid moiety of trienomycins was oxidized to the corresponding quinone using Fremy's salt [92]. Due to the low-yielding ring closure, alternative approaches were investigated to improve formation of the macrocyclic core of other ansatrienin congeners.
3.4.2 Total synthesis of (+)-mycotrienol I and (+)-mycotrienin I by Panek (1997)
Application of asymmetric crotylsilane bond construction methodology resulted in total syntheses of mycotrienin I 144a and mycotrienol I 145a in 1997 [99]. Installation of the triene moiety in mycotrienins was envisioned to come from Stille-type cross-coupling of bis-iodide 174 and enedistannane 175 (Scheme 45). This type of macrocyclization technique was previously applied in the synthesis of both rapamycin [100] and dynemycin [101]. Further disconnection at C16–C17 bond revealed dimethyl acetal containing fragment 176 and sulfone intermediate 177. The two subunits are closely related to fragments utilized in Smith's synthesis of trienomycins. The stereogenic centers of both the propionate unit 176 and aromatic sulfone 177 were installed using the crotylsilane reagent 31b [41].
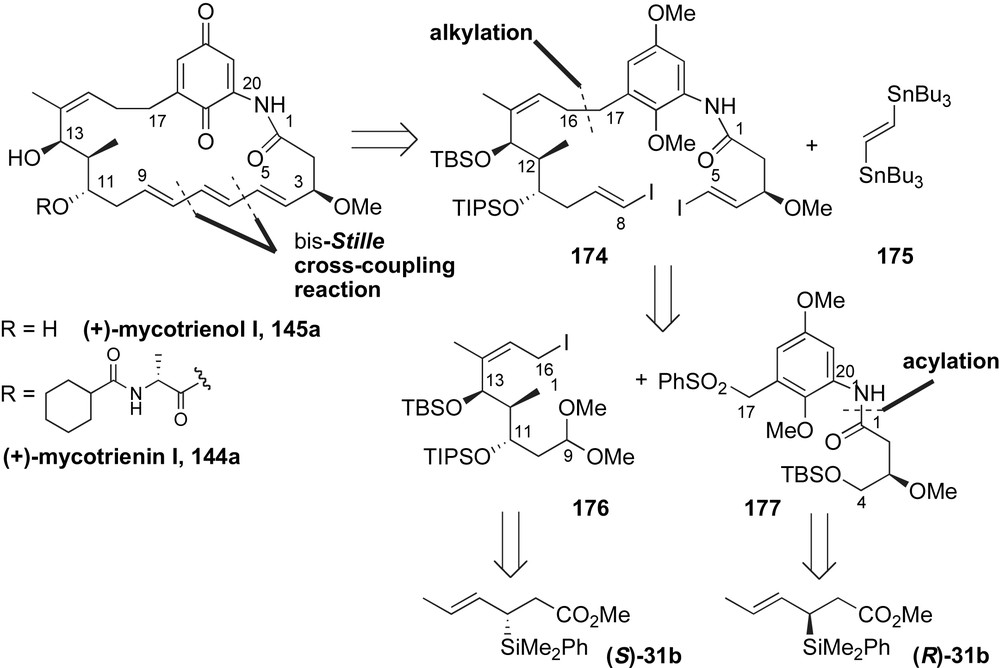
The preparation of the C9–C16 fragment 176 relied on use of enantioenriched silicon reagents to introduce the C11–C13 stereotriad (Scheme 46). Condensation of α-keto-dibenzylacetal 178 and (E)-crotylsilane reagent (S)-31b provided the homoallylic benzyl ether 179 in high yield and diastereoselectivity (80%, dr 30:1). The anti-SE′ mode of addition, illustrated in the antiperiplanar transition state, accounts for the high levels of relative and absolute stereocontrol. The crotylation product 179 was converted to α-methyl aldehyde 180 in 70% yield over three steps. Hosomi–Sakurai-type allylation [102] of 180 with TMS-allylsilane afforded the C11-alcohol with high diastereoselectivity. The 1,3-anti-relationship was rationalized by a nucleophilic addition to a titanium chelate between C13-benzyl ether and C11-aldehyde [103]. The newly emerged C11-alcohol was protected as TIPS ether and converted to dimethyl acetal containing fragment 181. Catalytic hydrogenation of benzyl groups effected simultaneous cyclization to γ-lactone. Elimination of the tertiary C14-acetoxy group with DBU afforded α,β-unsaturated lactone 182 in 93% yield over two steps. Reductive opening with LiAlH4 unveiled the (Z)-olefin and subsequent conversion of the primary alcohol to allylic iodide provided the C9–C16 fragment 176 in 14 steps (21% overall yield).
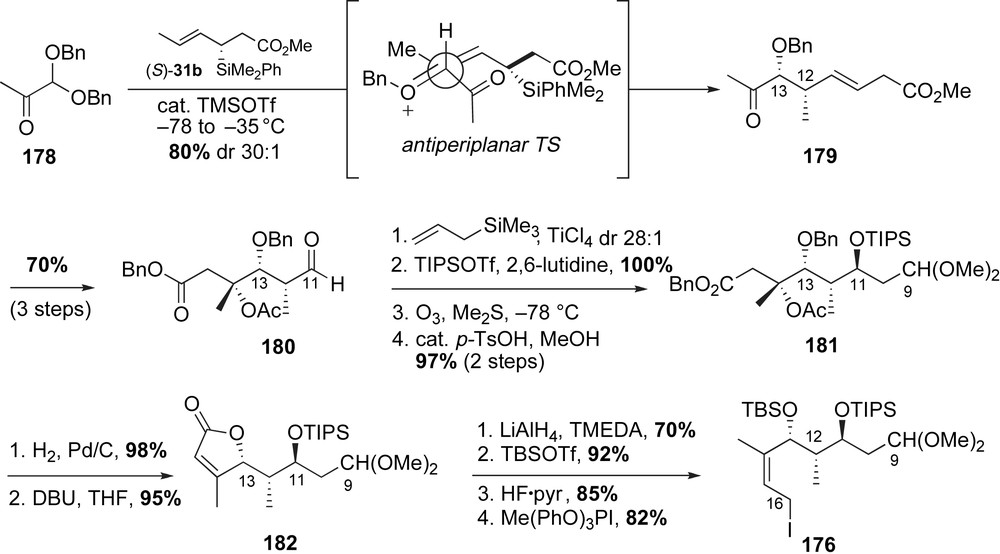
Completion of mycotrienin I and mycotrienol I relied on construction of the Stille-type precursor 174 utilizing aniline acylation, sulfone alkylation and Takai olefination reactions followed by macrolactamization (Scheme 47). Smith obtained the C3-methyl ether containing γ-lactone 167 from d-malic acid in four steps (refer to Scheme 44). Panek and co-workers modified their enantiopure crotylsilane reagent (R)-31b to prepare lactone 167 via Fleming–Tamao's procedure for silyl to hydroxyl interconversion. Acylation of 2,5-dimethoxy aniline 183 with lactone 167 in the presence of AlMe3 provided the benzylic sulfone fragment in 70% yield. The resulting primary alcohol was protected as a TBS ether to complete the preparation of C4–C16 fragment 177. The critical coupling of benzylic sulfone 177 with allylic iodide 176 followed by reductive desulfonylation afforded the dimethyl acetal containing subunit 184 in 78% yield (two steps). Formation of bis-aldehyde substrate was achieved in three steps (68% yield). Subsequent homologation to the bis-(E,E)-vinyl iodide 174 was achieved using the Takai olefination protocol [104]. The crucial Stille-type “double stitching” cyclization of 174 with enedistannane 175 successfully installed the desired (E,E,E)-triene of mycotrienins. Oxidation of the aromatic core followed by immediate global deprotection of all masked oxygen atoms furnished mycotrienol I 145a in 0.86% overall yield over 27 steps (longest linear sequence starting from pyruvate aldehyde dimethyl acetal). In addition, selective deprotection of the C11-secondary TIPS group over the C13-TBS silyl ether permitted regiospecific introduction of the side chain following Smith's precedent [89]. Oxidation of the aromatic core and subsequent removal of the remaining protecting group delivered mycotrienin I 144a in 0.40% overall yield over 31 steps.
The Stille-type cross-coupling strategy used in preparation of mycotrienins installed the configurationally pure (E,E,E)-triene. Since formation of other triene isomers was not observed, this method underscores the efficacy of this macrocyclization step.
3.4.3 Synthesis of the macrocyclic core of cytotrienins by Panek (2004)
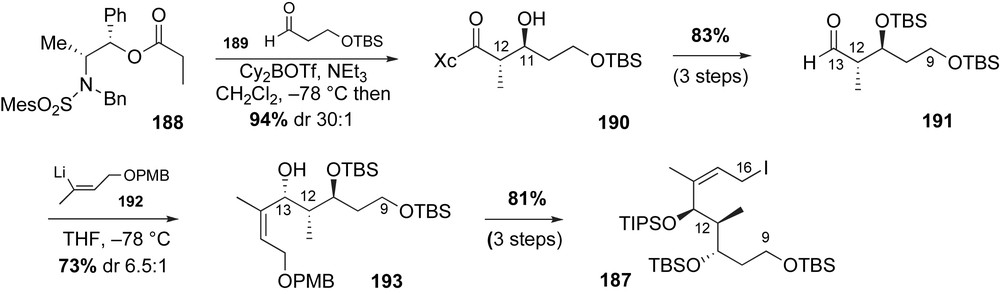
Inspired by recent advances in ring closing metathesis reactions [105], the Panek group initiated a revised route to the macrocyclic core of cytotrienins 185 in 2004 (Scheme 48) [106]. It was predicted that RCM of bis-diene 186 would install the C4–C9 (E,E,E)-triene and simultaneously effect macrocyclization. Disconnection at the C16–C17 bond of the triene precursor 186 revealed the prevalent allylic iodide 187 and sulfone 177 fragments. In contrast to their previous work on the mycotrienins, the stereogenic centers (C11–C13) of the stereotriad fragment 187 were introduced using aldol and Felkin-controlled addition reactions. Alternatively, construction of the aromatic sulfone 177 was attempted using a copper(I)-mediated amidation reaction.

Preparation of the C9–C16 subunit 187 with aldol and anion addition reactions resulted in a more convergent and efficient sequence (46% over eight steps) than their earlier approach (21% yield over 14 steps). The synthesis of the stereotriad commenced with the installation of the anti-C11–C12 relationship utilizing Abiko and Masamune's anti-aldol methodology (Scheme 49) [107]. Condensation of E(O)-boron enolate of (−)-norephedrine derivative 188 with aldehyde 189 afforded the anti-addition adduct 190 in 94% yield and with high selectivity (dr 30:1). The C11-alcohol was protected as a TBS ether and subsequent auxiliary cleavage yielded α-methyl aldehyde 191. Treatment of aldehyde 191 with the vinyllithium reagent 1922 produced the C13-alcohol as the major Felkin-adduct from a 6.5:1 epimeric mixture. Conversion of the stereotriad 193 to the primary iodide 187 completed the fragment synthesis (three steps, 81% yield).
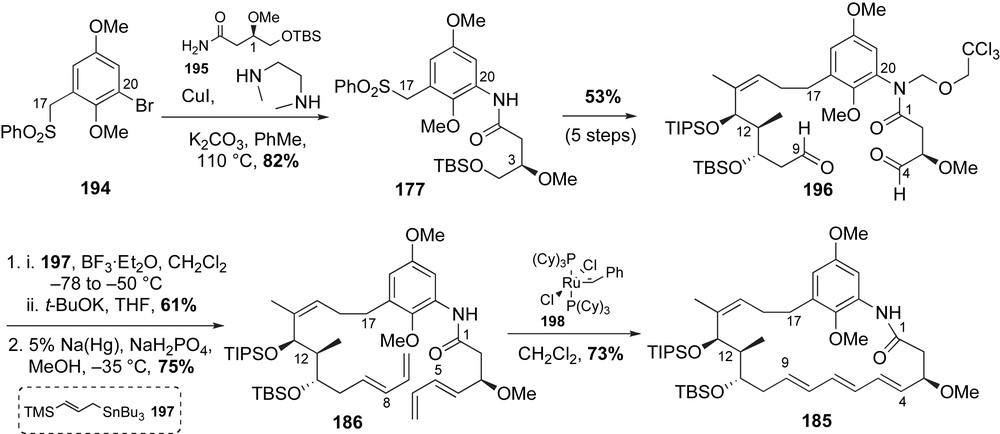
Fragment coupling, bis-diene installation and RCM were all that remained for completion of the macrocyclic core of cytotrienins (Scheme 50). The aromatic sulfone 177 was obtained utilizing Buchwald copper(I)-amidation [58]. Union of phenylbromide 194 and primary amide 195 in the presence of CuI afforded the desired fragment 177 in 82% yield. Following Smith's reported protocol for formation of bis-aldehyde 196, sulfone 177 was alkylated with allylic iodide 187 and the product amide was masked as the trichloroethylmethyl ether (five steps, 53%). Exposure of 196 to allyl tin reagent 197 in the presence of BF3·OEt2 and t-BuOK resulted in formation of the (E,E)-bis-diene product in 61% yield [108]. The desired RCM precursor 186 was obtained after deprotection of the amide protecting group (75% yield). The crucial macrocyclization and (E,E,E)-triene formation were accomplished using RCM. Subjection of 186 to Grubbs II catalyst 126 initially afforded the thermodynamically favoured (E,E)-diene product in 43% yield [69]. Use of less reactive Grubbs I catalyst 198, however, granted access to the desired (E,E,E)-cytotrienin core 185 in 73% yield. The macrocyclic core of cytotrienins was accomplished in just 18 steps with 6.6% overall yield beginning with 1,3-propane diol (longest linear sequence).
This last work highlights the synthetic utility of olefin metathesis for macrocyclization in contrast to more traditional carbon–carbon double bond construction methodologies [109]. To illustrate the efficiency of the RCM reaction, Blagg and co-workers recently prepared various trienomycin-based scaffolds employing this methodology [110].
3.5 Synthetic approaches involving a late union of the aromatic core and the C17-ansa chain
The second strategy employed for the synthesis of the triene-containing ansamycins involved final coupling of the aromatic fragment with a fully functionalized ansa chain. The advantage of this approach would allow for late-stage insertion of any aromatic fragment, providing facile access to a variety of triene-ansamycin natural products and structural analogues. Natural product synthesis of (+)-thiazinotrienomycin E 152e by Smith and preparation of the ansatrienol core 213 by Kirschning and co-workers are two successful applications of this synthetic strategy.
3.5.1 Total synthesis of (+)-thiazinotrienomycin E by Smith (1999)
Smith and co-workers reported the first successful execution of this strategy during their synthesis of thiazinotrienomycin E 152e [111]. Retrosynthetic analysis of this trienomycin planned for late-stage installation of the ester-linked side chain (C29–C38). Key disconnections of the macrocycle at C16–C17 and C1–N20 bonds revealed the aromatic fragment 199 and the aliphatic ansa chain containing fragment 200 (Scheme 51). It was envisioned that the macrocycle would be formed by alkylation of sulfone 199 with triene 200, followed by a Mukaiyama macrolactamization reaction. Construction of the (E,E,E)-triene 199 was achieved using a Kocienski-modified Julia olefination between tetrazole 201 and unsaturated aldehyde 202. The (E,E)-diene containing aldehyde 202 was proposed to arise from a Stille-cross-coupling reaction.
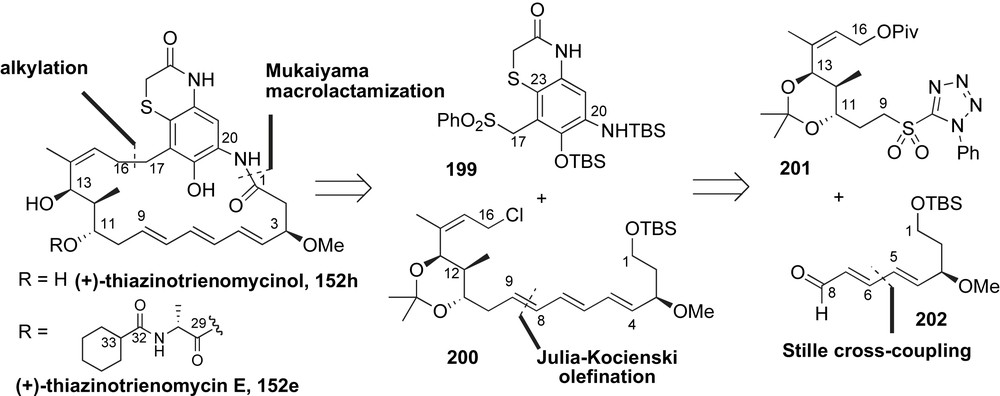
The (E,E,E)-triene of the ansa chain fragment 200 was installed using Stille coupling and a Julia olefination reaction. Synthesis of the C16–C9 tetrazole 201 started from readily available chiral homoallylic alcohol 203 (Scheme 52) [112]. After alcohol protection and reductive ozonolysis, the C13-aldehyde was alkylated using the in situ-generated vinyllithium species 204. The resulting diol 205 was obtained in 77% yield with modest diastereocontrol (dr 2:1). Sequential allylic oxidation and 1,3-directed β-hydroxyketone reduction furnished diol 206 with excellent diastereoselectivity (dr > 95%). Protecting group modification and installation of the required sulfone produced the phenyltetrazole 201 over six steps in 60% yield.
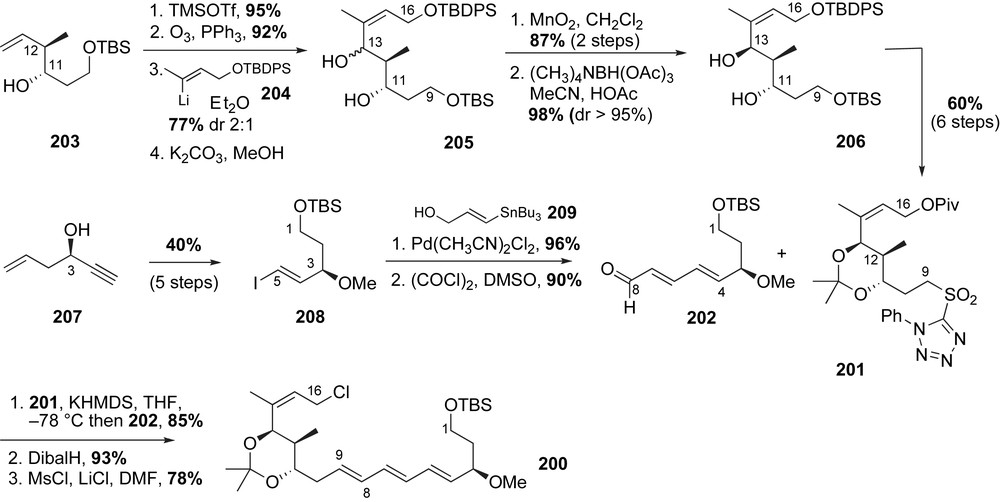
The aldehyde coupling partner 202 was accessed from secondary propargylic alcohol 207. Conversion of this alcohol to the vinyl iodide 208 was accomplished in 40% yield over 5-step sequence. Aldehyde fragment 202 was obtained in 87% yield over two steps via Stille coupling of iodide 208 with stannane 209 and subsequent Swern oxidation of the resulting alcohol. A Kocienski-modified Julia olefination reaction between tetrazole 201 and aldehyde 202 afforded the (E,E,E)-triene in 85% yield with moderate selectivity (10:1) [113]. The completed ansa chain fragment 200 was obtained after conversion of the protected allylic alcohol to the necessary chloride.
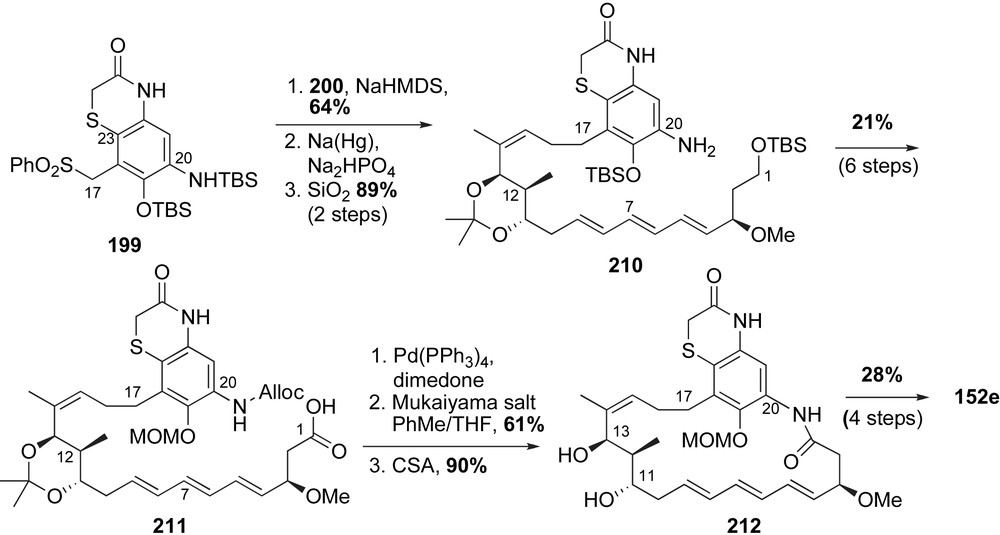
The remaining synthetic hurdles were incorporation of the aromatic ring into the macrocycle and attachment of the amino acid side chain (Scheme 53). The anion of sulfonate 199 was alkylated with allyl chloride 200. Following sulfonyl decomposition and acid deprotection aniline 210 was produced in 57% yield over three steps. Subsequent protecting group modifications and oxidation of the C1 alcohol provided the free acid 211. After unmasking of the aniline, a Mukaiyama salt-promoted macrolactamization [114] delivered the MOM-protected thiazinotrienomycinol derivative 212 in good yield. Smith and co-workers installed the side chain in a fashion reminiscent of their previous syntheses of trienomycin A 146a and F 146f. With this said, coupling of diol 212 to FMOC-alanine anhydride produced a 2:1 (C11/C13)-regioisomeric mixture of monoacylated products along with bis-acylated byproduct. Deprotection of the MOM-group and final HPLC purification furnished the natural product thiazinotrienomycin E 152e in 28% yield over four steps from diol 212. The first total synthesis of thiazinotrienomycin E was achieved in 35 steps with 0.27% overall yield starting with 3-buten-1-ol (longest linear sequence).
3.5.2 Kirschning's total synthesis of the ansatrienol Core (2007)
Kirschning and co-workers reported the second application of late-stage coupling of the aromatic fragment with the completed ansa chain in their synthesis of the ansatrienol macrocycle 213 [115]. Retrosynthetic analysis starts with disconnection of the C16–C17 and C1–N20 bonds revealing the aromatic aniline 183 and triene-containing vinyl iodide 214 (Scheme 54). Similar to the approach used by Smith for thiazinotrienomycin E, formation of the macrocycle was proposed to arise from sequential alkylation and macrolactamization reactions. A Duthaler–Hafner acetate aldol reaction was proposed to install the C3-stereocenter of the ansa chain 214. Disconnection of the C8–C9 olefin revealed the ester-containing fragment 215 and phosphonate 216. Formation of the (E,E,E) triene was to be completed using a Horner–Wadsworth–Emmons (HWE) olefination. The trisubstituted olefin of fragment 215 was planned to arise via a cross-metathesis reaction.

The ester-containing fragment 215 was prepared in a sequence that featured a β-chelation-controlled reduction, cross-metathesis and Evans aldol reaction as the key steps (Scheme 55). The C12-methyl stereocenter was installed via an Evans aldol reaction of the Z(O)-boron enolate (R)-44 to methacrolein 162. Removal of the chiral auxiliary and protection of the primary alcohol with pivaloyl chloride produced the vinyl alcohol 217 in 72% yield over three steps. Olefin cross-metathesis with allylic boronate 218 using Grubbs II generation catalyst 126, followed by hydroboration furnished allylic diol 219 in high yield [116].
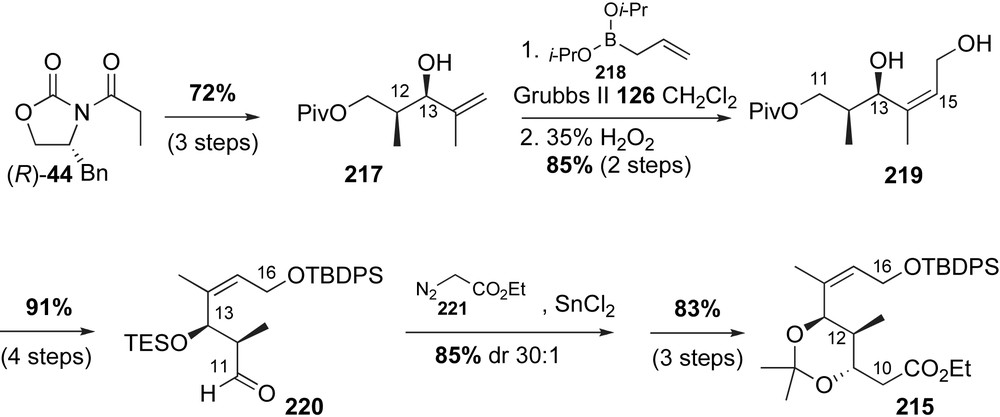
Protecting group modification and oxidation of the C11-alcohol eventually furnished protected aldehyde 220. Tin-mediated homologation with diazo ester 221 produced an intermediate β-keto ester in 85% yield with dr 30:1. Triethylsilyl ether deprotection was followed by a selective anti-1,3 reduction and subsequent diol protection to produce ester 215 in 83% yield. This sequence to access the C16–C9 fragment 215 proved to be more reliable than an earlier method developed by Kirschning that relied on an Evans aldol reaction to install the C12–C13 stereocenters of 223 using the (Z)-configured aldehyde 222 (Scheme 56) [117].
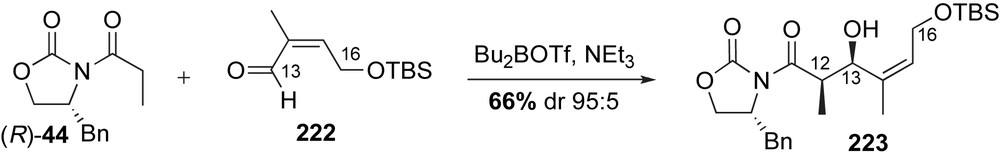
Completion of the ansamycin derivative relied on Horner–Wadsworth–Emmons olefination and Duthaler–Hafner aldol reactions to construct the ansa chain, whereas alkylation and macrolactamization reactions were used to close the 21-membered ring (Scheme 57). Ester 215 was reduced to the necessary aldehyde and combined with phosphonate 216 in an HWE alkenation that produced a 6:1 (E,E,E):(Z,E,E) triene mixture in 96% yield. Subsequent ester reduction and oxidation furnished aldehyde 224 in 89% yield over two steps. The C3-stereocenter was installed using a Duthaler–Hafner acetate aldol reaction [118]. Titanium enolate 227, derived from d-glucose diacetonide 225, was condensed with aldehyde 224 to give the corresponding alcohol 226 in 93% yield. This approach provided multigram quantities of pure ansa chain 226 and was transformed into the ansatrienol core 213 in 6% yield (15 steps) using similar alkylation and macrolactamization protocols employed by Smith during synthesis of (+)-thiazinotrienomycin [94]. Overall, the thiazinotrienomycin core was prepared in 34 linear steps (1.3% yield) beginning with methacrolein.
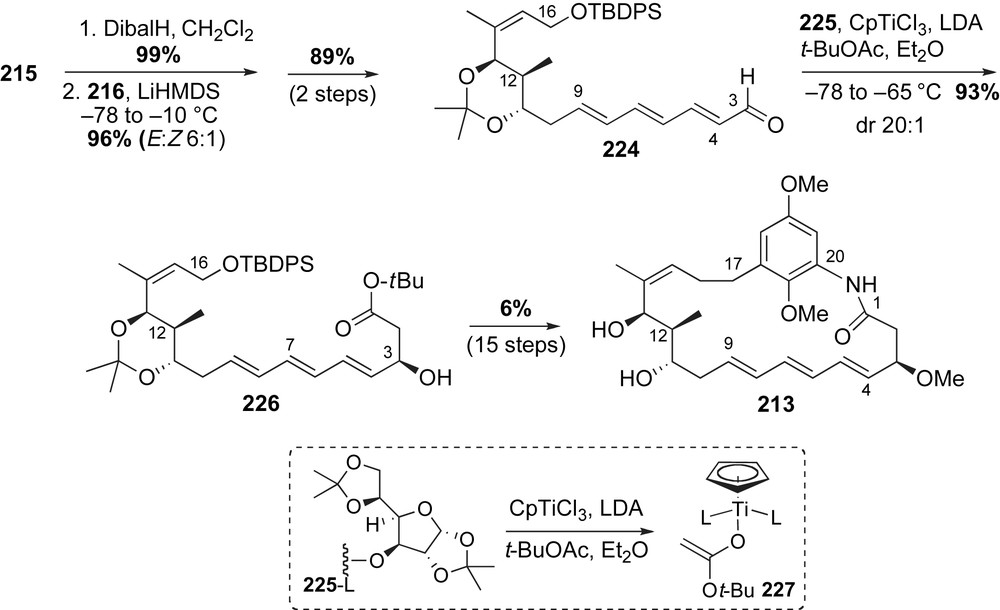
3.6 Conclusions
Two complementary general strategies have been employed for five successful total syntheses of triene-containing ansamycin antibiotics and derivatives. Introduction of the triene moiety via stitching approach to effect macrocyclization was reported by Smith and Panek. In this context, the most recent approach to the ansatrienin core utilized RCM as a high-yielding macrocyclization step. In a different and complementary approach, Smith and Kirschning proposed an easily scalable process to access the fully constructed ansa chain. Late incorporation of the aromatic core and subsequent ring closure by macrolactamization offers a highly convergent route to a variety of trienomycin natural product and structural analogues. This approach could be exploited to probe the structure–activity relationship of triene-ansamycins, allowing for deeper understanding of the associated biology.
Acknowledgments
Financial support for this work was obtained from NIH CA56304. I.E.W. acknowledges a Novartis Graduate Fellowship. The authors are grateful to Dr. Joshua R. Giguere (Boston University) for careful proof reading of this manuscript.
1 Aromatic sulfone 168 was obtained from 3-hydroxy-5-nitrobenzoic acid in five steps, see Ref. [87].
2 Addition of such vinyllithium-type reagent has been initially reported by Smith and co-workers in 1999. See paragraph 2.2.1.