

1 Introduction
Kinases are essential components of cellular signaling networks and, as such, play key roles in the regulation of many processes related to cell growth and differentiation, but also in the expression of pro- and anti-inflammatory mediators. It is, thus, not surprising that the functional dysregulation of many of these enzymes has been linked to a variety of disease states. As a consequence, the inhibition of kinase function over the last two decades has developed into a major paradigm for the discovery of new drugs, primarily in the area of oncology [1], but also against inflammatory diseases [2], and, more recently, for cardiovascular [3] and CNS-based disorders [4].
At this point in time, nine different low-molecular-weight protein kinase inhibitors have been approved for the clinical treatment of different types of cancers [5]; in addition, antibodies against cell surface receptors containing an intracellular tyrosine kinase domain or against the corresponding ligands have become an important part of the clinical armamentarium for cancer treatment [6].
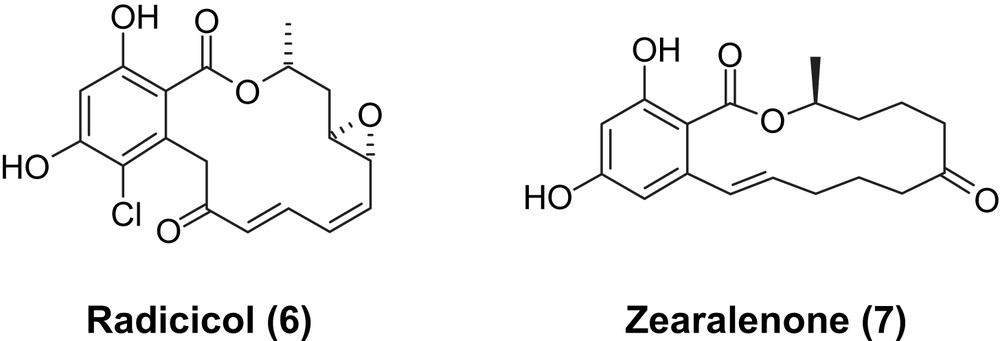
Almost all of the low-molecular-weight protein kinase inhibitors that have been approved for clinical use to date are synthetic small molecules, containing at least one aromatic heterocycle and a varying number of hydrogen bond donor (amide- or anilino-type NH groups) or acceptor (amide carbonyl groups or heteroaromatic N atoms) moieties [7]. The only natural-product-derived agent that has reached the market so far is the mTOR kinase inhibitor temsirolimus (CCI-779), which is a semisynthetic derivative of rapamycin [5f]. Other natural product derivatives that have been investigated in a number of clinical trials are the staurosporine derivatives UCN-01 [8] and midastaurin (PKC412) [9]. Staurosporine itself, although a potent kinase inhibitor, is too toxic to be therapeutically useful, which is likely the result of its indiscriminate action on a multitude of kinases. While a number of other natural products have been reported to be more or less potent kinase inhibitors [10], the most exciting group of natural products that have recently emerged as new lead structures for kinase inhibition are resorcylic acid lactones 1–4 and, to a somewhat lesser extent, also aigialomycin D (5) (Fig. 1) [11]. With the exception of aigialomycin (5) all of these compounds are characterized by the presence of a cis-enone moiety, which appears to be crucial for highly potent kinase-inhibitory activity and at the same time also represents a source of significant selectivity for a specific sub-group of kinases (see below). Although this review will strictly focus on the chemistry associated with compounds 1–5, it should be noted that these are part of a larger family of natural products that all contain a resorcylic acid moiety as a key structural element and which display a wide range of biological activities [11,12]. Perhaps the most widely studied representatives of this class of compounds are radicicol (6), which is an Hsp90 inhibitor [13], and zearalenone (7), which is an estrogenic mycotoxin (Fig. 2) [14]. As many protein kinases depend on functional Hsp90 for proper folding, radicicol (6) indirectly affects cellular protein kinase activity and, thus, had initially been thought to be a kinase inhibitor. However, although the compound inhibits Hsp90 function through direct interference with ATP binding (Hsp90 is an ATPase), radicicol (6) does not effectively compete with ATP binding in protein kinases, which is a direct reflection of the structural differences between the ATP binding sites in ATPases and protein kinases [15].
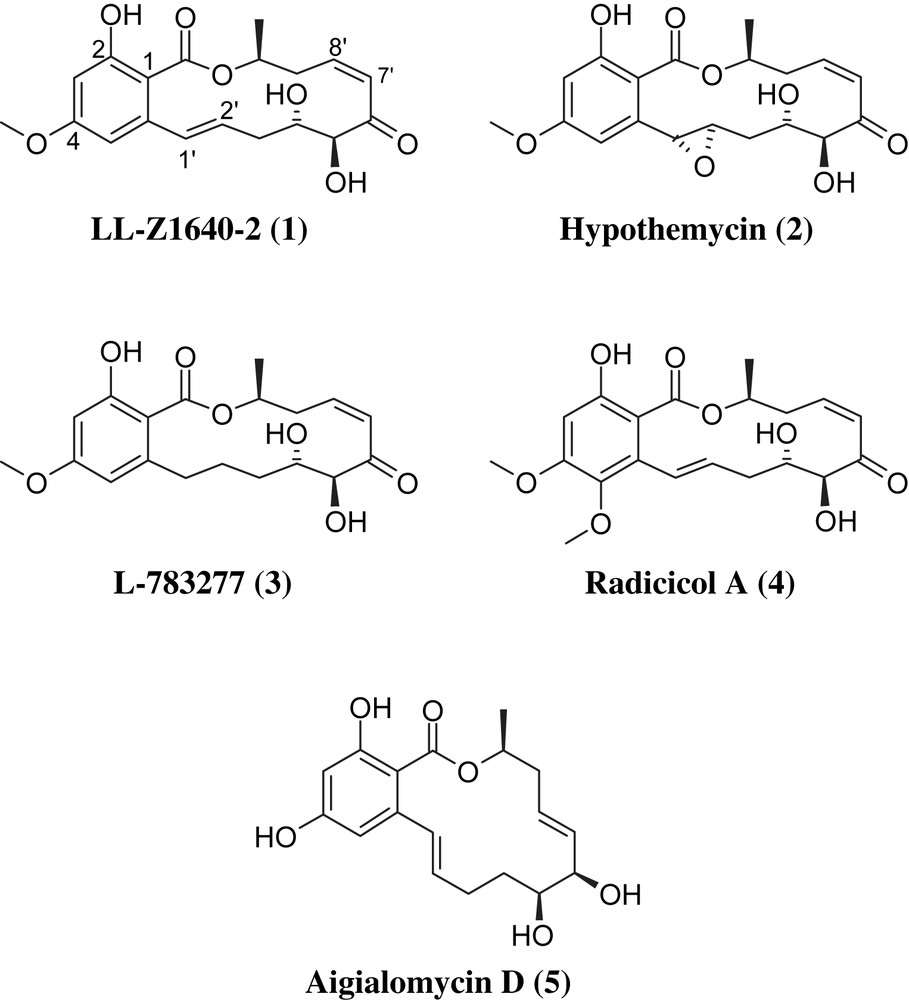
As the first cis-enone-containing resorcylic acid lactone, compound LL-Z1640-2 (1) was isolated in 1978 from an unidentified fungus (“Lederle culture Z1640”), but at the time was not found to be associated with any interesting biological activity [16]. Hypothemycin (2) was first isolated by Nair and Carey as a moderately active antibiotic from a strain of Hypomeces trichothecoides in 1980 [17]; however, the correct structure of the compound was only established after re-isolation from Coriolus versicolor by Agatsuma et al. in 1993 [18]. Hypothemycin (2) was shown to exhibit antiproliferative activity against a number of human cancer cell lines in vitro [18] and to inhibit tumor growth in vivo [19]. The antitumor activity of the compound was linked to the suppression of Ras-inducible gene expression, but the molecular mechanisms leading to this outcome were not understood at that time. In addition, hypothemycin (2) was also found to inhibit T-cell proliferation after stimulation with anti-CD3 mAb/PMA and to modulate the production of cytokines during T-cell activation [20]. Importantly, hypothemycin (2) was able to reduce the levels of phosphorylated ERK1/2 in PMA-treated T-cells, although it was not determined whether this effect was the result of direct kinase inhibition or else was secondary to the compound's interaction with other (non-kinase) targets. The latter finding was reminiscent of previous observations by the former Sandoz group with radicicol A (4), which had produced a significant reduction in tyrosine phosphorylation in LPS-stimulated THP-1 cells [21].
Direct kinase inhibition by cis-enone-containing resorcylic lactones was first demonstrated by Zhao et al. in 1999 for both hypothemycin (2) and L-783277 (3) [22], which were found to inhibit MEK1 in vitro with IC50 values of 15 nM and 4 nM, respectively. A more detailed analysis of the mode of action of L-783277 then revealed that MEK1 inhibition was ATP-competitive and, more importantly, was associated with the formation of a covalent adduct between the enzyme and the inhibitor. Interestingly, the inhibition of LCK by L-783277, although less effective than MEK1 inhibition, did not seem to involve covalent modification of the enzyme. In addition, in contrast to the cis-enone-containing L-783277, the corresponding 7′,8′-trans analog was found to be significantly less active against MEK1 (IC50 value of 300 nM). These findings have recently been corroborated in a more comprehensive study by Schirmer et al. on the mode of action and the kinase specificity of hypothemycin (2) [23]. Out of 124 kinases screened in this study at 200 nM hypothemycin (2) concentration, 21 were inhibited to an extent of >20%; of these 21 enzymes, 18 contained a conserved active site Cys residue corresponding to Cys166 in ERK1/2, i.e. all kinases with an active site cysteine residue that were present in the screening panel were inhibited by hypothemycin (2), with the sole exception of GSK3α. In all cases, the inhibition proved to be time-dependent, thus suggesting covalent inactivation of the enzymes, most likely through 1,4-addition of the protein thiol to the β-carbon atom of the α,β-unsaturated carbonyl system. In the case of ERK2, the formation of a covalent adduct between hypothemycin (2) and the enzyme (both in its active as well as its inactive form) was demonstrated by mass spectrometry of a tryptic protein digest after incubation with [3H]-hypothemycin. The paper by Schirmer et al. [23] also refers to unpublished crystallographic data for the covalent complex between ERK2 and hypothemycin that would provide direct proof for the attachment of Cys166 to C8′ of the hypothemycin macrocycle. (In contrast to these unpublished structural data, a crystal structure of the complex between ERK2 and LL-Z1640-2 (1) has been published recently by Ohori et al., which demonstrates covalent bond formation between Cys166 of ERK2 and the inhibitor [24]; vide infra.) For cancer cell lines carrying activating mutations in the hypothemycin-sensitive mitogen receptors c-kit, FLT3, and PDGFRa, which lead to activation of the ERK pathway, in vitro kinase inhibition by hypothemycin (2) also translates into potent growth-inhibitory activity at the cellular level [23]. Growth inhibition correlates with the inhibition of receptor autophosphorylation as well as the phosphorylation of downstream targets MEK1/2 and ERK1/2. As had been reported for the potent (synthetic) MEK inhibitors CI-1040 and PD0325901 [25], cell lines carrying the ERK-pathway-activating B-RAF V600E mutation (which is present in ca. 60% of melanomas and ca. 20% of colon cancers [26]) were found to be particularly sensitive to hypothemycin (2), although the mechanistic underpinnings of this phenomenon have not been clearly elucidated.
The first kinase shown to be inhibited by LL-Z1640-2 (1) was TAK1 (TGF-β-activated kinase 1 or MAPKKK; IC50 = 8.1 nM) [27], which has been implicated as a key molecule in pro-inflammatory signaling pathways and which may also be of interest as an anticancer target [28]. As discussed above for L-783277 (3) and hypothemycin (2) and their corresponding kinase targets, TAK1 inhibition by LL-Z1640-2 (1) was found to be ATP-competitive and time-dependent [27], again suggesting that enzyme inhibition is based on the covalent modification of an active site Cys residue. Subsequent work by Ohori et al. has also established LL-Z1640-2 (1) as a potent inhibitor of ERK2 (IC50 = 8.0 nM) [24]; at the same time, Ohori et al. have solved the crystal structure of the ERK2/LL-Z1640-2 (1) complex, which provides direct evidence for covalent bond formation between ERK2-Cys166 and C8′ of LL-Z1640-2 (1).
The most recent data on kinase inhibition by one of the natural products 1–5 have been reported by Winssinger and co-workers, who have shown that radicicol A (4), like hypothemycin (2), is a potent inhibitor of VEGFR2/R3, FLT3, and also PDGFRβ [29]. No inhibition was observed for a number of other kinases that are devoid of active site Cys residues. The same group has also demonstrated that aigialomycin (5) is an inhibitor of CDK1, CDK5 (cyclin-dependent kinases 1 and 5), and GSK3 [30]. However, compared to compounds 1–4, the kinase-inhibitory potential of aigialomycin D (5) appears to be rather modest (IC50 values against CDK1/5 and GSK are in the μM range). Nevertheless, as aigialomycin (5) belongs to the same family of natural products as 1–4 and has been established to be a kinase inhibitor, it will also be considered in this review article.
Based on the biological data summarized above for resorcylic acid lactones 1–5, it is clear that these compounds represent a promising and attractive new class of lead structures for kinase inhibition, especially when containing an appropriately positioned cis-enone moiety. Apart from being structurally distinct from the various traditional kinase inhibitor scaffolds, cis-enone-containing resorcylic acid lactones offer the advantage of “built-in” (group) specificity, as their inhibitory potential appears to be confined to kinases with an active site Cys residue (at least for inhibition at the nM concentration level). While this feature may also be regarded as a limitation, as it restricts the number of kinases that could be susceptible to inhibition to ca. 10% of the human kinome [23], many of the kinases from this group are highly relevant targets for drug discovery. It should also be noted that covalent enzyme inhibitors are often viewed with skepticism as potential drug candidates; however, as has been pointed out recently, covalent enzyme inhibition is not an infrequent mode of action even among marketed drugs [23].
Unsurprisingly, the intriguing biological properties of resorcylic acid lactone-based kinase inhibitors have also led to a growing interest in the chemistry of these systems and total syntheses have been reported for all compounds 1–5 (Fig. 1). In addition, a number of semisynthetic analogs of hypothemycin (2) have recently been described by a group at Kosan (vide infra). In this review article, we will provide a condensed overview on the synthetic chemistry of resorcylic acid lactones that have been established to be protein kinase inhibitors (i.e. compounds 1–5 and some closely related analogs). Other resorcylic acid lactones, even if equipped with interesting biological activities other than kinase inhibition, are not within the scope of this review.
2 Chemistry of resorcylic acid lactone kinase inhibitors
2.1 LL-Z1640-2 and hypothemycin
The first total synthesis of a cis-enone-containing resorcylic acid lactone was reported by Tatsuta and co-workers in 2001 for LL-Z1640-2 (1) [31]. As illustrated in Scheme 1, the key elements of the retrosynthesis conceived by Tatsuta for this target structure included the Mukaiyama macrolactonization reaction of a protected seco acid LL-I, introduction of the E double bond between C1′ and C2′ through reductive double bond isomerization of the Z-configured allylic carbonate LL-II, and the construction of the aliphatic/aromatic bond connection between C6 and C1′ through Sonogashira coupling between L-III and LL-IV. The latter was envisioned to be accessible from d-ribose, thereby obviating the need for the stereoselective construction of the chiral centers at C4′ and C5′ by chemical methods.
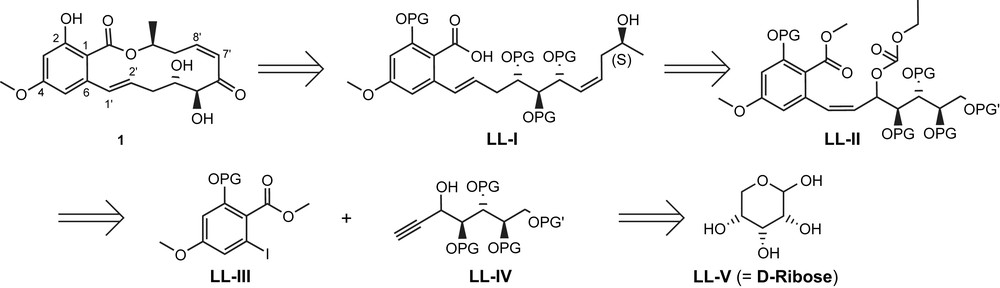
Retrosynthesis of LL-Z1640-2 (1) according to Tatsuta et al. [31].
The actual synthesis of 1 is summarized in Scheme 2, commencing with the addition of lithiated TMS–acetylene to tris–MOM-protected d-ribose 8. This was followed by pivaloylation of the primary hydroxyl group and removal of the TMS group with TBAF to provide propargylic alcohol 9 in 48% overall yield (from 8). Although the stereochemical outcome of the acetylide addition remains inconsequential, due to the reductive removal of the propargylic hydroxyl group later in the synthesis, it should be noted that the reaction proceeded with high selectivity (only “traces” of a second isomer were observed [31]). Alkyne 9 underwent smooth Sonogashira coupling with iodide 10 in the presence of Pd(OAc)2, CuI, and Ph3P to furnish, after ethoxycarbonylation of the free hydroxyl group, compound 11 in excellent overall yield (83%). The aromatic iodide 10 was prepared according to a procedure previously developed by Hegedus [32], presumably from 2-hydroxy-4-methoxy-benzoic acid methyl ester, but the preparation of 10 is not detailed in Ref. [31]. After partial reduction of the C1′/C2′-triple bond in 11, subsequent hydrogenolysis under Tsuji conditions [33] gave the desired E olefin in almost quantitative yield. Removal of the Piv-protecting group followed by Swern oxidation of the resulting free alcohol and dibromo-olefination of the corresponding aldehyde gave dibromo-olefin 13, which was further elaborated into propargylic alcohol 14 through treatment with nBuLi and reaction of the lithiated acetylene formed in situ with (S)-propylene oxide. Seco ester 14, which incorporates the entire carbon framework of LL-Z1640-2 (1), was then converted to the immediate macrocyclization precursor 15 via partial hydrogenation of the triple bond under Lindlar conditions and subsequent cleavage of the methyl ester with NaOH. Macrolactonization of 15 according to the method of Mukaiyama [34] followed by the cleavage of all protecting groups with 5% HCl/MeOH gave the fully deprotected macrolactone 17, which was obtained in 36% overall yield for the four-step sequence from 14. The transformation of this intermediate into LL-Z1640-2 (1) then required selective oxidation of the allylic hydroxyl group at C6′, which could be accomplished with Dess–Martin periodinane (62% yield) or DDQ, although the latter gave 1 in only 20% yield. Although this is not explicitly discussed in Ref. [31], it appears that several other oxidation methods failed to deliver the desired product. Overall, the synthesis depicted in Scheme 2 provided the target compound LL-Z1640-2 (1) in 3% overall yield for a longest linear sequence of 19 steps (from d-ribose).
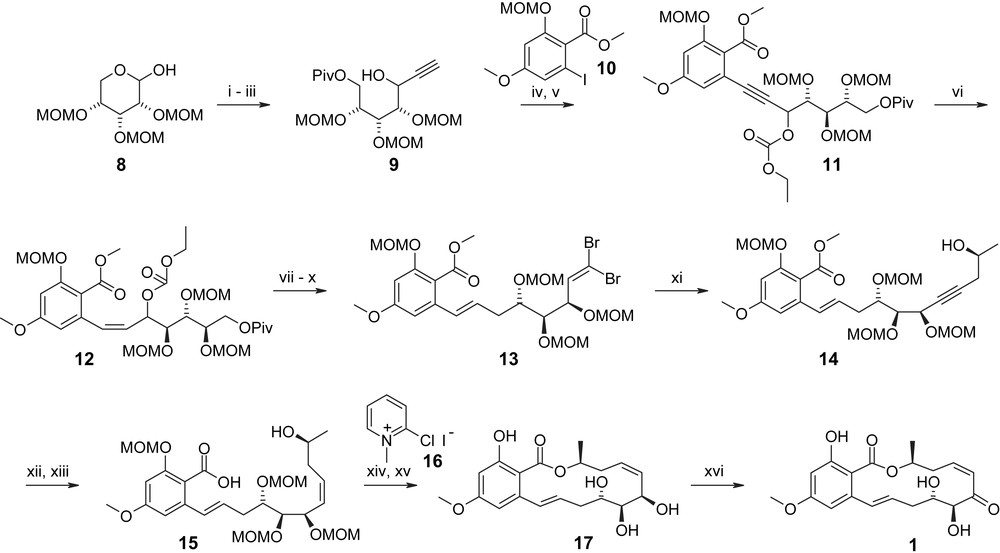
(i) TMS–acetylene, nBuLi, BF3·Et2O, THF, −78 °C → rt. (ii) PivCl, pyridine, 0 °C, 1 h, 48% (two steps). (iii) TBAF, AcOH/THF, 2 h, quant. (iv) Pd(OAc)2, CuI, Ph3P, Et3N, 2 h, 85%. (v) ClC(O)OEt, pyridine, 0 °C, 1 h, 98%. (vi) H2, Pd/BaCO3, quinoline, EtOH, 30 min. (vii) Pd(dba)2CHCl3, nBu3P, HCOONH4, dioxane, 95 °C, 1 h, 96% (two steps). (viii) MeONa, MeOH, 50 °C, 3 h, 95%. (ix) (COCl)2, DMSO, Et3N, DCM, −78 °C → rt. (x) CBr4, Ph3P, DCM, 0 °C, 15 min, 85% (two steps). (xi) (S)-Propylene oxide, nBuLi, BF3·Et2O, THF, −78 °C → rt, 45%. (xii) H2, Pd/BaCO3, quinoline, AcOEt, 30 min. (xiii) 2 M NaOH/MeOH–dioxane, 90 °C. (xiv) Et3N, MeCN, 50 °C, 1 h, 47% (three steps). (xv) 5% HCl/MeOH, 50 °C, 2 h, 76%. (xvi) 16, Dess–Martin periodinane, DCM, 15 min, 62% [31].
An alternative approach to the synthesis of LL-Z1640-2 (1) has been reported by Sellès and Lett, who also investigated the conversion of 1 into hypothemycin (2). According to Sellès and Lett's retrosynthetic analysis (Scheme 3) [35], the macrocyclic framework of hypothemycin (2) and LL-Z1640-2 (1) would be constructed either through Mitsunobu-based macrolactonization from seco acid H-I or, alternatively, through a Suzuki-type macrocyclization of ester H-I′. Both H-I and H-I′ would be derived from o-bromo benzoic acid H-II and either the 10′R or 10′S acetylene H-III Suzuki coupling or DDC-based esterification, respectively. The requirement for two diastereoisomeric versions of H-III is a consequence of the inversion at C10′ in the Mitsunobu-based macrolactonization step, which mandates that the configuration at this carbon in precursor H-I is opposite to that in the natural product (i.e. 10′R). The synthesis of H-III would be achieved in a convergent manner from two fragments comprising C1′–C6′ and C7′–C10′, respectively.
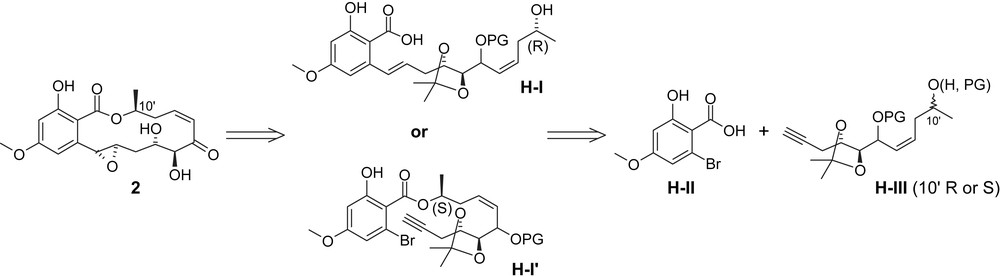
Retrosynthesis of hypothemycin (2) according to Sellès and Lett [35].
The synthesis of the aromatic precursor H-II, as in Tatsuta's case, was based on previous work by Hegedus and co-workers [32] and involved O-TBS protection of 4-methoxy salicylic acid 18 followed by conversion to the diethylamide and ortho bromination to provide the o-bromo diethylamide 19 in 74% overall yield (Scheme 4) [35]. The latter was transformed to the corresponding methyl ester 20 by treatment with Meerwein salt and subsequent hydrolysis of the resulting (O-deprotected) imino ester. Saponification of 20 with NaOH in DME at reflux then gave carboxylic acid 21 in quantitative yield, i.e. without any detectable decarboxylation (which occurs in salicylic acid derivatives with a free phenolic hydroxyl group under more forcing conditions [32]).
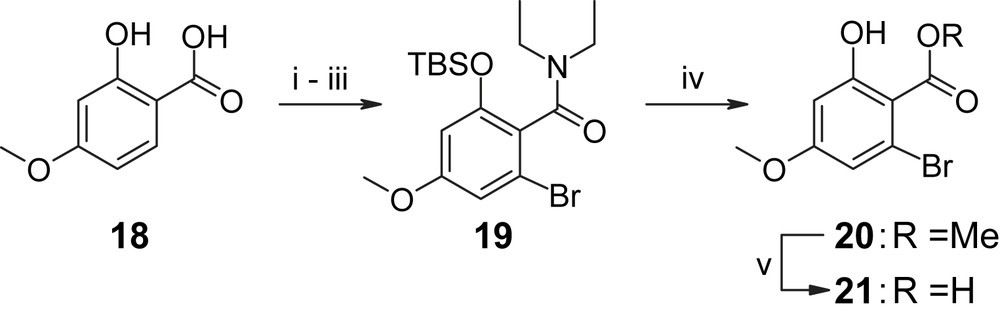
(i) TBSCl, iPr2NEt, DMF, 2 h. (ii) Et2NAlMe2, refl., overnight, 98% (two steps). (iii) tBuLi/pentane (1.08 equiv), Et2O, −78 °C, 10 min, then Br2 (1.06 equiv), 75%. (iv) (a) Me3OBF4 (1.3 equiv), DCM, overnight; (b) aq. sat. Na2CO3/MeOH 1/1, 6 h, 76%. (v) Concd NaOH/DME 1/1, refl., overnight; or TMSOK (2 equiv), DME, refl., overnight, quant. [35].
The synthesis of precursors H-III is summarized in Scheme 5 starting with mono-TBS-protected 2-butene-1,4-diol 22 (obtained from 2-butyne-1,4-diol via Red-Al reduction and TBS protection in 60% overall yield) [35]. Conversion of alcohol 22 to the corresponding iodide followed by alkylation of TMS–acetylene and selective cleavage of the TBS ether with DDQ gave allylic alcohol 23, which underwent Sharpless asymmetric epoxidation in 85% yield and with >90% ee. The epoxy alcohol was then transformed to the phenyl carbamate 24, which upon treatment with BF3·Et2O gave the cyclic carbonate 25, thus establishing the chiral centers at C4′ and C5′ (hypothemycin/LL-Z1640-2 numbering) in a highly efficient manner (78% overall yield for the three-step sequence from achiral allylic alcohol 23). Base-catalyzed carbonate methanolysis followed by appropriate protecting group manipulations then led to intermediate 26, which was further elaborated into aldehyde 27 via reprotection of the terminal acetylene, TBS removal and Swern oxidation. The protection of the alkyne moiety was required, in order to avoid side reactions in the addition of the lithiated vinyl iodide 34 to aldehyde 27, which proceeded in 55% and 77% yield for the S- and R-enantiomers of 34, respectively. Vinyl iodide 34 was obtained from TMS–acetylene and (R)- or (S)-propylene oxide in five steps and excellent overall yield (59%) through Lewis acid-catalyzed epoxide opening followed by TBS protection (to give 32), removal of the TMS group from the terminal alkyne moiety, conversion to the iodo alkyne 33, and finally hydroboration with Sia2BH and protonolysis (Scheme 6). The addition products 28 (Scheme 5) were then converted to the free terminal acetylenes 29 and 30, which correspond to the (C10′) hydroxyl-protected and hydroxyl-unprotected versions of precursor H-III in Scheme 3.
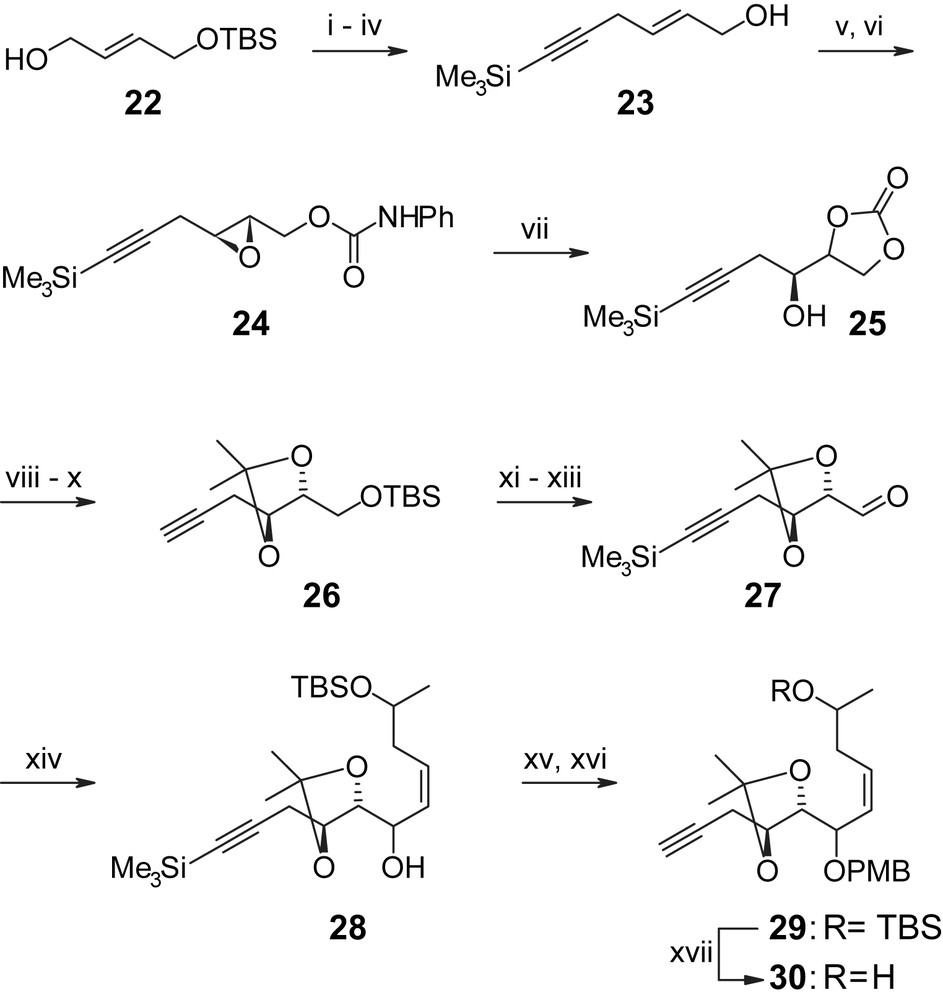
(i) MesCl, Et3N, DCM, −10 °C → rt, 30 min. (ii) NaI, acetone, 1 h, 86% (two steps). (iii) TMS–acetylene, nBuLi, THF/HMPA 10/1, −78 °C → rt, 4 h, 90%. (iv) DDQ (5 mol%), MeCN/H2O 9/1, 2 h, 74%. (v) Ti(OiPr)4, (+)-DET, DCM, tBuOOH, −25 °C, overnight, 85%, >90% ee. (vi) PhNCO, DCM/pyridine, 1 h, quant. (vii) BF3·Et2O, Et2O, −20 °C, 2 h, then 1 N H2SO4, overnight, 91%. (viii) MeONa, MeOH, 8 h, then Dowex 50 WX8 column eluted with MeOH, 93%. (ix) TBSCl, imidazole, DMF, 1 h. (x) 2-Methoxypropene, TsOH (cat), DCM, 1 h, 73% (two steps). (xi) nBuLi/hexane, Et2O, TMSCl (1.1 equiv), −30 → −10 °C. (xii) DDQ (8 mol%), MeCN/H2O 9/1, 2 h, 71% (two steps). (xiii) (COCl)2, DMSO, Et3N, DCM, −78 → 0 °C, 30 min, quant. (xiv) 34, tBuLi/pentane, Et2O, −78 → 0 °C, 56% (10′S); 77% (10′R). (xv) K2CO3 (1.4 equiv), MeOH, 5 h. (xvi) PMBO(NH)CCCl3, CF3SO3H (0.004 equiv), Et2O, 4 h, 72% (10′S); 74% (10′R) (two steps). (xvii) TBAF, THF, 10 h, 97% [35].
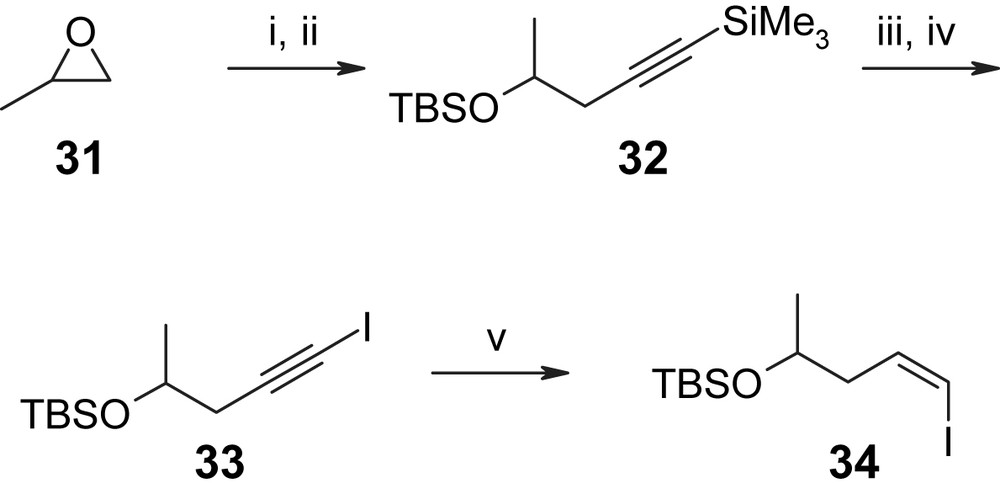
(i) TMS–acetylene, nBuLi, Et2O, −78 °C, then (R)- or (S)-propylene oxide (31R or 31S) and further addition of BF3·Et2O (1.0 equiv) in 50 min, −78 °C, 89%. (ii) TBSCl, imidazole, DMF. (iii) K2CO3, MeOH, 5 h, 86% (two steps). (iv) nBuLi, THF/hexane, −78 °C, 15 min, then I2, THF, 86%. (v) (a) Sia2BH, THF, −20 → 0 °C, 3 h; (b) AcOH, 65 °C, 3 h, 89% [35].
Esterification of alcohol 30 with carboxylic acid 21 in the presence of DCC proceeded smoothly and furnished the desired macrocyclization substrate 35 in 76% yield (Scheme 7) [36]. Unfortunately, the intramolecular Suzuki reaction with the vinyl-borane derived from 35 gave the desired macrolactone 36 only in low yield as part of complex reaction mixtures (15% under the optimized conditions shown for Scheme 7). Notably, model substrate 37 (Fig. 3) gave the corresponding macrolactone in only 9% yield, while none of the desired cyclization product was observed with the least constrained substrate 38 (Fig. 3).
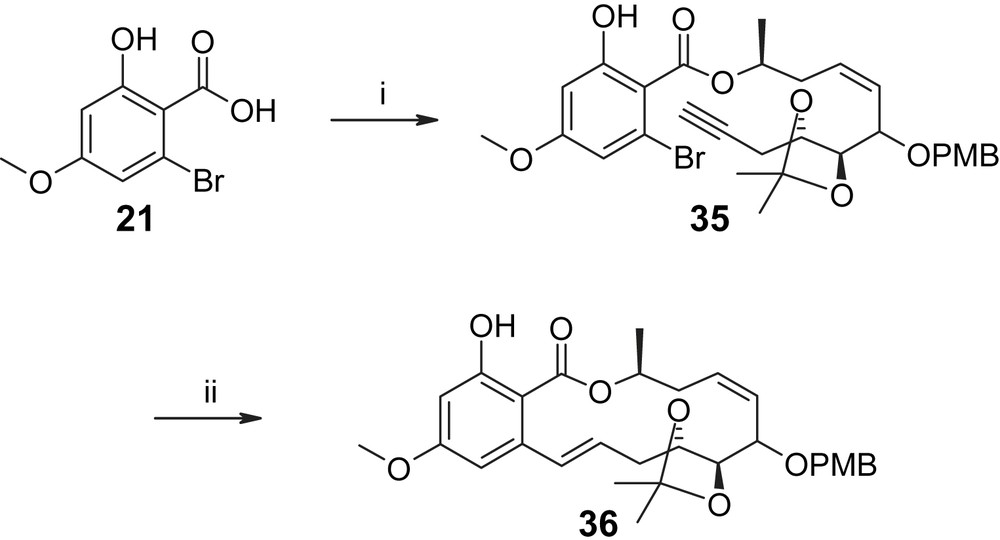
(i) 30 (1 equiv), DCC (2.2 equiv), DMAP (1.2 equiv), DCM, 5 h, 76%. (ii) (a) Sia2BH (3.9 equiv), THF, −20 °C → rt, 2 h; (b) addition of acetone (3 equiv), 2 M aq. K3PO4 (2 equiv), −10 °C → rt; (c) addition of the mixture to a solution of 4 mol% [Pd(OAc)2 + 4TFP] in DME (i.e. substrate concentration = 0.034 M in DME/H2O ∼ 30/1), refl., 6 h, 15% [35,36].

More satisfactory results were obtained by Sellès and Lett for their macrolactonization-based approach to the hypothemycin/LL-Z1640-2 macrocycle (Scheme 8) [36]. In contrast to the intramolecular setting, the intermolecular Suzuki reaction between the aryl bromide 20 and the vinyl-borane derived in situ from acetylene 29 proceeded very efficiently and provided the coupling product in 76% yield. Subsequent TBS removal and ester saponification gave partially protected seco acid 39, which was cyclized to 36 with Ph3P/DEAD in 67% yield. Removal of the PMB protecting group from 36 gave an inseparable 60/40 mixture of diastereoisomeric alcohols 40, whose oxidation to the required ketone proved to be a significant challenge. Oxidation could not be achieved with either activated MnO2 or DDQ, but was finally accomplished with PCC in the presence of 2,5-dimethyl-pyrazole [37] in 62% yield for the mixture of diastereoisomers 40. Interestingly, the major isomer was quantitatively converted to the enone, whereas a large part of the minor isomer was recovered unchanged (23% yield based on the starting mixture of diastereoisomers). In subsequent experiments the minor isomer was shown to react only slowly under the above Parish conditions [37] and after 24 h gave only the 7′,8′-E-isomer 41 in 50% yield (Scheme 9). On the other hand, Jones oxidation of the 60/40 mixture of 40 gave the desired enone in 74% yield (on a millimolar scale). It is also noteworthy that Sellès and Lett were unable to achieve selective allylic oxidation of the free triol 17 (derived from 40 by treatment with TosOH/MeOH; see also Scheme 2) either under Parish conditions or with MnO2. The latter finding is in contradiction with the work of Tatsuta (vide supra). Conversion of 40 into LL-Z1640-2 (1) was accomplished in 74% yield with TosOH/DCM/MeOH at rt for 3.5 h. Virtually no isomerization of the C7′/C8′ double bond was observed under these conditions and the remainder of the material was recovered unchanged (23% yield of recovered starting material). The final transformation of LL-Z1640-2 (1) into hypothemycin (2) was severely hampered by the low reactivity of the C1′/C2′ double bond and could only be achieved in 17% yield with MCPBA/NaHCO3 (30% of starting material recovered, Scheme 8) [36]. However, as had been predicted by Sellès and Lett at the outset of their work [35], the epoxidation reaction was highly stereoselective (and also regioselective) and provided hypothemycin (2) as the only isolable epoxide isomer. In summary, the synthesis developed by Sellès and Lett provided LL-Z1640-2 (1) and hypothemycin (2) in overall yields of 1% and 0.17%, respectively, in 26 (27) steps for the longest linear sequence (from 2-butyne-1,4-diol). Interestingly, although Sellès and Lett's approach to LL-Z1640-2 (1) would appear to be more convergent than the Tatsuta strategy, the former includes a significantly higher number of steps for the longest linear sequence and is characterized by a lower overall yield. The higher efficiency of the Tatsuta synthesis of LL-Z1640-2 (1) mostly reflects the use of d-ribose as a chiral starting material, which more than compensates for the less convergent character of the overall strategy.
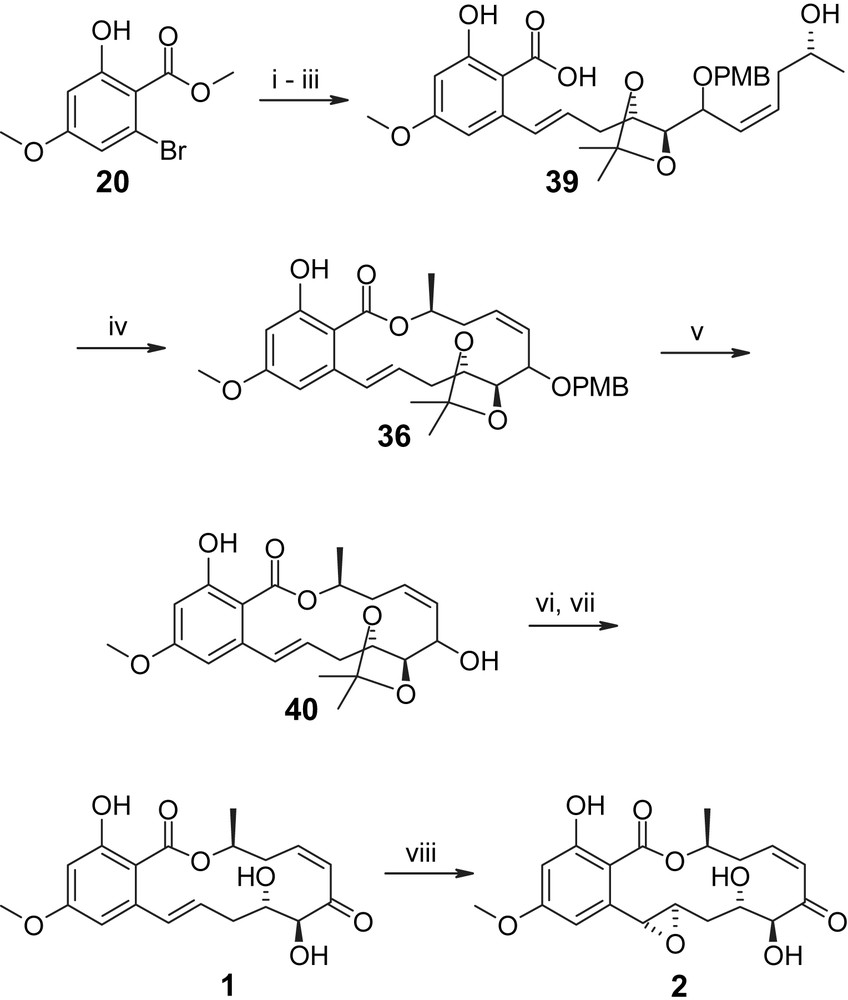
(i) (a) 29, Sia2BH (2 equiv), THF, −25 °C → rt, 2 h; (b) addition of 2 M aq. K3PO4 (2 equiv); (c) addition of the mixture to a solution of 20 (1.2 equiv) and 15 mol% [Pd(OAc)2 + 4TFP] in DME (DME/H2O ∼ 7/1), refl., 8 h, 71%. (ii) TBAF, THF, 6 h, 93%. (iii) 2 N aq. NaOH (13 equiv)/MeOH 1/3, refl., overnight, 71%. (iv) 39 (0.007 M), toluene, Ph3P (2 equiv), DEAD (2 equiv), rt, 15 min, 67%. (v) DDQ, DCM/pH 7 buffer 9/1, 30 min, 94% (3/2 mixture of isomers at C6′). (vi) PCC (3 equiv), 2,5-dimethyl-pyrazole (10 equiv), DCM, 0 °C, 6 h, 62%. (vii) pTsOH (0.5 equiv), DCM/MeOH 1/1, 3.5 h, 76%. (viii) MCPBA/NaHCO3 (3 equiv), −20 → 0 °C, 4 h, then pH 7 work-up, 17% [35,36].
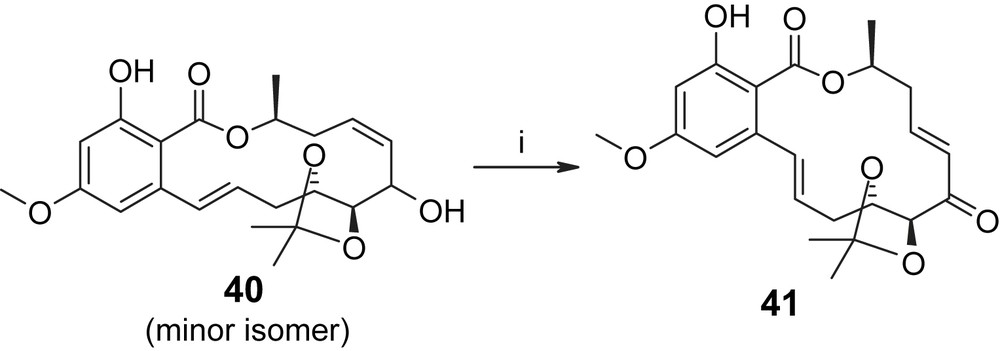
(i) PCC (3 equiv), 2,5-dimethyl-pyrazole (10 equiv), DCM, 0 °C, 24 h, 50% [36].
Only one total synthesis of hypothemycin (2) has been reported in the literature so far, but a group at Kosan has recently reported a number of semisynthetic derivatives of this natural product and their biological assessment [38]. For example, methylation of 2 with TMS–diazomethane produced the methyl ethers 42a, 42b, and 42c in a 3/1/2 ratio (Scheme 10; unfortunately, no yields for these transformations are reported in Ref. [38], including the Supplementary information for the paper). The evaluation of compounds 42a–c in cancer cell proliferation assays revealed that the methylation of the hydroxyl group on C5′ does not significantly affect antiproliferative activity (compared to hypothemycin (2)), while monomethylation of the C4′ hydroxyl group or dimethylation lead to a profound loss in potency [38]. The Kosan group has also isolated a naturally occurring derivative of hypothemycin (from Hypomyces subiculosus), which lacks the methyl substituent on the C4 hydroxyl group (Scheme 11; 4-O-demethylhypothemycin (43)) [39]. Employing Mitsunobu chemistry, 43 was converted to a series of aryl–alkyl ether derivatives 44 with enhanced polarity (and, presumably, improved aqueous solubility) over hypothemycin (2) (Scheme 11) [38]. Interestingly, derivatives 44a–d, like 4-O-demethylhypothemycin (43), showed similar antiproliferative activity as hypothemycin (2) in most cases investigated, which indicates that substituents on C4–O do not interfere with target protein binding and may thus be used to modulate the physico-chemical properties of 2 without impairing the desired biological activity. (It should be noted, however, that no kinase inhibition data have been reported for 43 or any of the semisynthetic derivatives shown in Schemes 10 and 11.)
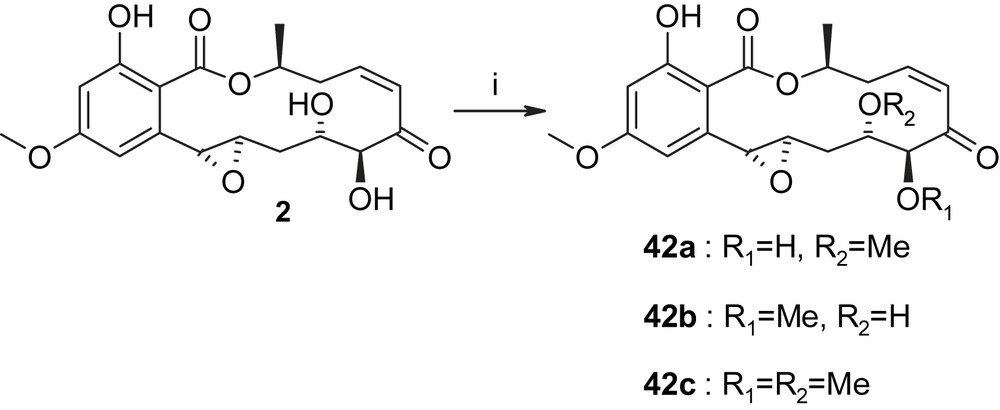
(i) TMSCHN2, HBF4, DCM, 42a:42b:42c = 3/1/2 [38].
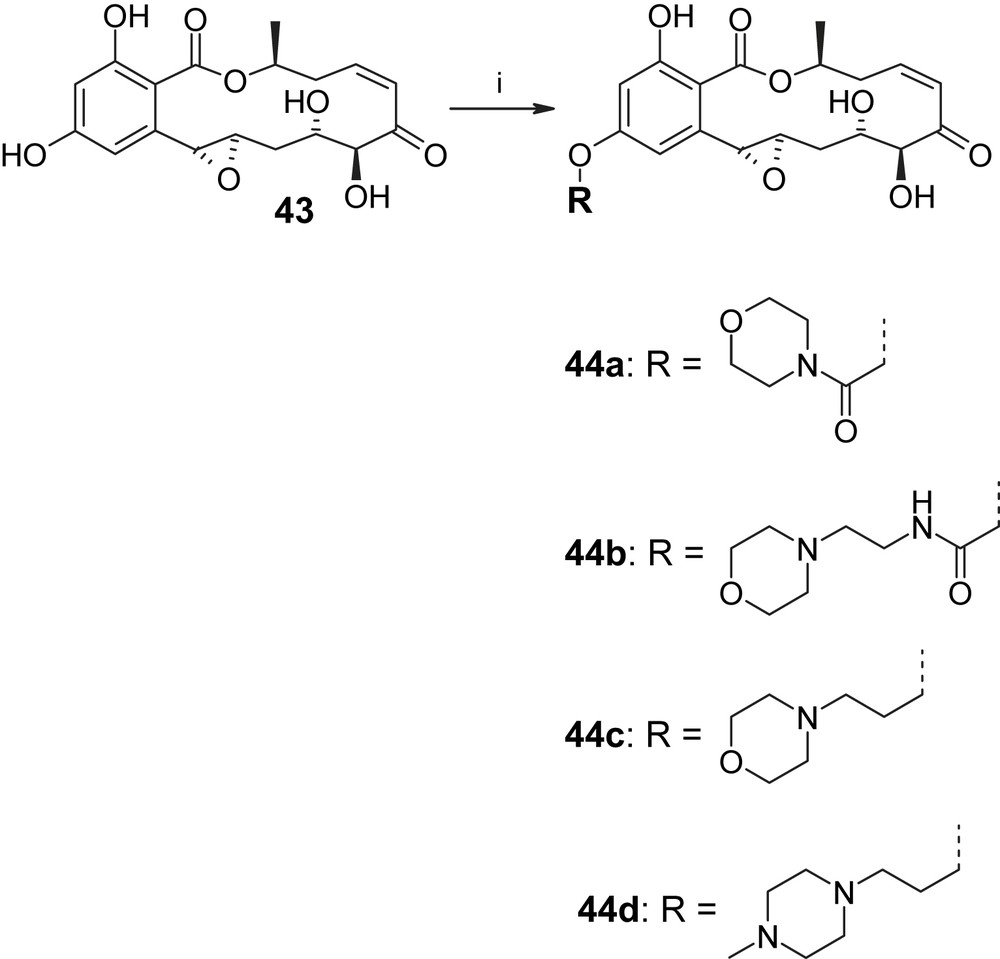
(i) ROH, Ph3P, DEAD, THF [38].
In addition to the syntheses discussed above, a third macrolactonization-based approach to LL-Z1640-2 (1) has been reported by Winssinger and co-workers in the context of their work on radicicol A (4), which will be discussed in detail below. In contrast, a conceptually different strategy for the synthesis of 1 has been proposed by Henry et al. that would rely on ring closure between C1′ and C2′ through ring-closing olefin metathesis (RCM) [40]. While this strategy has not been fully implemented so far, Henry et al. have reported the synthesis of the requisite triene precursor for the RCM-based ring closure (51), which relies on tartaric acid as the source of chirality at C4′ (Scheme 12) [40]. Thus, protected tartaric acid derivative 45 was first transformed into the known aldehyde 46 [41] through LAH reduction, mono-TBS protection of the resulting diol and oxidation of the free hydroxyl group under Swern conditions in 76% overall yield. Compound 46 was then converted to acetylene 47, which, after lithiation with nBuLi, was reacted with (S)-propylene oxide; subsequent TBS protection of the resulting secondary alcohol gave bis-TBS-protected diol 48. The latter was elaborated into diene 49 via partial hydrogenation of the triple bond followed by selective cleavage of the primary TBS ether with HF·Pyr, oxidation of the alcohol to the aldehyde and allylation with CH2CHCH2MgBr in 62% overall yield. Protection of the hydroxyl group formed in the allylation step as a PMB ether and removal of the TBS group from C10′–O (hypothemycin/LL-Z1640-2 numbering) then furnished the free secondary alcohol 50, whose DCC-mediated esterification with carboxylic acid 54 provided the desired triene 51 in 67% yield. Acid 54 was obtained in five steps from commercially available methyl 2,4,6-trihydroxybenzoate (52), which was initially converted to triflate 53 through selective methylation at O-4 with TMS–diazomethane, mono-TBS protection, and reaction of the unprotected hydroxyl group with triflic anhydride (Scheme 13). Subsequent Stille coupling with Bu3SnCHCH2 followed by ester saponification with NaOH gave 54 in 69% overall yield (based on 52). No attempts on RCM with triene 51 have been reported so far and it remains to be seen how efficient this reaction can be in light of potential competition from six-membered ring formation through RCM between the terminal double bond in the aliphatic portion of 51 and the double bond between C7′ and C8′ (hypothemycin/LL-Z1640-2 numbering).
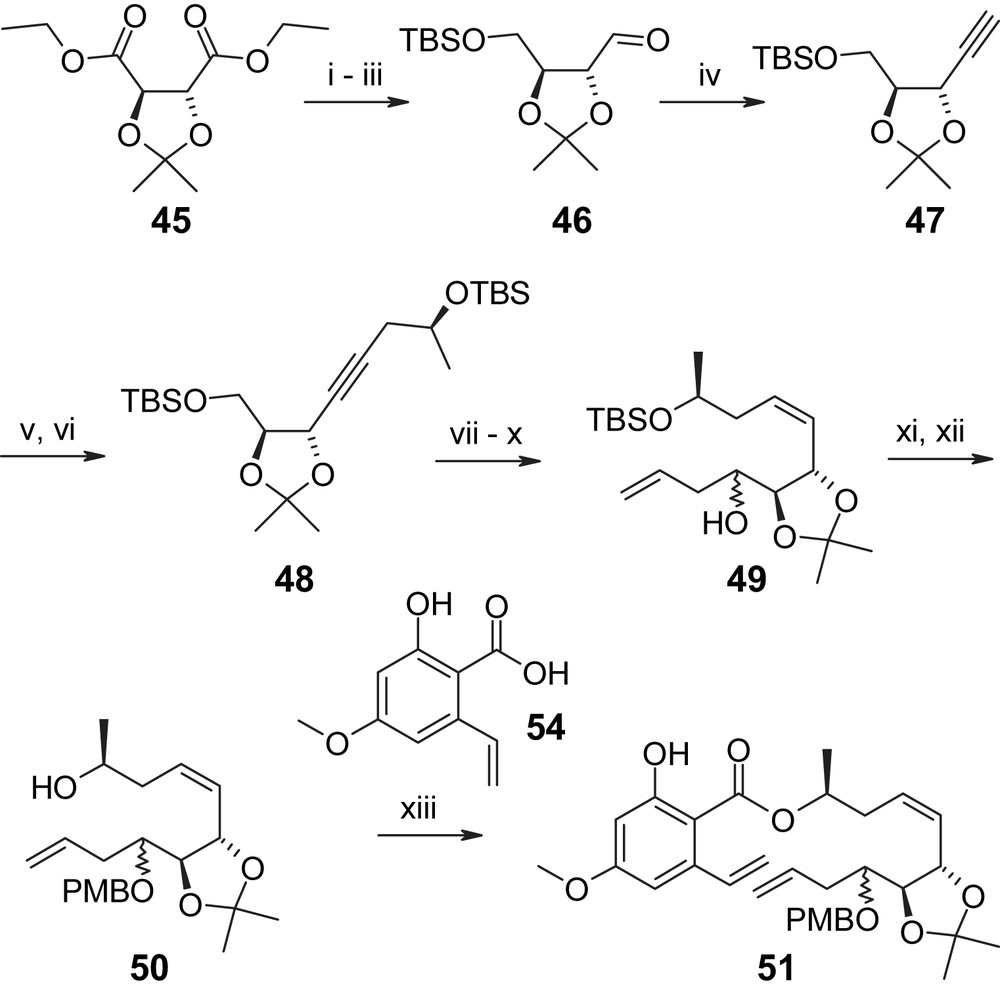
(i) LAH, THF. (ii) NaH, TBSCl, THF, 89% (two steps). (iii) Swern oxidation, 100%. (iv) (a) CBr4, Ph3P, DCM; (b) nBuLi, Et2O, 79%. (v) (S)-Propylene oxide, nBuLi, BF3·Et2O, THF, 76%. (vi) TBSCl, Et3N, DCM, 97%. (vii) H2, Pd/BaSO4, quinoline, 100%. (viii) HF·Pyr/pyridine, 65%. (ix) Swern oxidation. (x) CH2CHCH2MgBr, THF, 96% (two steps). (xi) NaH, PMBCl, TBAI, THF, 100%. (xii) HF·Pyr/pyridine, 97%. (xiii) 54, DCC, 67% [40].
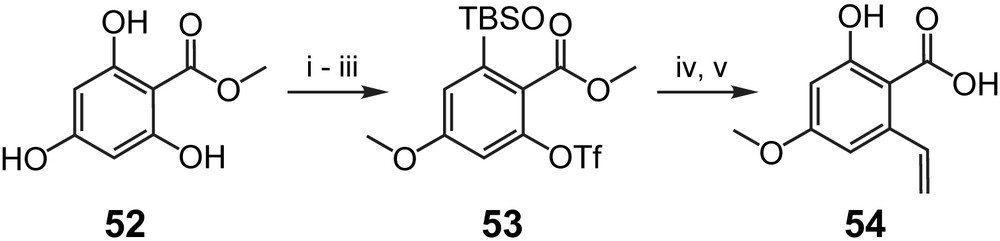
(i) TMS–CH2N2, Et2O, 90%. (ii) TBSCl, Et3N, DCM, 80%. (iii) Tf2O, pyridine, 100%. (iv) nBu3SnCHCH2, Pd(Ph3P)4, DCM, 100%. (v) NaOH, dioxane, 96% [40].
2.2 Radicicol A
The first total synthesis of radicicol A (4) was reported only recently by Winssinger and co-workers based (in part) on methodology that they had previously developed in the course of their work on aigialomycin (5) (see below) [29]. As illustrated by the retrosynthesis depicted in Scheme 14, Winssinger's approach to radicicol A (4), as for the various syntheses discussed above for LL-Z1640-2 (1), was to rely on macrocyclization through ester bond formation under Mitsunobu conditions. However, in contrast to all previous syntheses of cis-enone-containing resorcylic acid lactones, the construction of the seco acid RA-I (Scheme 14) would not involve formation of a C6–C1′ aryl–alkyl bond; rather, C1′ would be part of the aromatic building block RA-II and the C1′–C2′ E double bond would be formed through alkylation of this precursor with an alkyl iodide intermediate RA-III and subsequent oxidation/elimination. The C2′–C10′ fragment RA-III was envisioned to be assembled from appropriately protected building blocks RA-IV and RA-V, thus leading to a highly convergent synthesis of radicicol A (4). An additional important feature of Winssinger's strategy was the projected use of a fluorinated protecting group for fragment RA-V, which would enable the application of fluorous isolation technologies [42] up to the penultimate step before macrolactonization.
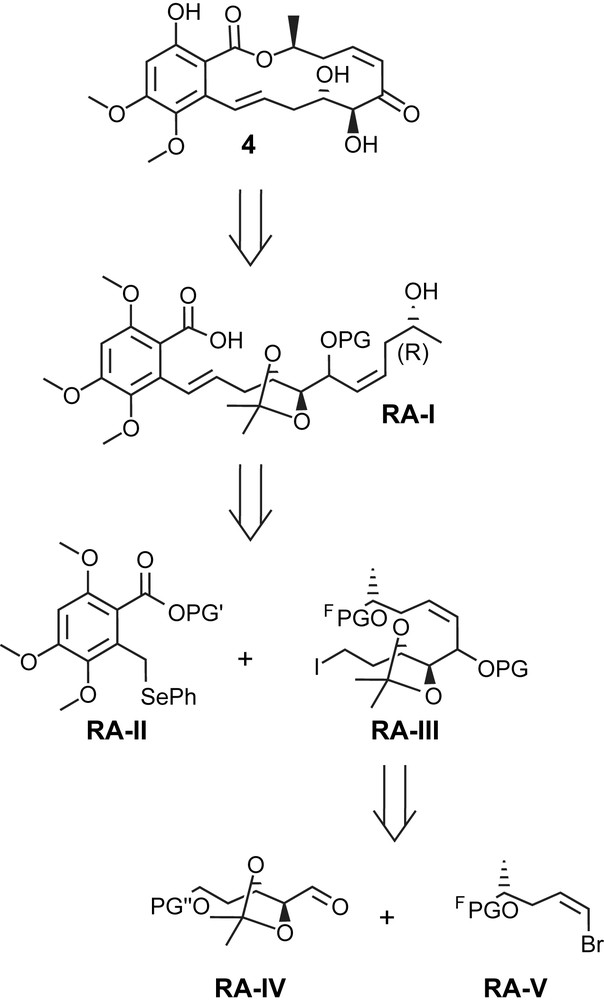
Retrosynthetic analysis for radicicol A (4) according to Winssinger [29].
As shown in Scheme 15, the aromatic building block 56 was obtained from benzoic acid 55 via esterification with 2-TMS–ethanol followed by deprotonation at the benzylic position (LDA) and reaction with (PhSe)2 in 88% overall yield. It should be noted that the 2-methoxy substituent in 56 functions as a reversibly protected OH group that needed to be liberated at a later stage in the synthesis. The protection of the 2-hydroxyl group as a methyl ether was dictated by the enhanced acid sensitivity of commonly used acid-labile protecting groups (such as MEM, EOM, PMB) in the presence of a 5-methoxy substituent (which also makes the aryl moiety highly prone to oxidation) [29]. This problem does not exist in resorcylic lactones that are unsubstituted at the 5-position.
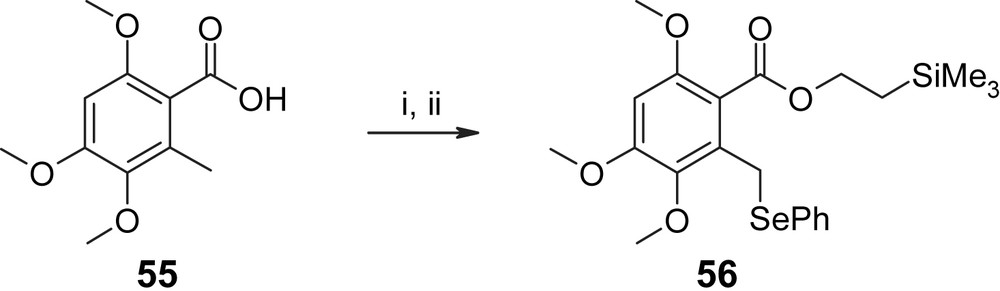
(i) (a) (COCl)2, DMF (cat), DCM, 0 °C, 1 h; (b) 2-(trimethylsilyl)ethanol, Et3N, DMAP (cat), 23 °C, 1 h, 98%. (ii) LDA (2.0 equiv), (PhSe)2 (1.0 equiv), THF, −78 °C, 1 h, 90% [29].
The synthesis of an appropriately protected C7′–C10′ fragment RA-V started with the protection of homoallylic alcohol 57 with a fluorous-tagged version of the conventional PMB group [43] followed by cross-metathesis with a 2-fold excess of vinyl-borolane 58 and subsequent conversion of the resulting E-vinyl-borolane 59 into the desired Z-vinyl-bromide 60 [44] (Scheme 16; 72% overall yield from 57). Addition of lithiated 60 to aldehyde 66 (obtained in three steps from acetal-protected 2-deoxy-d-ribose 65, Scheme 17) gave an 88% yield of a 3/1 mixture of allylic alcohols that was further elaborated into the corresponding mixture of alkyl iodides 61 (Scheme 16) through EOM protection of the newly formed hydroxyl group, cleavage of the primary TBS ether, and treatment of the free alcohol with I2/Ph3P/imidazole. Alkyl iodide 61 was then reacted with the lithiated selenide 56; subsequent oxidation of the phenylselenyl ether and elimination led to the desired E olefin 62 in high yield (64% for the three-step sequence from 61). Consecutive removal of the fluorous-tagged PMB and the TMS-ethyl protecting groups with DDQ and TBAF, respectively, led to seco acid 63, which could be cyclized under Mitsunobu conditions in excellent yield (81%). Quite remarkably, no conventional work-up was required in any of the steps leading from 57 to 62 and all products could be recovered by a simple washing/elution sequence on fluorous silica gel. Likewise, the use of fluorous-tagged versions of Ph3P as well as DEAD as Mitsunobu reagents enabled easy separation of by-products formed in the cyclization step. Treatment of the cyclization product with BCl3 in CH2Cl2 for 15 min at 0 °C allowed the removal of all remaining protecting groups in a single step, including the selective cleavage of the aromatic methyl ether in the 2-position of the aromatic ring. Completion of the synthesis then required the selective oxidation of the allylic hydroxyl group in triol 64, which was accomplished with polymer-bound IBX [45] to provide the target compound radicicol A (4) in excellent yield (90%). Overall, radicicol A (4) was obtained from (R)-4-hydroxy-1-pentene (57) in 14 steps with a total yield of 15%. Substituting aromatic building blocks 67 and 68 (Fig. 4) for 56, the process outlined in Scheme 16 for the synthesis of radicicol A (4), has also been used by Winssinger and co-workers to prepare LL-Z1640-2 (1) and a 5-chloro analog of 4, respectively, with similar efficiency [29].
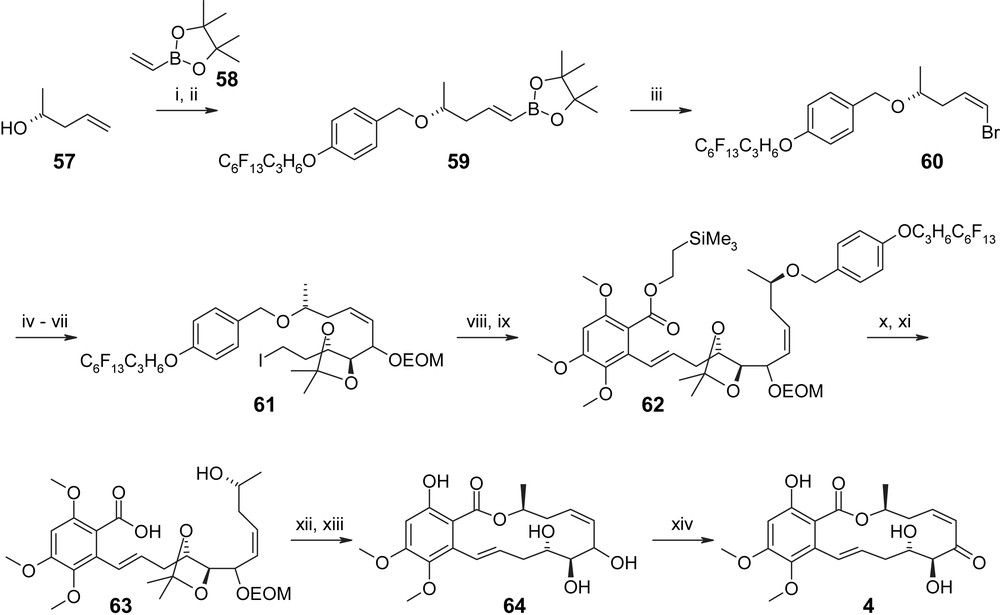
(i) p-(C6F13C3H6O)BnO(NH)CCl3, CSA (cat), DCM, 12 h, 92%. (ii) 58 (2.0 equiv), Grubbs II (2.5 mol%), toluene, 80 °C, 12 h, 92%. (iii) Br2 (1.0 equiv, 1 M in CH2Cl2), Et2O, −20 °C, 10 min, then NaOMe (2.2 equiv, 1 M in MeOH), −20 °C, 30 min, 89%. (iv) tBuLi, THF/Et2O, −100 °C, 15 min, then 66 (1.0 equiv), −100 °C, 15 min, 88%. (v) EOMCl (8.0 equiv), iPr2NEt (8.0 equiv), TBAI (cat), DMF, 12 h, 96%. (vi) TBAF, THF, 12 h, 92%. (vii) Ph3P, I2, imidazole, THF, 0 °C, 1 h, 91%. (viii) 56 (1.0 equiv), LDA (2.0 equiv), THF/HMPA 10/1, −78 °C, 20 min, 89%. (ix) H2O2 (2.0 equiv), THF, 2 h, 80%. (x) DDQ, DCM/H2O 2/1, 2 h, 77%. (xi) TBAF, THF, 2 h, 87%. (xii) RF–Ph3P (2.0 equiv), RF–DEAD (2.0 equiv), toluene (10 mM), 2 h, 81% (>80% purity). (xiii) BCl3, DCM, 0 °C, 15 min, 88% (purified by PTLC). (xiv) PS-IBX (3.0 equiv), DCM, 1 h, 90%. RF = C6F13C3H6; PS = polystyrene [29].
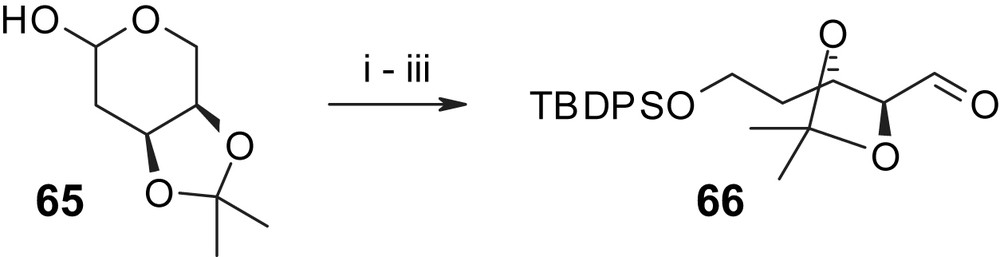
(i) LAH, THF, 0 °C → rt, 2 h, 95%. (ii) TBDPSCl, imidazole, DMF, 2 h, 66%. (iii) PS-IBX (3.0 equiv), DCM, 2 h, 100%. PS-IBX = polymer-supported 2-iodoxybenzoic acid [29].
2.3 L-783277
The first (and so far only) total synthesis of L-783277 (3) has recently been reported by our own group and is based on building blocks 72, 73, and 75 as key precursors for macrocycle assembly (Schemes 18 and 19) [46]. As shown in Scheme 18 the synthesis departed from commercially available isopropylidene-d-erythrono-1,4-lactone (69), which was elaborated into the C1′–C10′ fragment 74 via ester 70, alcohol 71, and aldehyde 72. The latter underwent smooth addition reaction with the anion derived from O-TBS-protected (R)-1-pentyn-4-ol 73 to produce an 87% yield of an inseparable 1.9/1 mixture of secondary alcohols (epimers at C6′) that was converted into the corresponding mixture of C6′–O–MOM ethers 74 by reaction with MOM–Cl in the presence of catalytic amounts of TBAI. As the hydroxyl group formed upon addition of 73 to 72 was to be oxidized to a C6′-ketone in the natural product, the stereochemical outcome of the addition reaction at this point was thought to be inconsequential and no attempts were made to improve the selectivity of the acetylide addition reaction. Olefin 74 underwent smooth Suzuki–Miyaura coupling with ortho-bromo ester 75 to provide the fully protected seco acid 76 in high yield (79%). The protection of the carboxyl group in the aromatic building block 75 as a 2-TMS–ethyl ester was based on prior optimization work on substrates closely related to 76; in particular, these experiments had shown that the saponification of the methyl ester corresponding to 76 might be highly problematic, if not impossible without substantial degradation [46]. A 2-TMS-ethyl ester group has also been employed independently by Winssinger and co-workers in their synthesis of radicicol A (4) [29]. Hydrogenation of 76 under Lindlar conditions gave the desired Z olefin in 94% yield. At this stage, the C6′ epimers (originating from the addition reaction of 73 to 72, vide supra) were separated and advanced through the remainder of the synthesis individually. After simultaneous cleavage of the TMS-ethyl ester and the TBS-ether moieties with TBAF the resulting seco acids 77 (obtained in 91% and quantitative yield for the major and minor C6′-isomers, respectively) were cyclized under Mitsunobu conditions; subsequent deprotection with sulfonic acid resin gave the fully deprotected macrolactones 78. Somewhat surprisingly, the final oxidation of these isomers with polymer-bound IBX [29] produced L-783277 (3) as the major product only in one of the two cases. Thus, the major C6′ isomer of 78 led to a 1/4 mixture of L-783277 (3) and a second mono-oxidized product, whose exact structure has not been assigned (82% total yield). In contrast, the minor C6′ isomer of 78 gave L-783277 (3) in 93% yield and 91% HPLC purity, together with 8% of a second mono-oxidation product that could not be separated by preparative TLC or flash chromatography. Purification of this material by RP-HPLC gave L-783277 (3) with >95% final purity. It is interesting to note at this point that no differences in oxidation selectivity between C6′ isomers are operative in the case of radicicol A (4), for which Winssinger and co-workers have reported a >90% yield for the polymer-bound IBX-mediated oxidation of the C6′-isomeric mixture of triol precursors corresponding to 78 [29].
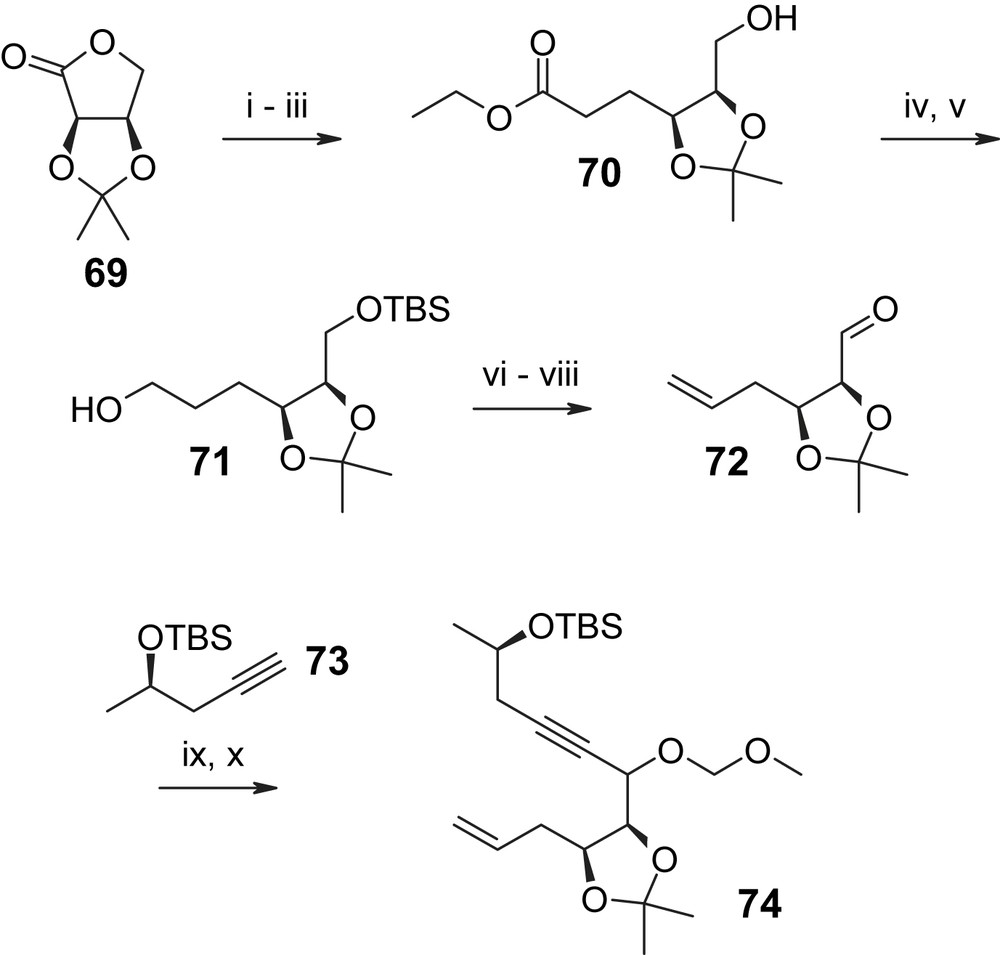
(i) DIBAL-H, Et2O, −78 °C, 2 h, 95%. (ii) Ph3PCHCOOEt, dioxane, refl., 7 h, 91% (E/Z = 1.1/1). (iii) H2, Pd/C, EtOH, 4 h, quant. (iv) TBSOTf, CH2Cl2, 93%. (v) DIBAL-H, toluene, 2 h, 85%. (vi) (a) 2-NO2–PhSeCN, Bu3P; (b) NaHCO3, 30% H2O2, 19 h, 81%. (vii) TBAF, THF, 0 °C → rt, 0.5 h, 90%. (viii) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C, 1 h, 74%. (ix) 73, nBuLi, −78 °C, 1.5 h, 87%. (x) MOM–Cl, (iPr)2NEt, TBAI, DMF, 19 h, 87% (1.9/1 mixture of isomers) [46].
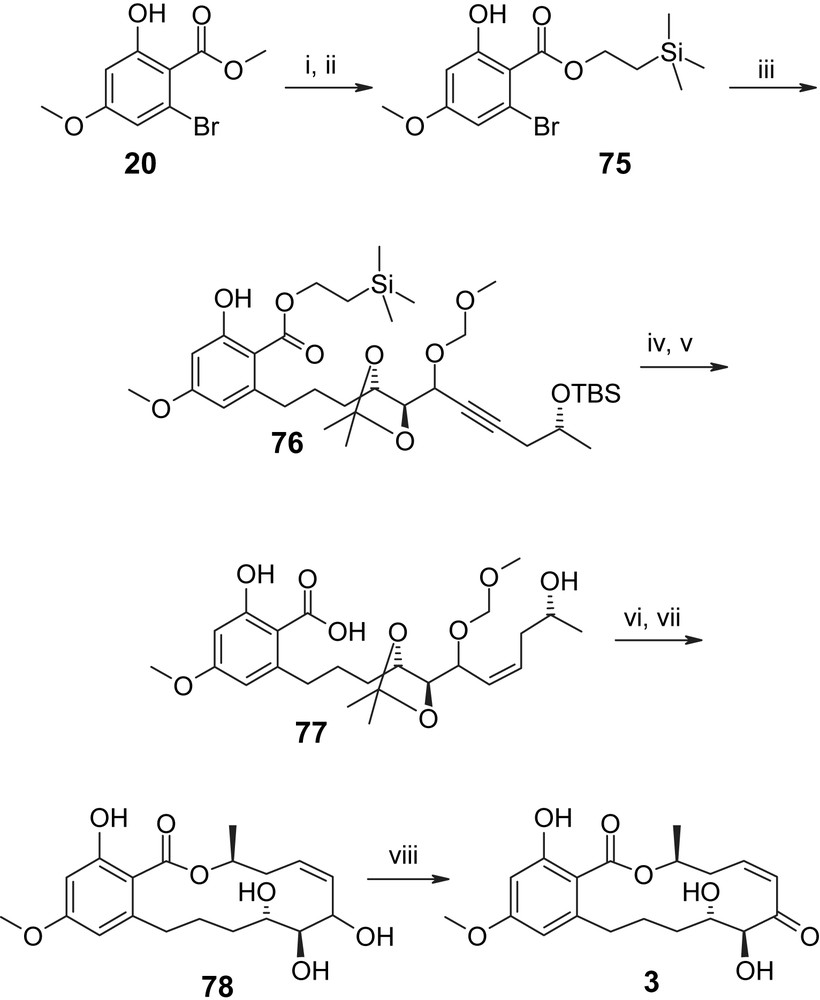
(i) TMSOK, DME, MW, 110 °C, 2 h, acidic work-up, 85%. (ii) (CH3)3SiCH2CH2OH, DCC, DMAP, CH2Cl2, 15 h, 88%. (iii) (a) 74, 9-BBN, THF, rt, 2 h; (b) 2 M K3PO4, [Pd(OAc)2 + 4TFP], DME, refl., 5.5 h, 81%. (iv) (a) H2, Lindlar catalyst, AcOEt, 3 h, 94%; (b) separation of isomers (1.6/1). (v) TBAF, THF, rt, 15 h, quant (major isomer); quant (minor isomer). (vi) DIAD, Ph3P, toluene, 25 min, 59% (major isomer); 74% (minor isomer). (vii) Sulfonic acid resin, MeOH, refl., 6 h, 78% (major isomer), 46% (minor isomer). (viii) IBX (polymer supported), CH2Cl2, rt, 6 h; from major isomer of 78: 1/4 product mixture of L-783277 (3) and a second mono-oxidized species (major product), 82% (total yield); from minor isomer of 78: 93% of 3 (91% purity) [46].
2.4 Aigialomycin
As indicated above, part of the methodology employed by Winssinger and co-workers in their synthesis of radicicol A (4) had already been developed in the course of their work on aigialomycin D (5), which has led to the recognition of this natural product as a protein kinase inhibitor [30]. However, the first synthesis of aigialomycin D (5) had been reported by Danishefsky and co-workers two years earlier, when the kinase-inhibitory properties of the compound had not yet been uncovered [47]. Danishefsky's synthesis of aigialomycin (5) is summarized in Scheme 20 and features an “ynolide” Diels–Alder reaction between alkyne 84 and diene 85 for the build-up of the aromatic ring as a key defining element, i.e. in contrast to all other syntheses described above, the aromatic moiety was not introduced as part of a discrete separate building block, but was only generated after macrocycle formation. The synthesis of macrolactone 84 departs from 2-deoxy-d-ribose which was first converted to acetonide 65 and then further elaborated into aldehyde 80 through Wittig olefination and pivaloylation (to give terminal olefin 79) followed by hydroboration and oxidation. Subsequent Reformatsky-type nucleophilic propargylation with propargyl-bromide and Zn gave a mixture of secondary alcohols that were protected as TBS ethers. Subsequent removal of the pivaloyl protecting group, oxidation, and Wittig olefination furnished acetylene 81, whose deprotonation and subsequent reaction with dry ice gave the crucial carboxylic acid 82. Mitsunobu esterification of 82 with (R)-4-hydroxy-1-pentene (57) followed by reaction of the ester with Co2(CO)8 provided the protected diene 83, which underwent smooth RCM with 2nd generation Grubbs catalyst to provide the desired E olefin in 70% total yield (combined yield of the individual diastereoisomeric alcohols that were separated at this stage). It should be noted here that Co-complexation of the alkyne moiety proved to be absolutely crucial for the success of the RCM, as no product formation was observed with the uncomplexed (“free”) triple bond. Decomplexation of the RCM products with CAN proved to be straightforward and furnished the desired ynolide(s) 84 in excellent yield(s). Acetylene(s) 85 underwent highly efficient Diels–Alder reaction with diene 85, resulting in the formation of resorcylic acid lactone(s) 86 in high yield(s). Protection of the phenolic hydroxyl groups as MOM ethers followed by TBS removal and dehydration with Martin sulfurane [48] gave protected aigialomycin D derivative 87, which was converted to aigialomycin D (5) by treatment with 0.5 N HCl/MeOH in 69% yield. Overall, the total yield of aigialomycin (5) for the 19-step sequence from 2-deoxy-d-ribose was 5%.
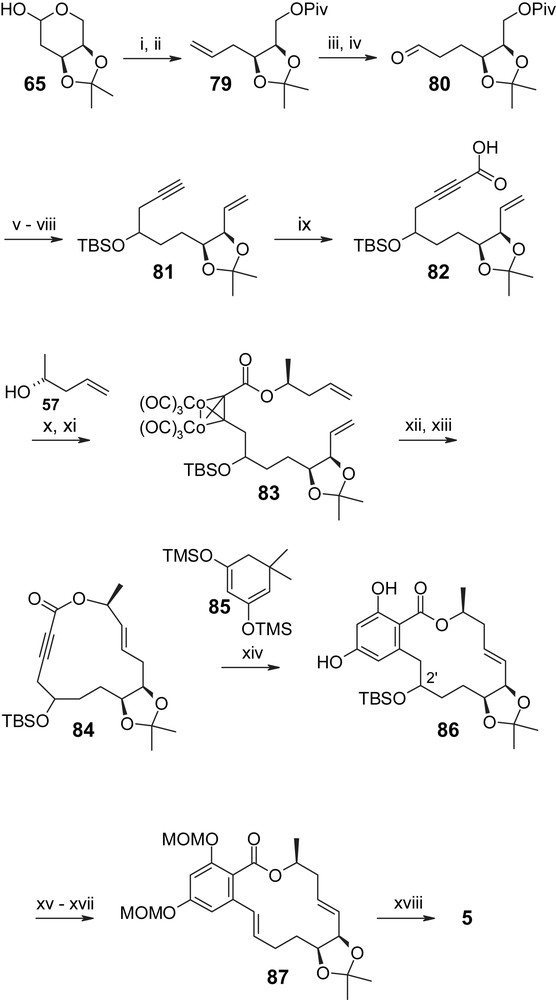
(i) KHMDS, Ph3P+CH3I−, THF, −78 °C → rt, 68%. (ii) PivCl, Et3N, DMAP, DCM, 90%. (iii) (a) 9-BBN, 0 °C → rt; (b) NaOH, H2O2, 88%. (iv) SO3·Pyr, DMSO, DCM, Et3N. (v) (a) Propargyl-bromide, Zn, THF; (b) TBSOTf, 89% (three steps). (vi) MeONa, 88%. (vii) SO3·Pyr, DMSO, DCM, Et3N. (viii) KHMDS, Ph3P+CH3I−, THF, −78 °C → rt, 86% (two steps). (ix) nBuLi, dry ice, −78 °C → rt. (x) 57, DIAD, Ph3P, toluene, 85% (two steps). (xi) (Co)2(CO)8, toluene, 94%. (xii) Grubbs II, DCM, 2′-(R)-isomer, 38%; 2′-(S)-isomer, 42%. (xiii) CAN, −10 °C, 2′-(R)-83, 94%; 2′-(S)-83, 95%. (xiv) 140 °C, neat 85, 2 d, 2′-(R)-84, 74%; 2′-(S)-84, 84%. (xv) MOMCl, iPr2NEt, 2′-(R)-isomer, 78%; 2′-(S)-isomer, 83%. (xvi) HF·Pyr, 2′-(R)-isomer, 78%; 2′-(S)-isomer, 87%. (xvii) [PhC(CF3)2O]2SPh2, 90% from 2′-(R)-isomer; 87% from 2′-(S)-isomer. (xviii) 0.5 N HCl, 69% [47].
As illustrated in Scheme 21, Winssinger's synthesis of aigialomycin D (5) follows a more conventional strategy for introduction of the resorcylic acid moiety than Danishefsky's approach, but it also relies on RCM for macrocycle formation [30]. As indicated above, the build-up of the requisite diene precursor is based on similar principles as have already been discussed above for the synthesis of radicicol A (4). Specifically, this involves the alkylation of benzyl selenide 89 (obtained from benzoic acid 88 through Mitsunobu-based esterification with (R)-2-hydroxy-1-pentene (57) followed by EOM protection of the phenolic hydroxyl groups and phenylselenylation at the benzylic position) with alkyl bromide 96 to furnish the phenylselenide 90. The synthesis of compound 96 is summarized in Scheme 22 and the initial step involves cross-metathesis of 5-bromo-1-pentene (92) with 2-butene-1,4-diol (93) to produce allylic alcohol 94 with an E/Z ratio of >25/1. The latter was submitted to Sharpless asymmetric epoxidation, which proceeded in excellent yield (85%), and this was followed by oxidation and Wittig olefination to give intermediate 95. Lewis acid-catalyzed (Sc(OTf)3) epoxide hydrolysis then furnished the desired anti-diol, which was converted to acetonide 96 as the desired electrophile for the alkylation of selenide 89 (Scheme 21). The resulting alkylation product 90 was then cyclized with 2nd generation Grubbs catalyst under equilibrating conditions, which produced the desired C7′–C8′ E alkene 91 with >10/1 selectivity in 92% yield. Oxidation of the selenide followed by base-induced elimination of phenylseleninic acid gave fully protected aigialomycin, which was converted to aigialomycin D (5) by treatment with polymer–sulfonic acid in quantitative yield (23% total yield for the longest linear sequence (13 steps from 88)). Interestingly, if the RCM and oxidation/elimination steps were carried out in the reverse order, i.e. if RCM was attempted on a substrate that already incorporated a double bond between C1′ and C2′, significant amounts of the undesired 6-membered cyclization product were observed, arising from RCM between the C1′–C2′ double bond and the terminal double bond adjacent to the dioxolane ring.
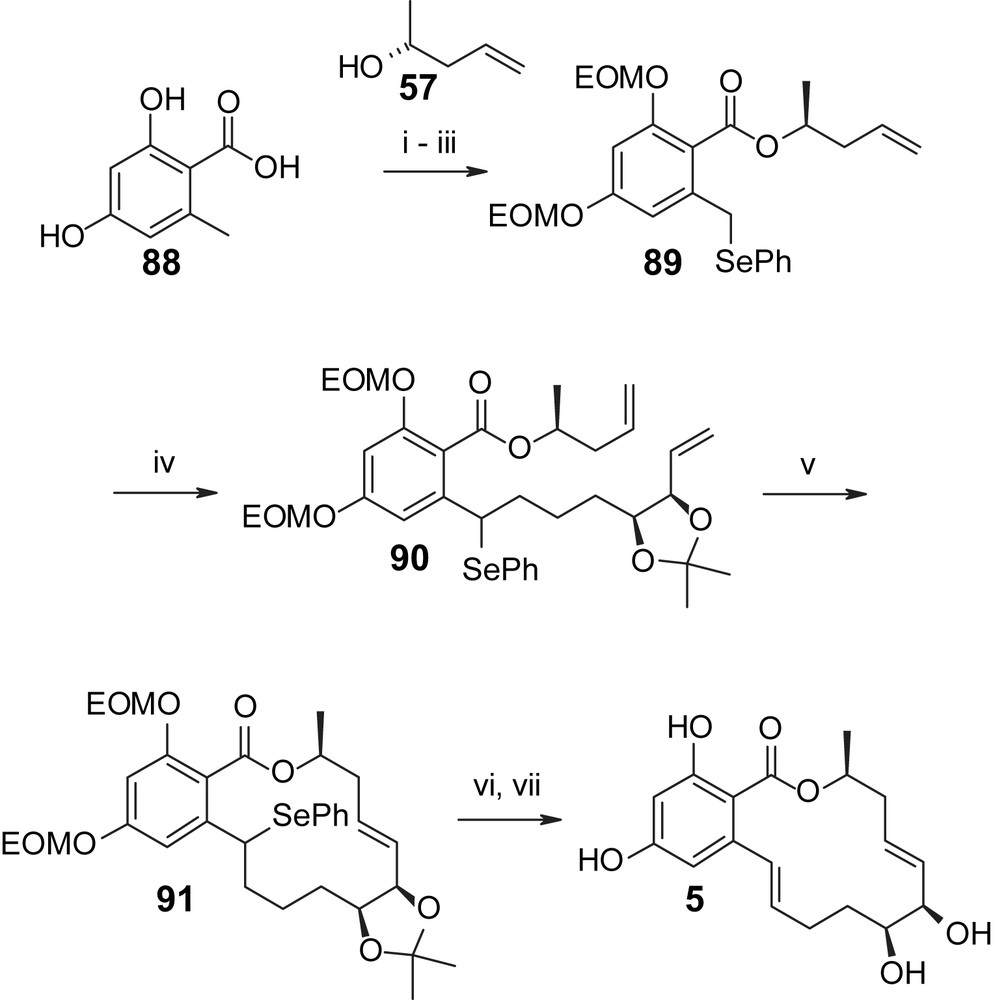
(i) PS-DEAD, (R)-(+)-4-hydroxy-1-pentene (57), m-ClPh3P, DCM, 0.5 h, 83%. (ii) EOMCl, iPr2EtN, TBAI (cat), DMF, 80 °C, 5 h, 95%. (iii) (a) LDA, THF, −78 °C; (b) (PhSe)2, 2 h, 75%. (iv) LDA (2.0 equiv), 96 (1.0 equiv), THF/HMPA 10/1, −78 °C, 20 min, 75%. (v) Grubbs II (0.05 equiv), toluene, 80 °C, 12 h, 92%. (vi) H2O2 (2.0 equiv), THF, 23 °C, 3 h, 82%. (vii) PS-SO3H, MeOH, 50 °C, 2 h, quant. PS = polymer-supported [30].
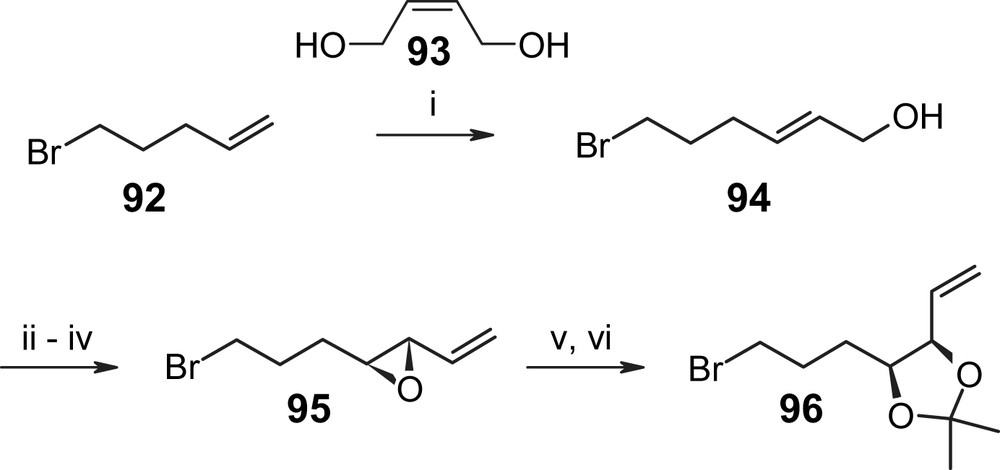
(i) 92 (1.0 equiv), 2-buten-1,4-diol (93) (2.0 equiv), Hoveyda–Grubbs catalyst II (0.01 equiv), DCM, 4 h, 97%. (ii) (a) l-(+) diethyl tartrate (0.12 equiv), Ti(Oi-Pr)4 (0.1 equiv), tBuOOH (1.52 equiv), DCM, −40 °C, 30 min; (b) 94 (1.0 equiv), −24 °C, 12 h, 85%. (iii) SO3·Pyr, DCM/DMSO, 0 °C, 30 min. (iv) Ph3PCH2, THF, −10 °C, 10 min, 70% (two steps). (v) Sc(OTf)3 (0.2 equiv), THF/H2O 10/1, 2.5 h, 100%. (vi) Dimethoxypropane, TsOH·H2O (cat), DCM, 12 h, 70%. Grubbs–Hoveyda catalyst II = 1,3-(bis-(mesityl)-2-imidazolidinylidene)dichloro-(o-isopropoxyphenyl-methylene)ruthenium [30].
Having successfully completed the total synthesis of aigialomycin D (5) by solution methods, Winssinger and co-workers have also extended the chemistry depicted in Scheme 21 to the solid-phase synthesis of a small library of aigialomycin analogs (Scheme 23) [30]. The enabling intermediate for this solid-phase approach is the polymer-bound sulfide 99 (which corresponds to intermediate 89 in Scheme 21), which could be elaborated into polymer-bound macrocycles in a way completely analogous to that depicted in Scheme 21 and employing a series of different alkyl bromides 100 (Scheme 23). Cleavage of the cyclization products from the solid-phase could be achieved either through oxidation/elimination or through radical reduction, thus leading to two different product series, 102 and 103, with or without a double bond between C1′ and C2′, respectively. Unfortunately, and in contrast to aigialomycin (5) itself, none of the analogs investigated showed any meaningful inhibition of CDK1, CDK5, or GSK3 [30].
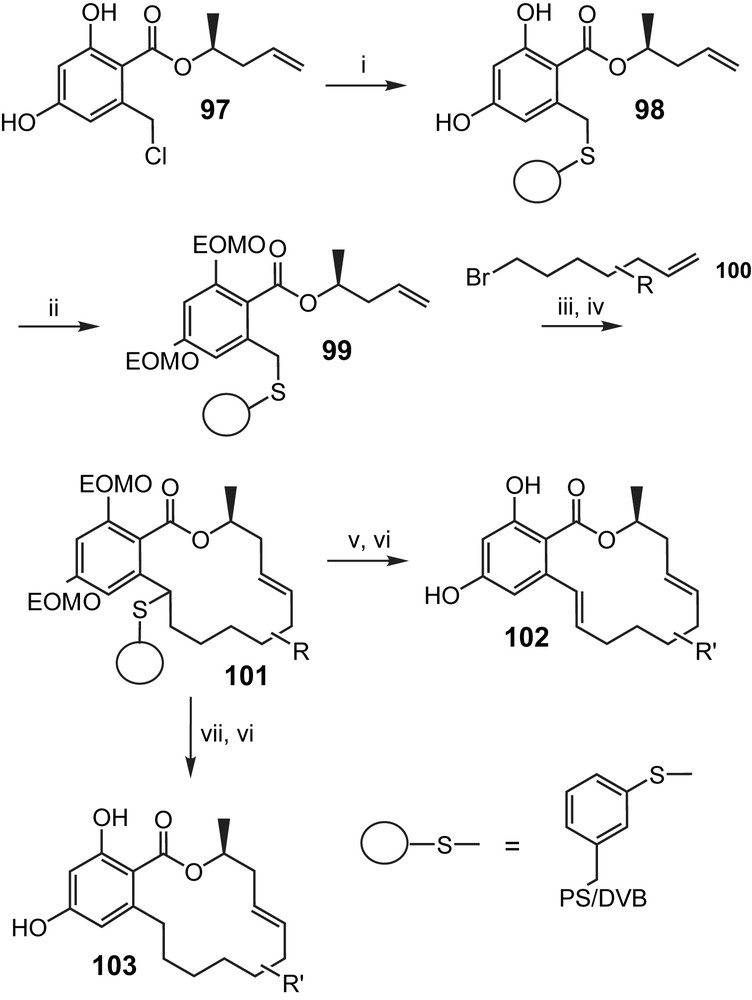
(i) PS-SH (1.0 equiv SH), 97 (1.1 equiv), iPr2NEt (1.0 equiv), DMF, 60 °C, 12 h, 82% (mass gain). (ii) EOMCl, DBU, TBAI (cat), DMF, 12 h, ∼96% (mass gain; 80% over two steps based on radical cleavage: AIBN (cat), nBu3SnH (5.0 equiv), toluene, 150 °C, microwave, 10 min). (iii) LDA (6.0 equiv), 100 (2.0 equiv), THF/HMPA 10/1, −78 °C, 20 min, quant. (based on radical cleavage or oxidation/elimination (H2O2 (4.0 equiv), DCM/HFIP 1/1, 12 h; then toluene, 80 °C, 3 h)). (iv) Grubbs II, DCM, 120 °C, microwave, 25 min, 100% (based on radical cleavage). (v) H2O2 (4.0 equiv), DCM/HFIP 1/1, 12 h; then toluene, 80 °C, 3 h, >90%. (vi) PS-SO3H, MeOH, 45 °C, 3 h, >90%. (vii) AIBN (cat), Bu3SnH (5.0 equiv), toluene, 150 °C, microwave, 10 min, >98%. For substituents R that constitute or contain protected functional groups R′ represents the deprotected substituent. Otherwise R′ = R [30].
More recently, total syntheses of aigialomycin D (5) have also been reported by Lu et al. [49] and Vu et al. [50] and a total synthesis of epi-aigialomycin has been published by Bajwa and Jennings [51]. While a detailed discussion of these additional syntheses of aigialomycin D (5) is beyond the scope of this review, a few of their key aspects shall still be highlighted. Thus, the synthesis of Vu et al. [50] is conceptually similar to Winssinger's approach, with ring closure being achieved through RCM with diene 106 (Scheme 24). The latter was obtained by benzylic acylation of ester 104 with Weinreb-amide 105. Remarkably, diene 106 could be cyclized to 107 under microwave conditions in 98% yield and with complete E selectivity, while conventional RCM conditions led to a 5.7/1 mixture of E/Z-isomers (Scheme 24) [50]. Reduction of the keto group followed by dehydration with Martin sulfurane [48] and global deprotection under acidic conditions then completed the synthesis of aigialomycin D (5). In contrast to the RCM-based approaches of Winssinger and Vu, Lu et al. [49] have prepared aigialomycin D (5) via macrolactonization of seco acid 109 under Yamaguchi conditions, which furnished protected aigialomycin (5) in 51% yield (Scheme 25). Interestingly, and in contrast to all other macrolactonization-based approaches discussed in this review, the carboxyl group of the resorcylic acid portion of 109 was introduced only late in the synthesis through metalation of bromine 108 and subsequent carboxylation with CO2. Global deprotection under standard acidic conditions gave aigialomycin (5).
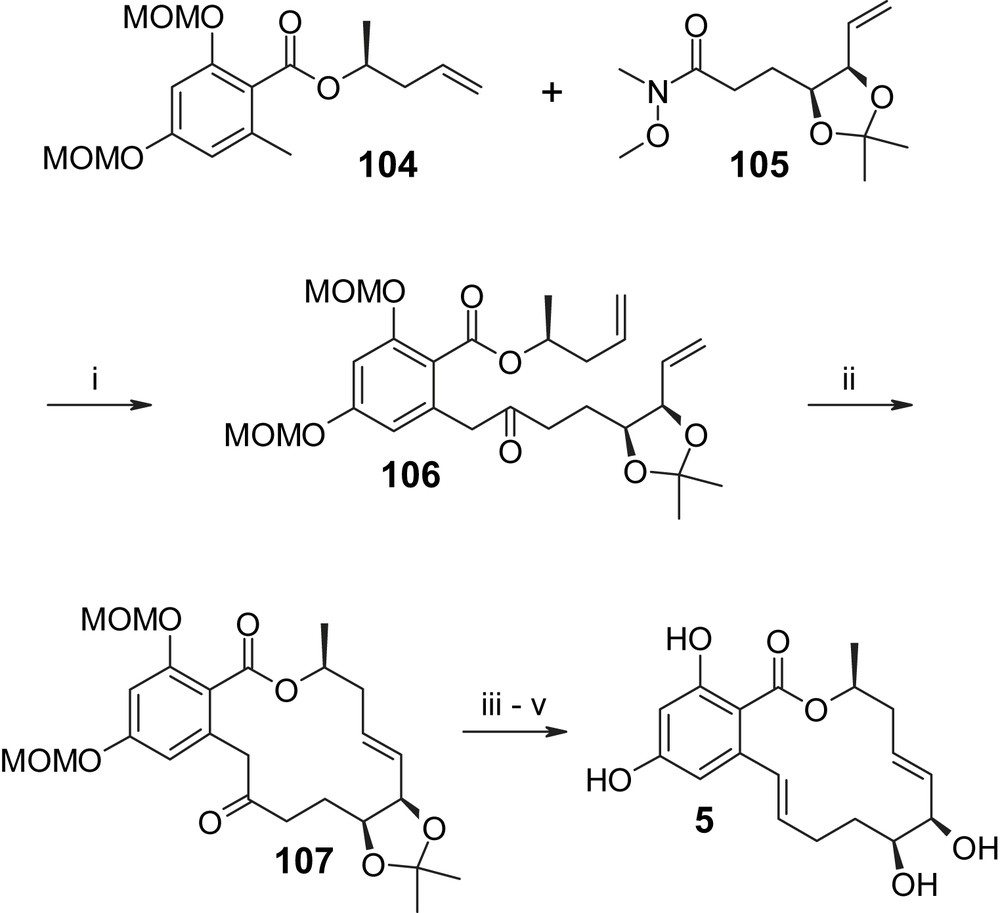
(i) LDA, THF, −78 °C, then 105, 82%. (ii) 106, DCM (0.005 M), Grubbs II catalyst (10 mol%), MW irradiation, 100 °C, 30 min, 98%, E only. (iii) NaBH4, MeOH/H2O 4/1, quant. (iv) (a) MsCl, Et3N, DMAP, DCM; (b) DBU, toluene, refl., 74% (two steps). (v) 1 N HCl/MeOH 1/1, 48 h, 91% [50].
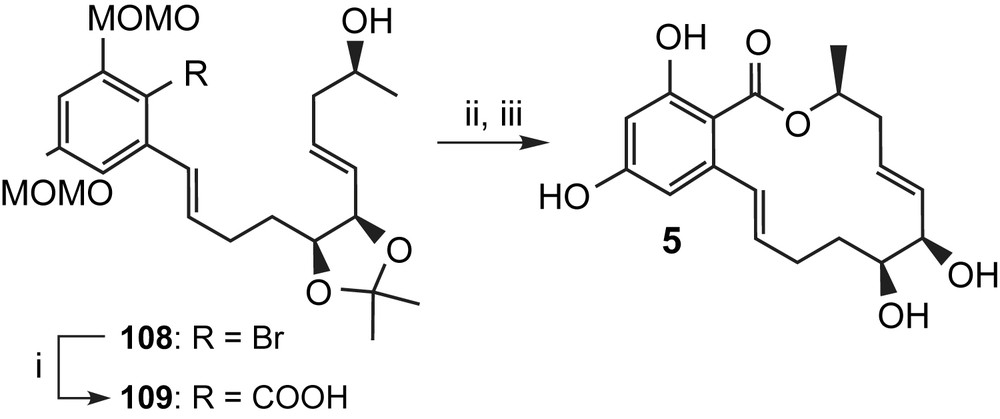
(i) nBuLi, CO2, THF, −78 °C → rt, 83%. (ii) 2,4,6-Trichlorobenzoylchloride, Et3N, THF, rt, 2 h, then DMAP, toluene, refl., 36 h, 51%. (iii) 0.5 M HCl, H2O/MeOH, 70% [49].
Finally, it is worth noting that RCM with a 6/1 mixture of trienes 110a/110b has been reported by Bajwa and Jennings [51] to produce none of the 14-membered macrocycle derived from 110a, while 110b was almost completely converted to macrolide 112 (Scheme 26) [51]. The major product isolated from the reaction mixture was diene 111, which was obtained in 84% yield and which is the result of 6-membered (rather than 14-membered) ring formation in the RCM. For triene 110a this is the sole reaction path, whereas 14-membered ring formation is clearly preferred for 110b, thus highlighting the importance of conformational effects in RCM-based cyclization reactions.
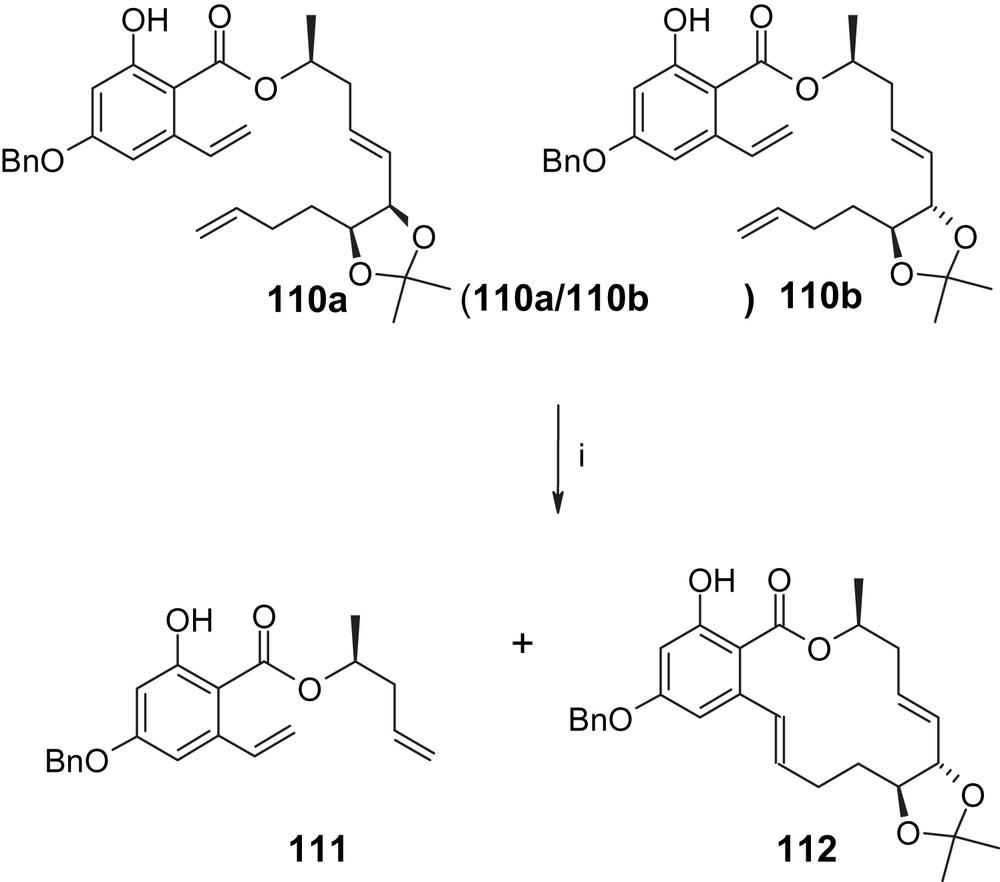
(i) 110a/b, DCM, Grubbs II catalyst (5 mol%), 50 °C, 16 h, 111: 83%; 112: 13% [51].
3 Conclusions
The discovery of the kinase-inhibitory activity of a number of natural resorcylic acid lactones has led to a broadened interest in the chemistry of these systems that extends beyond the older members of the class like radicicol or zearalenone. Of particular interest in this context are the cis-enone-containing compounds 1–4, which have all been shown to be highly potent protein kinase inhibitors. Total syntheses have now been developed for all four of these natural products, which should provide the foundation for the future synthesis of improved analogs and more extensive structure–activity studies. Interestingly, all successful approaches to the synthesis of cis-enone-containing resorcylic acid lactones to date are macrolactonization-based and alternative modes of ring closure have rarely been investigated. In particular, attempts on ring closure through RCM have not been described (in contrast, e.g., to aigialomycin D (5)), although the synthesis of a triene precursor for RCM-based macrocyclization between C1′ and C2′ has been reported recently. However, successful RCM with this substrate has not yet been demonstrated. It remains to be seen, whether efficient non-macrolactonization-based strategies can be developed for cis-enone-containing resorcylic acid lactones and whether the kinase-inhibitory potential of these structures will eventually lead to resorcylic acid lactone-based drug development candidates.