

1 Introduction
There is currently intense interest in the chemistry of dithiolate-bridged diiron complexes, [Fe2(CO)6(μ-SXS)], resulting from the realisation that they closely resemble the two-iron unit of the H-cluster active site of iron-only hydrogenases. A key step in the electrocatalytic conversion of protons to hydrogen at the active centre is the coordination of the proton(s). The hexacarbonyls themselves are generally not basic enough to bind a proton and the main approach adopted to circumvent this problem is to substitute one or more carbonyls for more basic phosphine ligands. In this way a large number of phosphine-substituted derivatives of [Fe2(CO)6(μ-pdt)] (pdt = SCH2CH2CH2S) have been prepared.
Recent theoretical studies have suggested that the asymmetry of the diiron centre may be a desirable feature of biomimetic models of iron-only hydrogenases [1]. One way to build in both steric and electronic asymmetries is to coordinate diphosphines in a chelating manner. With this in mind, we [2–4] and others [5–8] have begun exploring the reactivity of [Fe2(CO)6(μ-pdt)] (1) and related dithiolate-bridged complexes with a range of bidentate phosphines. From these studies it is becoming clear that formation of bridge and chelate isomers [Fe2(CO)4(μ-diphosphine)(μ-pdt)] and [Fe2(CO)4(κ2-diphosphine)(μ-pdt)] can occur, the latter existing as a mixture of dibasal and basal–apical isomers. The nature of products formed and their relative amounts depend upon the reaction conditions employed and the nature of the diphosphine backbone. For example with dppm, the formation of monodentate [Fe2(CO)5(κ1-dppm)(μ-pdt)] initially results [4,8]. This in turn converts under more forcing conditions into the bridged tetracarbonyl complex [Fe2(CO)4(μ-dppm)(μ-pdt)] with only very small amounts of the chelate isomer [Fe2(CO)4(κ2-dppm)(μ-pdt)] being formed [4]. Further, since the latter subsequently converts rapidly to the bridged isomer upon heating this precludes its development as a biomimetic model.
We thus sought to probe the use of other diphosphines towards preparation of asymmetric chelate complexes [Fe2(CO)4(κ2-diphosphine)(μ-pdt)]. In this context, cis-Ph2PCHCHPPh2 [4,5] and 1,2-(Ph2P)2C6H4 [4] have been shown to react with 1 to exclusively yield the desired chelate complexes and both are currently under investigation as models. As an extension of our work in this area we sought to probe the coordination to 1 with the widely utilized diphosphines, Ph2P(CH2)nPPh2 (n = 2, dppe; n = 3, dppp; n = 4, dppb), which have much more flexible backbones than dppm (n = 1). While this work was in progress two independent reports appeared covering aspects of this work. Thus, Sun and co-workers reported that addition of dppe to 1 under mild conditions affords tetranuclear [{Fe2(CO)5(μ-pdt)}2(μ,κ1,κ1-dppe)] [8], while under more forcing conditions Schollhammer detailed the isolation of the chelate complex, [Fe2(CO)4(κ2-dppe)(μ-pdt)] [6]. This latter work is particularly relevant to ours since it is also shown that low-temperature protonation of [Fe2(CO)4(κ2-dppe)(μ-pdt)] affords terminal hydride complexes, which rearrange to give the more stable bridged hydride [(μ-H)Fe2(CO)4(κ2-dppe)(μ-pdt)]+ [6]. This discovery may be important since, in context of biomimetic models, it is widely accepted that terminal hydride species are key intermediates in the electrocatalytic proton reduction.
2 Results and discussion
2.1 Diphosphine-linked tetranuclear complexes [{Fe2(CO)5(μ-pdt)}2(μ,κ1,κ1-diphosphine)] (2a–d)
Hexacarbonyl [Fe2(CO)6(μ-pdt)] (1) is unreactive towards phosphines at room temperature in the absence of either the carbonyl-activating reagent Me3NO or UV–visible irradiation. Addition of diphosphines Ph2P(CH2)nPPh2 (n = 2–4) and trans-Ph2PCHCHPPh2 to 2 equiv of 1 in acetonitrile with added Me3NO·2H2O resulted in the immediate formation of the diphosphine-linked tetranuclear complexes [{Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-diphosphine)] (2a–d) in high yields. If 1 equiv of 1 is employed then formation of 2a–d is again clean leaving some unreacted 1. Formation of 2a–d is readily seen by changes in the IR spectrum the characteristic low frequency carbonyl vibration of 1 at 2074 cm−1 being replaced by a band at ca. 2045 cm−1. All display a singlet in the 31P NMR spectra consistent with a symmetric structure, while 1H NMR data are generally not very informative. While this work was in progress, Sun, Akermark and co-workers reported an analogous synthesis of the dppe complex 2a [8].
In order to elucidate the binding mode of the diphosphine in these complexes a crystallographic study was carried out on 2d and the results of which are shown in Fig. 1. The molecular structure closely resembles that of 2a characterised by Song. The diphosphine links the two diiron centres by binding to each in an apical manner as defined by the Fe(1)–Fe(2)–P(1) angle of 151.81(3)°. Tetranuclear 2d was the only product isolated from the reaction of 1 and trans-Ph2PCHCHPPh2 under all reaction conditions employed in this study. In contrast, the nature of the products obtained from the more flexible diphosphines Ph2P(CH2)nPPh2 (n = 2–4) was found to be highly dependent upon the reaction conditions employed. These studies are described below.

Molecular structure of [{Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-trans-Ph2PCHCHPPh2)] (2d) with selected bond lengths (Å) and angles (°): Fe(1)–Fe(2) 2.5233(6), Fe(2)–P(1) 2.2199(8), Fe(1)–S(1) 2.2618(8), Fe(1)–S(2) 2.2757(8), Fe(2)–S(1) 2.2653(8), Fe(2)–S(2) 2.2640(8), Fe(1)–Fe(2)–P(1) 151.81(3).
2.2 Chelate complexes [Fe2(CO)4(κ2-diphosphine)(μ-pdt)] (3a and b)
As stated in Section 1, a key goal of our work in this area is the synthesis of biomimetic models in which the diiron centre is both electronically and sterically unsymmetrical. Accordingly, chelate complexes [Fe2(CO)4(κ2-diphosphine)(μ-pdt)] are key targets. While this work was in progress, Schollhammer and co-workers reported the synthesis and crystallographic characterization of [Fe2(CO)4(κ2-dppe)(μ-pdt)] (3a) formed in moderate yields upon heating a toluene solution of 1 and dppe toluene at 70 °C in the presence of Me3NO [6]. We have also prepared 3a in an analogous manner, heating the same component mixture in acetonitrile at 70 °C for ca. 5–8 h. Our data for 3a correspond closely with that published and in light of Schollhammer's work we did not investigate the reactivity of 3a any further.
Heating an acetonitrile solution of 1 and dppp in the presence of 2 equiv of Me3NO·2H2O for 5–8 h leads to the formation of a dark brown solution. After removal of volatiles a 31P NMR spectrum revealed the formation of a number of products, including the diphosphine-linked 2b. Extraction of the mixture with diethyl ether gave a dark brown solution from which [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b) was isolated as dark red-brown crystals after cooling at 0 °C. The latter was also formed upon heating a toluene solution of 1 and dppp at reflux for 14 h. In this case, column chromatography afforded 3b as a dark brown solid. Formation of the chelate complex is immediately apparent from the characteristic nature of the carbonyl region of the IR spectrum which for 3b displays bands at 2019vs, 1946s and 1890s cm−1. In the 31P NMR spectrum a large singlet is observed at δ 53.3 ppm together with a smaller singlet at δ 48.6 ppm. These are ascribed to basal–apical and dibasal isomers of 3b, respectively, approximately in a 12:1 ratio. Such isomerism has been widely observed in complexes of this type [2–7]. It is noteworthy that the major basal–apical isomer is observed as a sharp singlet suggesting the rapid interconversion of the two inequivalent phosphorus nuclei at room temperature, while the also sharp nature of the dibasal signal shows that this exchange does not occur via this isomer. At 198 K in CD2Cl2, the major room temperature singlet resonance attributed to the apical–basal isomer (δ 55.09 ppm) splits into two broad resonances at δ 59.7 and 54.9 ppm, while the resonance attributed to the dibasal isomer (δ 51.02 ppm) remains as a singlet. These observations are fully consistent with those reported by Schollhammer and co-workers for the dppe complex 3a [6].
In order to fully elucidate the solid-state structure of 3b and to relate this to the isomeric mixture observed in solution an X-ray crystallographic study was carried out and the results of which are displayed in Fig. 2 and Table 1. In the solid state the diphosphine binds in an apical–basal manner with one phosphorus atom lying approximately trans to the iron–iron vector [Fe(2)–Fe(1)–P(2) 151.996(15)°], and the second one lies trans to one of the thiolate atoms [P(1)–Fe(1)–S(2) 163.544(18)°]. The two iron–phosphorus bonds differ slightly [Fe(1)–P(1) 2.2272(4), Fe(1)–P(2) 2.2077(5) Å], while the dithiolate ligands bridge the diiron vector approximately symmetrically. As can be clearly seen in Fig. 2, the six-membered chelate ring adopts an approximate chair conformation. In an earlier communication we compared the structure of 3b to five other crystallographically characterised complexes of the type [Fe2(CO)4(κ2-diphosphine)(μ-pdt)] [3]. The structure of 3b is very similar to that of 3a (Table 1) although the bite-angle [P(1)–Fe(1)–P(2)] of 94.96(2)° found in 3b is significantly larger than that of 87.7(4)° observed in the dppe analogue. The larger bite-angle of the dppp ligand in 3b may account for the increased amount of apical–basal vs dibasal isomer in solution; a ratio of 12:1 in 3b as compared to ca. 5:1 in 3a. Crystal structures of related dibasal complexes [3] reveal much smaller bite-angles of ca. 71–75°. For example, the diphosphine bite-angle in the dibasal complex [Fe2(CO)4(κ2-dppm)(μ-pdt)] is 74.55(4)° [3,4]. Thus, exchange between apical–basal and dibasal isomers requires a significant change in the bite-angle of the diphosphine and may account for the slow interconversion of the different isomers in solution.

Two views of the molecular structure of [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b).
Selected metric data for [Fe2(CO)6(μ-pdt)] and diphosphine derivatives
| Subs. type | Fe–Fe (Å) | Fe–S (Å) | Fe–Fe–P (°) | Fe–P (Å) | P–Fe–P (°) | Reference | |
| [Fe2(CO)6(μ-pdt)] (1) | 2.510(1) | 2.252(2) | [9] | ||||
| [Fe2(CO)4(κ2-dppm)(μ-pdt)] | Basal–basal | 2.5879(7) | 2.224(2)a | 107.51(3) | 2.2123(11) | 74.55(4) | [3,4] |
| 2.263(2)b | 113.56(3) | 2.2217(11) | |||||
| [Fe2(CO)4(κ2-dppe)(μ-pdt)] (3a) | Basal–apical | 2.547(7) | 2.256(2)a | 110.9(3)c | 2.231(1)c | 87.7(4) | [6] |
| 2.271(2)b | 158.4(3)d | 2.190(1)d | |||||
| [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b) | Basal–apical | 2.5614(3) | 2.258(1)a | 110.23(2)c | 2.2272(4)c | 94.96(2) | This work |
| 2.262(1)b | 152.00(2)d | 2.2077(5)d | |||||
| [Fe2(CO)4(μ-dppm)(μ-pdt)]e | Basal–basal (cisoid) | 2.5076(9) | 2.256(2) | 97.44(4) | 2.213(1) | [4] | |
| 2.254(2) | 97.86(4) | 2.210(1) | |||||
| 2.5095(12) | 2.244(2) | 97.36(3) | 2.217(1) | ||||
| 2.5130(12) | 2.253(2) | 98.03(3) | 2.215(1) | ||||
| [Fe2(CO)4(μ-dppe)(μ-pdt)]f (4a) | Basal–basal (cisoid) | 2.5266(4) | 2.261(2) | 101.90(2) | 2.2341(5) | This work | |
| 2.266(2) | 110.56(2) | 2.2097(5) | |||||
| 2.5304(4) | 2.260(2) | 106.87(2) | 2.2262(6) | ||||
| 2.259(2) | 106.31(2) | 2.2072(6) | |||||
| [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c) | Basal–basal (cisoid) | 2.6246(3) | 2.259(1) | 122.64(1) | 2.2388(4) | This work | |
| 2.244(1) | 118.09(1) | 2.2344(4) |
a Fe(CO)–S.
b Fe(CO)3–S.
c Basal P.
d Apical P.
e One full and two half molecules in asymm. unit.
f Two molecules in asymm. unit.
In all reactions between 1 and dppb, no evidence was noted for the formation of the chelate complex [Fe2(CO)4(κ2-dppb)(μ-pdt)]. Given the propensity for dppp to form the chelate complex 3b this is somewhat surprising. However, it is well known that seven-membered chelate complexes are less stable than analogous five or six-membered rings and it may be that under the relatively harsh reaction conditions employed conversion of [Fe2(CO)4(κ2-dppb)(μ-pdt)] to the bridge isomer 4c may be rapid.
2.3 Bridge complexes [Fe2(CO)4(μ-diphosphine)(μ-pdt)] (4a–c)
We have previously reported that thermolysis of the small bite-angle diphosphines, R2PCH2PR2 (R = Ph, Cy) and 1 in toluene affords the diphosphine-bridged complexes [Fe2(CO)4(μ-R2CH2PR2)(μ-pdt)] as the major products [4]. Refluxing equimolar amounts of 1 and dppe in toluene for 14 h resulted in the clean formation of [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a) in good yield after chromatography. Characterization as a bridge isomer is easily made on the basis of the very distinctive nature of the carbonyl region of the IR spectrum with absorptions observed at 1990s, 1953vs, 1920s and 1902m cm−1, which can be compared to the analogous chelate complex 3a which shows absorptions at 2019s, 1949s and 1904w cm−1. Other spectroscopic data were relatively uninformative, the appearance of a singlet at δ 60.5 ppm in the 31P NMR spectrum confirming the symmetric nature of the diiron centre.
While this manuscript was in preparation, Schollhammer and co-workers reported the independent synthesis of 4a [7]. This resulted from the electron-transfer-catalysed (ETC) isomerisation of the chelate isomer 3a. We have also shown independently that 3a rearranges upon heating in toluene to the bridge isomer 4a clearly showing that the latter is the thermodynamic product. Indeed, in a wide range of other [Fe2(CO)4(diphosphine)(μ-pdt)] complexes this is also found [10]. We were able to grow large orange block crystals of 4a suitable for X-ray crystallography upon slow diffusion of methanol into a saturated dichloromethane solution. The structure contains two independent molecules in the asymmetric unit. One of these is displayed in Fig. 3 while key metric parameters are given in Table 1. Gross structural characteristics are very similar to those found in [Fe2(CO)4(μ-dppm)(μ-pdt)] (which crystallizes with one full and two half molecules in the asymmetric unit) [4,8]. The diphosphine spans the iron–iron vector, lying cis to one sulfur and trans to the second. While independent molecules of 4a are very similar there are some significant differences. Most notable is the large difference in Fe–Fe–P angles in the two independent molecules. Hence in molecule A (shown) these vary by nearly 9°, while in contrast, in molecule B they are approximately the same. In both independent molecules, these angles are significantly greater than those found [97.36(3)–98.03(3)°] in the dppm analogue. These observations suggest that diphosphine coordination in 4a is considerably more flexible than that in [Fe2(CO)4(μ-dppm)(μ-pdt)].

Two views of the molecular structure of [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a) (molecule A).
As previously alluded to, heating a toluene solution of 1 and dppp at reflux for 14 h leads to the formation of a complex mixture from which the chelate complex [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b) was isolated in low yield after chromatography. When a similar reaction was carried out at 60 °C in acetonitrile with 2 equiv of Me3NO·2H2O, 3b was isolated in somewhat higher yields after extraction with diethylether. This left a dry red solid which has been tentatively assigned to the bridge isomer [Fe2(CO)4(μ-dppp)(μ-pdt)] (4b). In the 31P NMR spectrum a broad singlet is observed at δ 54.0 ppm, and while all resonances in the 1H NMR spectrum are broad, integration of the aryl and alkyl proton signals gives a relative ratio of ca. 5:3 as expected. The carbonyl region of the IR spectrum shows a pattern similar to that observed for 4a but with more features. Thus major bands are seen at 1994s, 1949vs, 1928s and 1900m cm−1, but two of the four bands have significant shoulders at 1985 and 1920 cm−1. All attempts to crystallize 4b have to date been unsuccessful. The extra features in the IR spectra and broad nature of the NMR data might be due to different conformations able to be adopted by the flexible dppp ligand.
Heating an acetonitrile solution of 1 and dppb at 70 °C for 6 h only gave rise to the isolation of the diphosphine-linked complex 2c. However, thermolysis of a toluene solution of 1 and dppb for 14 h afforded after chromatography the bridge complex [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c) in good yield. Formation of the bridge isomer was again easily shown by IR spectroscopy and no evidence was seen for the chelate isomer. A singlet resonance at δ 56.3 ppm in the 31P NMR spectrum confirmed the equivalence of the phosphorus atoms at this temperature, and while the 1H NMR spectrum was relatively uninformative, the observation of six resonances in the aliphatic region was consistent with the proposed structure. Three of these between δ 2.41 and 2.21 are associated with the pdt ligand, and the others between δ 1.99 and 1.53 are assigned to the tetramethylene sub-unit of the diphosphine.
In order to confirm the bridging nature of the dppb ligand an X-ray study was carried out and the results of which are shown in Fig. 4 and Table 1. The overall solid state structure is superficially similar to that noted for [Fe2(CO)4(μ-dppm)(μ-pdt)] and [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a), with the diphosphine adopting the expected cisoid dibasal geometry. However, some key metric parameters differ significantly. The iron–iron bond at 2.6246(3) Å is significantly longer than those found in other bridged diphosphine complexes of this type and is more akin to those found in the unsymmetrically substituted dibasal complexes. For example, the longest iron–iron bond we have previously encountered during our work in this area is 2.6236(5) Å in [Fe2(CO)4{κ2-Ph2PN(Pri)PPh2}(μ-pdt)] [3]. The Fe–Fe–P bond angles of 122.64(1) and 118.09(1)° also vary considerably from those in the analogous dppm and dppe complexes, being much larger. Thus, a not unexpected trend is apparent whereby the longer diphosphine backbone allows the bond angles within the Fe2P2(μ-S)2 core to adopt their preferred positions. This leads to a longer Fe–Fe bond and we believe these factors may be important when seeking to justify the observed differences in stabilities of the resulting protonated complexes (see below).
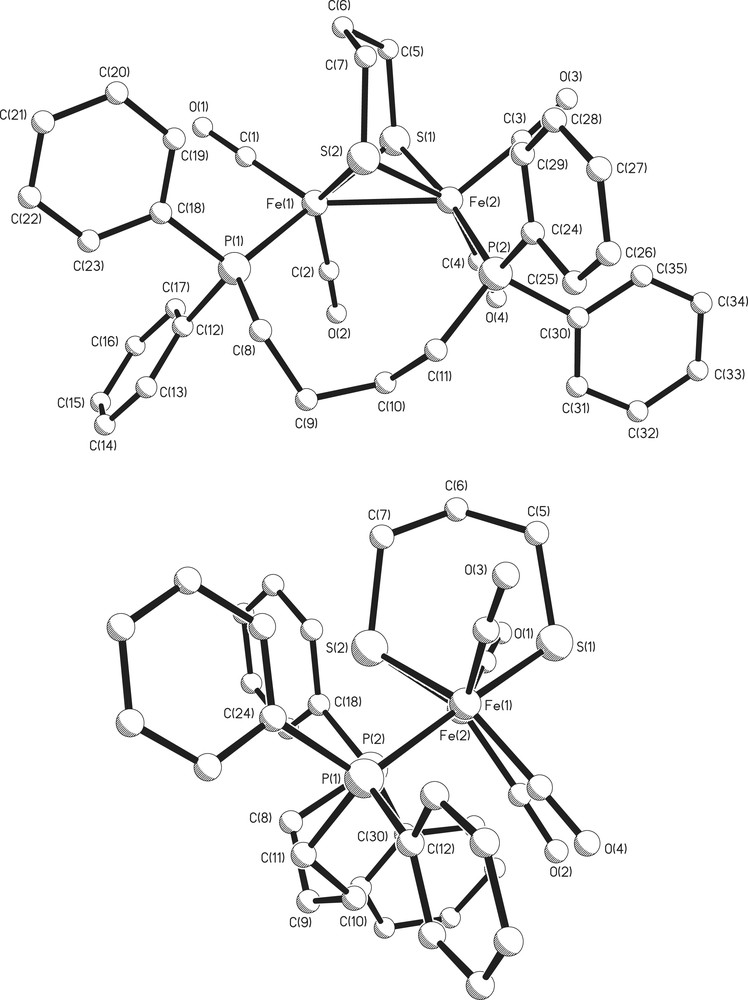
Two views of the molecular structure of [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c).
While the 1H and 31P spectra of 4c are straightforward at room temperature, upon both raising and lowering the temperature significant changes occur to both indicative of fluxionality. No significant changes occur to the 31P NMR spectrum upon raising the temperature; however, this is not the case with the 1H NMR spectrum. At room temperature the aromatic region is simple consisting of four well-defined signals (two accidentally overlap) associated with the inequivalence of the two phenyl groups on each phosphorus atom as a result of their relative disposition to the dithiolate bridges. Upon warming these broaden and collapse (ca. 323 K) into two sharp signals (333 K) suggesting that all four phenyl groups are now equivalent. Associated changes also occur with the aliphatic protons but these are more difficult to interpret. From an analysis of the data using the modified Eyring equation a free energy of activation for this process is estimated at 67 ± 1 kJ mol−1. We attribute these changes to the movement of the diphosphine from one side of the molecule to the other; that is between adjacent cisoid dibasal sites. We have proposed that a similar process occurs in the more constrained dppm analogue, [Fe2(CO)4(μ-dppm)(μ-pdt)] [4] which occurs with a free energy of activation of ΔG# 61 ± 1 kJ mol−1. Such a process in [Fe2(CO)4(μ-dppm)(μ-pdt)] must be concerted in nature since a transoid dibasal intermediate is inaccessible due to steric strain. In 4c, as a result of the highly flexible tetramethylene backbone, this need not be the case. This may account for the small (but significant) differences in the measured free energies of activation.
Further, given the flexibility of the dppb ligand, we considered that at low temperature more than one relative conformation of Fe2P2C4 ring might be accessible. In order to probe this we monitored both the 1H and the 31P NMR spectra upon cooling a CD2Cl2 solution. While significant changes occurred to the 1H NMR spectra, these were difficult to interpret. The 31P NMR spectra changed little until below 253 K, when the singlet resonance broadens considerably collapsing and appearing as five broad signals at 183 K. These signals fall into three sets of singlet pairs (two are coincident) approximately in a 5:2:1 ratio. We have previously shown that the phosphorus nuclei in [Fe2(CO)4(μ-dppm)(μ-pdt)] become inequivalent at low temperature, being associated with the freezing out of the “flipping” of the central methylene of the pdt ligand [4]. This is also clearly the case in 4c but if this alone is occurring then it should lead to only two low-temperature resonances in the 31P NMR spectrum. Thus, we believe that the presence of six inequivalent phosphorus sites shows that different Fe2P2C4 ring conformations are possible. We presume that the major low-temperature species in solution is that adopted in the solid state but unfortunately because of the broadness of the low-temperature spectra we cannot easily comment further on this.
2.4 Protonation studies
A key function of biomimetic models of the iron-only hydrogenase enzyme is their ability to bind a proton and consequently we have studied protonation reactions of the new complexes reported herein. While this manuscript was in preparation, Schollhammer and co-workers independently prepared [Fe2(CO)4(κ2-dppe)(μ-pdt)] (3a) and showed that it protonated readily upon addition of HBF4·Et2O to give the bridging hydride [(μ-H)Fe2(CO)4(κ2-dppe)(μ-pdt)][BF4] which was crystallographically characterised as the dibasal isomer [6]. Further, monitoring the protonation by 1H and 31P NMR at low temperature allowed a clear reaction pathway to be elucidated (Scheme 1). At 203 K in CD2Cl2, two terminal hydride species are initially observed differing in the location of the hydride at the phosphine (proximal) or non-phosphine (distal) bound iron atom. Notably these complexes are characterised by a triplet at δ −2.47 (JPH 69 Hz) and a singlet at δ −4.4 in the 1H NMR spectrum. Upon warming they convert to a bridging hydride complex with a basal–apical arrangement of the diphosphine, which in turn rearranges to give the isolated dibasal bridging hydride complex.
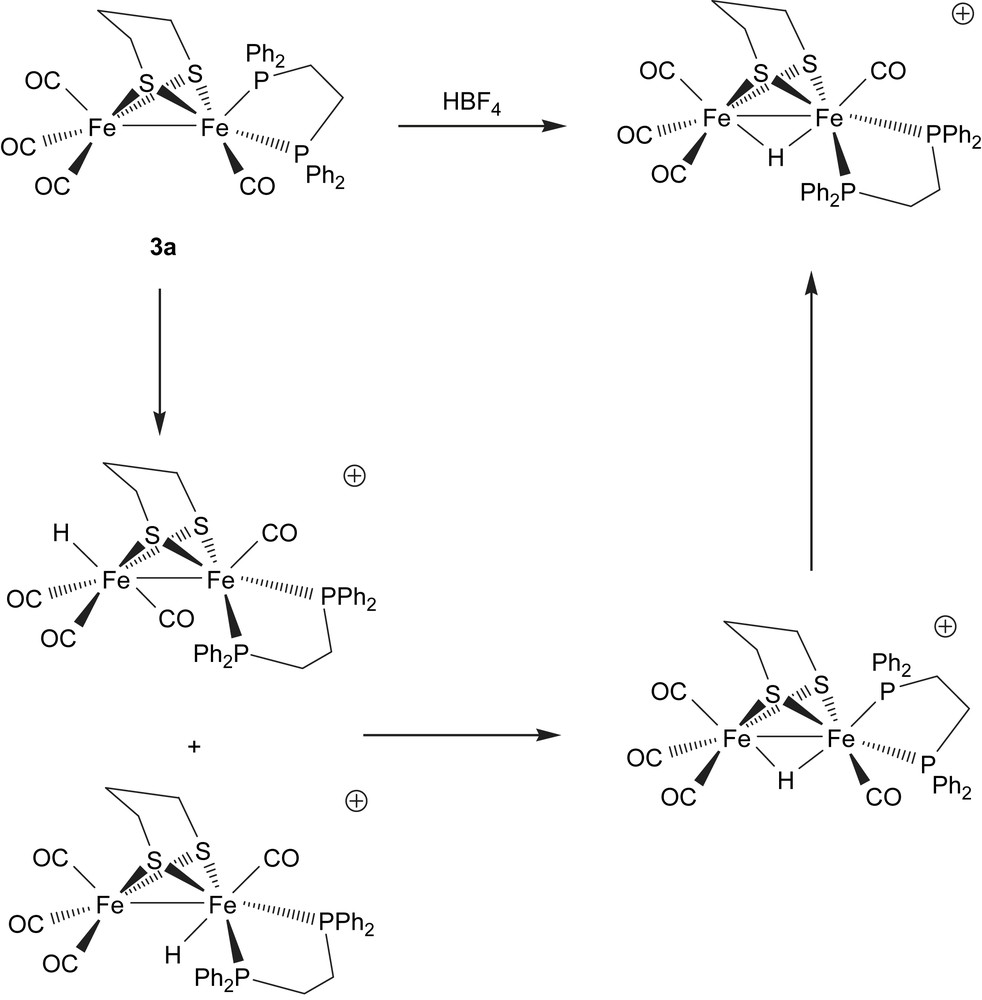
We have carried out a very similar protonation study of [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b). Addition of a slight excess of HBF4·Et2O to a dichloromethane solution of 3b in air results in a rapid darkening of the initial brown solution. After stirring for 2 h the protonated complex [(μ-H)Fe2(CO)4(κ2-dppp)(μ-pdt)][BF4] (5) was isolated in high yield. The presence of a bridging hydride and dibasal arrangement of the diphosphine were easily shown by the presence of a high-field triplet at δ −12.65 in the 1H NMR spectrum and a doublet at 44.8 ppm in the 31P NMR spectrum (JPH 16.4 Hz). Protonation was also clearly shown with the shift to higher wavenumbers of the carbonyl absorptions in the IR spectrum; from 2019, 1947 and 1890 cm−1 for 3b to 2096, 2039 and 1954 cm−1 for 5. Addition of excess NEt3 to a dichloromethane solution of 5 resulted in no discernable change as monitored by IR spectroscopy, showing that the diiron centre is extremely basic.
When monitoring the addition of HBF4·Et2O to 3b by IR spectroscopy at room temperature, it was noted that in the early stages of the reaction (30–60 s) an extra peak appeared at 2080 cm−1 while the low frequency absorption was very broad being centred at ca. 1960 cm−1. Within minutes these features were lost suggesting the formation of an intermediate species. In order to probe this further, a CD2Cl2 solution of 3b was protonated at low temperature (frozen solution in liquid nitrogen) whilst monitoring by 1H and 31P NMR spectroscopies. After addition of one drop of acid at 203 K the high-field (hydride) region of the 1H NMR spectrum clearly showed the formation of five hydridic species. These appeared as: a triplet at δ −1.21 (J 70.8 Hz), a triplet at −12.82 (J 16.4 Hz), a singlet at −14.39, a doublet at −14.98 (J 20.8 Hz) and a broad signal at −15.3 approximately in a 2:1:1:4:1 ratio. Upon warming to 298 K, four of these disappeared to be replaced by a triplet at δ −12.65 associated with 5. The low-field triplet is assigned to the terminal hydride 6, and corresponds closely to that assigned to the proximal dibasal complex of Schollhammer et al. [6]. We saw no evidence for any further terminal hydride complexes. On the basis of the 31{1H} NMR spectrum (see below) we assign the doublet at δ −14.98 to 7, in which the hydride bridges the iron–iron vector and the diphosphine adopts a basal–apical coordination.
Even greater complexity is seen in the 31P NMR spectrum at 203 K which we also tentatively assign to the presence of five species. A singlet at 66.1 ppm is assigned to the terminal hydride 6 (since in later spectra its disappearance coincided with that of the low-field hydride resonance); a set of doublets at 54.3 and 38.5 ppm (JPP 55.5 Hz) are assigned to the basal–apical bridging hydride complex 7 and a doublet at 45.8 ppm (JPH 16 Hz) is associated with 5. Two small and (approximately) equal intensity singlets at 55.8 and 52.1 ppm remain unassigned. These are presumably associated with hydride signal at δ −14.39 and −15.3. Such extra resonances were not seen by Schollhammer in the analogous dppe chemistry and this leads us to consider that they may be associated with different orientations of the six-membered dppp chelate ring. Warming to 223 K again results in little change but above this temperature all species convert to 5. In light of Schollhammer's work (Scheme 1), our interpretation of these observations is shown in Scheme 2.

It should be recalled that [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b) exists as a mixture of dibasal (3b′) and basal–apical (3b) isomers (ca. 1:12) in solution. Since the terminal hydride we observe correlates closely with that attributed to the dibasal–proximal hydride species by Schollhammer et al. [6] we propose that this results from protonation of the dibasal isomer of 3b′. We do not observe any further terminal hydride species suggesting that for 3b either terminal protonation of the major basal–apical isomer is unfavourable or that this complex is unstable and rapidly rearranges to another species. Unfortunately we have no way of differentiating these possibilities. The major species (>50%) we see upon protonation at 203 K is the apical–basal bridging hydride 7. We favour a scenario whereby this is directly formed upon protonation of the major basal–apical isomer of 3b, however, again this cannot be proven. The basal–apical arrangement of the diphosphine is clearly established from the appearance of the set of doublets at 54.3 and 38.5 ppm. We also attribute the extra peaks observed in the IR spectrum to the initial formation of this complex since it is the major component during early reaction times.
We have previously reported that protonation of [Fe2(CO)4(μ-dppm)(μ-pdt)] by HBF4·Et2O affords only an unstable complex which was believed to be the cationic bridging hydride, while in contrast under similar conditions the more basic diiron centre in [Fe2(CO)4(μ-dcpm)(μ-pdt)] (dcpm = Cy2PCH2PCy2) readily protonates to give [(μ-H)Fe2(CO)4(μ-dcpm)(μ-pdt)][BF4] which was crystallographically characterised [4]. On the basis of IR data, the diiron centres in [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a) and [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c) have comparable basicity to that in [Fe2(CO)4(μ-dppm)(μ-pdt)] and thus similar behavior might be expected upon proton addition. Indeed, this is the case with 4a. Addition of a slight excess of HBF4·Et2O resulted in a lightening of the solution and analysis of the resultant IR spectrum revealed the development of new absorptions tentatively associated with a cationic bridging hydride. However, the reaction is not clean, a significant amount of 4a remains and the stability of the purported cation is poor. As a result, no further characterization was possible. In contrast, addition of a slight excess of HBF4·Et2O to 4c resulted in an immediate colour change from dark orange to pale yellow resulting from the complete consumption of 4c (1986s, 1946vs, 1916s, 1893m cm−1) and clean generation of a new cationic complex (2058m, 2040s, 2001vs cm−1) as shown by IR spectroscopy. This species showed good stability in dichloromethane in air, the solution only decolorizing with loss of all carbonyl absorptions over ca. 1 d. When monitored by 31P NMR spectroscopy, similar addition of acid to a CD2Cl2 solution of 4c resulted in the complete loss of the singlet at 57.5 ppm associated with 4c and concomitant clean formation of a doublet resonance at 42.7 ppm (JPH 19.6 Hz), while in the 1H NMR spectrum a high-field triplet was observed at δ −13.8. When carried out on a preparative scale, the clean formation of a yellow solid with identical spectroscopic data was noted and this is characterised as the cationic bridging hydride [(μ-H)Fe2(CO)4(μ-dppb)(μ-pdt)][BF4] (8). Unfortunately, all our attempts to grow single crystals of 8 failed.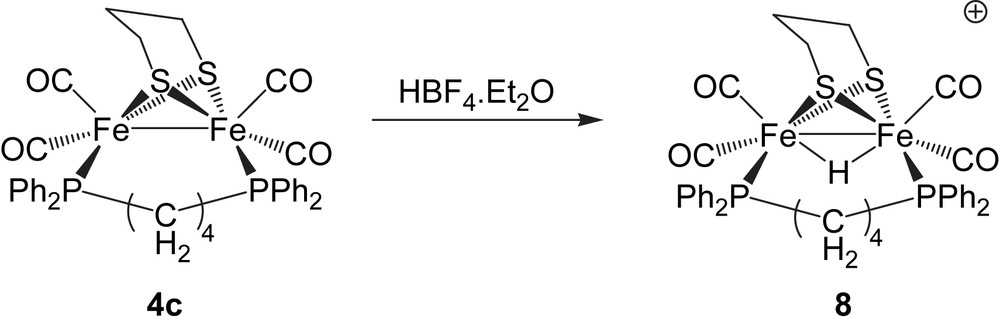
The ready protonation of 4c as compared to 4a or [Fe2(CO)4(μ-dppm)(μ-pdt)] is striking. All three have very similar IR spectra although 4c (1986s, 1946vs, 1916s, 1893m cm−1) is slightly more basic than 4a (1990s, 1953vs, 1920s, 1902m cm−1) and [Fe2(CO)4(μ-dppm)(μ-pdt)] (1988s, 1957vs, 1921s, 1908m cm−1) and it may be that these small differences tip the balance in favour of hydride formation for the more basic dppb complex. However, we feel that this may be only part of the answer. As clearly seen from a comparison of the solid-state structures of the three, while superficially very similar the increase in flexibility of the diphosphine resulting from the increasing number of methylene linking groups potentially allows a relaxation of the diiron core as seen by the longer Fe–Fe bond length and greater Fe–Fe–P angles. The former is likely to be especially important as the iron–iron bond is expected to elongate upon protonation, while the flexibility of the diphosphine may allow access to the most favourable conformation of the cationic hydride, being otherwise inaccessible for the more rigid diphosphines. Both of these factors might be expected to affect both the thermodynamic (overall stability) and the kinetic (rate of formation) basicities of the diiron centre – both observations being made above. In support of this, we have also found that a series of analogous bridged small bite-angle diphosphine complexes [Fe2(CO)4{μ-Ph2PN(R)PPh2}(μ-pdt)] also form unstable cationic hydrides [10].
3 Conclusions
We have shown in this and related [4] work that the nature of the products formed upon reaction of [Fe2(CO)6(μ-pdt)] (1) with diphosphines Ph2P(CH2)nPPh2 (n = 1–4) depends significantly on the flexibility of the methylene backbone. Significantly in light of our interest in the synthesis of unsymmetrically substituted biomimetics of the iron-only hydrogenase enzyme, chelating tetracarbonyl complexes are accessible for both dppe (n = 2) [6] and dppp (n = 3). Further, both react with HBF4·Et2O at low temperature to yield terminal hydride complexes. The stability of these is, however, poor and both convert to the thermodynamically favourable bridging hydride complexes with dibasal diphosphine coordination via a process that involves a basal–apical bridging intermediate. If our supposition (Scheme 2) that it is the dibasal isomer of the unsymmetrical diphosphine complex which reacts rapidly to form the proximal terminal hydride, then it may be that the use of a diphosphine which greatly favours this orientation over the basal–apical geometry (preferred by dppe and dppp complexes) may allow better access and stability to such terminal hydride complexes. We are currently pursuing this strategy with low-temperature protonation studies of [Fe2(CO)4{κ2-Ph2PN(R)PPh2}(μ-pdt)] [5,10] and [Fe2(CO)4{κ2-Ph2PC(Me)2PPh2}(μ-pdt)] [10] which strongly favour dibasal diphosphine coordination.
4 Experimental section
4.1 Methods and materials
All reactions were carried out using standard Schlenk-line techniques under N2 and reaction solvents were purified on alumina columns. Work-up was done in air using standard bench reagents. All diphosphines were purchased from Aldrich and used without further purification, and [Fe2(CO)6(μ-pdt)] (1) was prepared by a standard procedure [11]. NMR spectra were run on a Bruker AMX400 spectrometer and referenced internally to the residual solvent peak (1H) or externally to P(OMe)3 (31P). Infrared spectra were run on a Nicolet 205 FT-IR spectrometer in a solution cell fitted with calcium fluoride plates, subtraction of the solvent absorptions being achieved by computation. Fast atom bombardment mass spectra were recorded on a VG ZAB-SE high resolution mass spectrometer and elemental analyses were performed in-house. Chromatography was carried out on deactivated alumina support. The crude mixtures were dissolved in a small amount (ca. 5 ml) of dichloromethane, absorbed onto alumina (ca. 10 g) and dried. This was then added to the top of the wet chromatography column.
4.2 Synthesis of [{Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-diphosphine)] (2)
To a solution of 1 (0.206 g, 0.534 mmol) and dppe (0.108 g, 0.267 mmol) in MeCN (20 cm3) was added a solution of Me3NO·2H2O (0.076 g, 0.684 mmol) in MeCN (15 cm3). The dark red mixture was stirred at room temperature for 2 h and then heated to 60 °C for 4 h. After cooling to room temperature, removal of volatiles gave a red solid which was washed with hexane (ca. 50 cm3) and diethyl ether (ca. 25 cm3) to afford 2a as a bright orange solid (0.268 g, 90%). Complexes 2b–d were prepared in an analogous fashion. Compound 2a [8] IR ν(CO)(CH2Cl2): 2044s, 1981s, 1921w cm−1; 31P{1H} NMR (CDCl3): δ 59.8 (s) ppm; compound 2b IR ν(CO)(CH2Cl2): 2044s, 1982s, 1950sh, 1925w cm−1; 31P{1H} NMR (CDCl3): δ 55.9 (s) ppm; compound 2c IR ν(CO)(CH2Cl2): 2044s, 1982s, 1965sh, 1921w cm−1; 31P{1H} NMR (CDCl3): δ 56.4 (s) ppm; compound 2d IR ν(CO)(CH2Cl2): 2046s, 1985s, 1933w cm−1; 31P{1H} NMR (CDCl3): δ 60.9 (s) ppm; 1H NMR (CDCl3): δ 7.65 (s, 8H, Ph), 7.45 (s, 12H, Ph), 6.89 (t, J 19.7, 2H, CH), 1.67 (br s, 4H, CH2), 1.50 (br s, 2H, CH2), 1.34 (br, 6H, CH2); FAB (+ve) m/z = 1112.3 (M+); Anal. calc. for (found): C42H34Fe4O10P2S2: C, 45.35 (44.95); H, 3.08 (3.20).
4.3 Synthesis of [Fe2(CO)4(dppe)(μ-pdt)]
A toluene solution (80 cm3) of 1 (0.200 g, 0.52 mmol) and dppe (0.215 g, 0.54 mmol) was refluxed for 16 h leading to a colour change from orange to dark red. After cooling to room temperature volatiles were removed by rotary evaporation to give an oily red solid which was washed with hexane (3 × 10 cm3) to remove a small amount of unreacted 1 and excess dppe. After drying an orange-red solid was obtained and chromatography on alumina gave an orange band eluting with hexane which gave a small amount (0.03 g) of unreacted 1. Eluting with diethyl ether:hexane (1:1) gave a red band which afforded [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a) as a bright orange solid (0.245 g, 77%). Crystallization from dichloromethane–methanol afforded [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a) as large orange blocks. To a solution of 1 (0.206 g, 0.534 mmol) and dppe (0.218 g, 0.534 mmol) in MeCN (20 cm3) was added a solution of Me3NO·2H2O (0.152 g, 1.368 mmol) in MeCN (15 cm3). This was heated to 80 °C for 14 h resulting in the formation of a very dark brown solution. After removal of volatiles, the red-brown solid was washed with hexane (ca. 2 × 10 cm3) and dried. The mixture was then extracted with diethyl ether (ca. 50 cm3) to give a dark brown solution which was cooled to −5 °C. This resulted in the deposition of [Fe2(CO)4(κ2-dppe)(μ-pdt)] (3a) as a fine brown solid (0.103 g, 27%). compound 3a [6]: IR ν(CO)(C6H14): 2023s, 1962s, 1953s, 1919w cm−1; 31P{1H} NMR (CDCl3): δ 90.6 (s, 77%, basal–apical), 76.9 (s, 23%, basal–basal); compound 4a: IR ν(CO)(CH2Cl2): 1990s, 1953vs, 1920s, 1902m cm−1; 1H NMR (CDCl3): δ 7.80–7.30 (m, br, 20H, Ph), 2.34 (br, 6H, CH2), 2.10 (br, 4H, CH2); 31P{1H} NMR (CDCl3): δ 60.5 (s); Anal. calc. for (found) Fe2P2S2O4C33H30: C, 54.40 (53.85); H, 4.12 (4.13).
4.4 Synthesis of [Fe2(CO)4(dppp)(μ-pdt)]
A toluene solution (80 cm3) of 1 (0.200 g, 0.52 mmol) and dppp (0.213 g, 0.52 mmol) was refluxed for 16 h leading to a colour change from orange to dark red. The crude reaction mixture was dissolved in a minimum of dichloromethane (ca. 5 cm3) and absorbed onto deactivated alumina. Chromatography gave an orange band eluting with hexane which gave a small amount (0.01 g) of unreacted 1. Eluting with diethyl ether:hexane (1:20) gave a brown band which afforded [Fe2(CO)4(κ2−dppp)(μ-pdt)] (3b) as dark brown solid (0.055 g, 15 %). Crystallization of 3b from diethyl ether–hexane afforded fragile red blocks. To a solution of 1 (0.200 g, 0.52 mmol) and dppp (0.220 g, 0.534 mmol) in MeCN (20 cm3) was added a solution of Me3NO·2H2O (0.127 g, 1.144 mmol) in MeCN (15 cm3). This was heated to 60 °C for 4 h resulting in the formation of a very dark brown solution. After removal of volatiles, the red-brown solid was washed with hexane (ca. 2 × 5 cm3) and dried. The mixture was then extracted with diethyl ether (ca. 20 cm3) to give a dark brown solution which was cooled to −5 °C. This resulted in the deposition of 3b as a brown solid (0.120 g, 31%). After extraction with diethyl ether the remainder was dried to give a dark red solid (0.16 g). All attempts to crystallize this resulted only in decomposition. On the basis of spectroscopic data it is tentatively identified as [Fe2(CO)4(μ-dppp)(μ-pdt)] (4b). Compound 3b: IR ν(CO)(CH2Cl2): 2019vs, 1946s, 1890m cm−1; 31P{1H} NMR (CDCl3): δ 53.3 (s, 91%, basal–apical), 48.7 (s, 9%, basal–basal); 31P{1H} NMR (CD2Cl2, 298 K): δ 55.1 (s, 94%, basal–apical), 50.3 (s, 6%, basal–basal); 31P{1H} NMR (CD2Cl2, 198 K): δ 59.7 (br, basal–apical), 54.9 (br, basal–apical), 51.0 (s, basal–basal); 1H NMR (CDCl3): δ 7.63–7.17 (m, 20H), 2.67 (br, 2H, CH2), 2.53 (t, 2H, J 13.8, CH2), 2.34 (m, 1H, CH2), 1.86 (m, 1H, CH2), 1.50 (br, 4H, CH2), 0.85 (br, 2H, CH2); Anal. calc. for (found) Fe2P2S2O4C34H32: C, 54.99 (54.49); H, 4.31 (4.39); [Fe2(CO)4(μ-dppp)(μ-pdt)] (4b): IR ν(CO)(CH2Cl2): 1994s, 1985sh, 1949vs, 1928s, 1920sh, 1901m cm−1; 1H NMR (CDCl3): δ 7.55 (br, 8H, Ph), 7.29 (br, 12H, Ph), 2.47 (br, 6H, CH2), 1.61 (br, 2H, CH2), 0.94 (br, 4H, CH2); 31P{1H} NMR (CDCl3): δ 54.0 (br s).
4.5 Synthesis of [Fe2(CO)4(μ-dppb)(μ-pdt)]
A toluene solution (80 cm3) of 1 (0.200 g, 0.52 mmol) and dppb (0.221 g, 0.54 mmol) was refluxed for 16 h leading to a colour change from orange to dark red. Chromatography on alumina in hexane gave an orange band eluting with hexane which gave a small amount (0.03 g) of unreacted 1. Eluting with diethyl ether:hexane (1:5) gave a red-brown band which afforded [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c) as a dark red solid (0.166 g, 50 %). Crystallization from a diethyl ether and hexane mixture afforded large red blocks. Compound 4c: IR ν(CO)(CH2Cl2): 1986s, 1946vs, 1916s, 1893m cm−1; 1H NMR (CDCl3, 298 K): δ 7.72 (br s, 4H, Ph), 7.61 (br s, 4H, Ph), 7.43 (s, 6H, Ph), 7.35 (s, 6H, Ph), 2.39 (m, 2H, CH2), 2.29 (s, 2H, CH2), 2.23 (m, 2H, CH2), 1.99 (s, 2H, CH2), 1.94 (s, 2H, CH2), 1.53 (br, 4H, CH2); 1H NMR (CDCl3, 333 K): δ 7.68 (s, 8H, Ph), 7.38 (s, 12H, Ph), 2.27–1.20 (br, 14H, CH2); 31P{1H} NMR (CDCl3, 298 K): δ 56.3 (s); 31P{1H} NMR (CD2Cl2, 298 K): δ 57.5 (s); 31P{1H} NMR (CD2Cl2, 183 K): δ 64.7 (br, B), 58.8 (s, A), 53.1 (br, B + C), 50.5 (br, C), 45.6 (s, A) ratio A:B:C ca. 3:2:1; Anal. calc. for (found) Fe2P2S2O4C35H34: C, 55.55 (55.72); H, 4.50 (4.63).
4.6 Protonation of [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b)
To a dichloromethane (5 cm3) solution of 3b (0.015 g, 0.020 mmol) in air was added one drop of HBF4·Et2O. The solution immediately darkened. After 2 h volatiles were removed and the resulting solid was washed with diethyl ether (ca. 2 × 0.5 cm3) and dried. It was then redissolved in dichloromethane (ca. 2 cm3) and layered with diethyl ether (ca. 2 cm3). After mixing, basal–basal [(μ-H)Fe2(CO)4(κ2-dppp)(μ-pdt)][BF4] (5) (0.015 g, 90%) was isolated as a dark yellow microcrystalline solid. Compound 5 IR ν(CO)(CH2Cl2): 2096vs, 1939s, 1954m cm−1; 31P{1H} NMR (CD2Cl2): δ 44.8 (d, J 16.4); 1H NMR (CD2Cl2): δ 7.70–7.30 (m, 20H), 2.77–2.61 (m, 7H, CH2), 2.28 (m, 2H, CH2), 1.97 (m, 1H, CH2), 1.62 (m, 2H, CH2), −12.65 (t, J 16.4, μ-H); Anal. calc. for (found) Fe2P2S2O4C34H33BF4: C, 49.09 (48.07); H, 3.97 (3.92). When the same reaction was monitored by IR spectroscopy extra absorptions at 2080 and 1965 cm−1 were observed in the early stages (ca. 1–2 min). These disappeared completely over ca. 10 min. Addition of a drop of HBF4·Et2O to a CD2Cl2 solution of 3b (0.020 g) was carried out at low temperature (203 K) monitoring by NMR. Spectral changes were complex and only those in the high-field region of the 1H NMR spectrum were monitored. Upon immediate addition, five hydride resonances were observed; δ −1.21 (t, J 70.8), −12.82 (d, J 16.4), −14.39 (s), −14.98 (d, J 20.8), −15.3 (br) approximately in a 2:1:1:4:1 ratio. Upon warming above 233 K, four of these diminished in intensity rapidly to be replaced by the single doublet associated with basal–basal 5. At 203 K, the 31P{1H} NMR spectrum was complex and consisted of at least four or probably five different species: 66.1 (s), 55.8 (s), 54.3 (d, J 55.5), 52.1 (s), 45.8 (d, J 16) and 38.5 ppm (d, J 55.5). Due to the close proximity of a number of resonances it was not possible to easily discern their relative abundances but the doublet resonances at 54.3 and 38.5 ppm dominated the spectrum. Upon warming to room temperature all species converted cleanly into 5.
4.7 Protonation of [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c)
To a dichloromethane (5 cm3) of 4c (0.020 g) in air was added one drop of HBF4·Et2O. The orange solution turned pale yellow immediately. After 1 h volatiles were removed and the resulting solid was washed with diethyl ether (ca. 1 × 0.5 cm3) and dried to give [(μ-H)Fe2(CO)4(μ-dppb)(μ-pdt)][BF4] (8) (0.016 g, 72%). All attempts to crystallize 8 resulted in decomposition. Compound 8 IR ν(CO)(CH2Cl2): 2058m, 2040s, 2001vs cm−1; 31P{1H} NMR (CD2Cl2): δ 42.7 (d, J 19.6); 1H NMR (CD2Cl2): δ 7.80–7.40 (m, 20H), 2.84 (br, 4H, CH2), 2.66 (br, 1H, CH2), 2.40 (br, 1H, CH2), 1.61 (s, 4H, CH2), 1.34 (s, 4H, CH2), −13.8 (t, J 19.6, μ-H).
4.8 X-ray data collection and solution
Single crystals were mounted on glass fibres and all geometric and intensity data were taken from these samples using a Bruker SMART APEX CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 150 ± 2 K. Data reduction was carried out with SAINT PLUS and absorption correction applied using the programme SADABS. Structures were solved by direct methods and developed using alternating cycles of least-squares refinement and difference-Fourier synthesis. All non-hydrogen atoms were refined anisotropically. For 4c and 3b hydrogens were located from difference maps and refined isotropically while for 2d and 4a they were placed in calculated positions (riding model). Structure solution used SHELXTL PLUS V6.10 program package.
Crystallographic data for [{Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-trans-Ph2PCHCHPPh2)] (2d): orange block, dimensions 0.16 × 0.16 × 0.16 mm, monoclinic, space group P21/c, a = 9.3084(12), b = 17.031(2), c = 13.8980(18) Å, β = 90.0030(10)°, V = 2203.2(5) Å3, Z = 2, F(000) 1128, dcalc = 1.677 g cm−3, μ = 1.610 mm−1; 11 184 reflections were collected, 3750 unique [R(int) = 0.0435] of which 2944 were observed [I > 2.0σ(I)]. At convergence, R1 = 0.0350, wR2 = 0.0649 [I > 2.0σ(I)] and R1 = 0.0460, wR2 = 0.0669 (all data), for 280 parameters. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 673894.
Crystallographic data for [Fe2(CO)4(κ2-dppp)(μ-pdt)] (3b): red block, dimensions 0.28 × 0.14 × 0.12 mm, triclinic, space group , a = 10.4973(8), b = 11.1050(8), c = 16.5294(12) Å, α = 73.6890(10), β = 84.6700(10), γ = 61.8330(10)°, V = 1628.5(2) Å3, Z = 2, F(000) 764, dcalc = 1.514 g cm−3, μ = 1.155 mm−1; 14 343 reflections were collected, 7449 unique [R(int) = 0.0160] of which 6774 were observed [I > 2.0σ(I)]. At convergence, R1 = 0.0273, wR2 = 0.0675 [I > 2.0σ(I)] and R1 = 0.0306, wR2 = 0.0694 (all data), for 525 parameters. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 639868.
Crystallographic data for [Fe2(CO)4(μ-dppe)(μ-pdt)] (4a): orange block, dimensions 0.36 × 0.26 × 0.24 mm, orthorhombic, space group Aba2, a = 18.9912(10), b = 30.2113(26), c = 22.3356(16) Å, V = 12815.0(1.6) Å3, Z = 8, F(000) 5984, dcalc = 1.514 g cm−3, μ = 1.175 mm−1; 55 201 reflections were collected, 15 194 unique [R(int) = 0.0205] of which 14 683 were observed [I > 2.0σ(I)]. At convergence, R1 = 0.0262, wR2 = 0.0667 [I > 2.0σ(I)] and R1 = 0.0273, wR2 = 0.0673 (all data), for 775 parameters. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 673896.
Crystallographic data for [Fe2(CO)4(μ-dppb)(μ-pdt)] (4c): red block, dimensions 0.24 × 0.21 × 0.18 mm, monoclinic, space group P21/c, a = 13.3073(10), b = 14.5984(11), c = 17.9408(13) Å, β = 107.1810(10), V = 3329.7(4) Å3, Z = 4, F(000) 1560, dcalc = 1.509 g cm−3, μ = 1.131 mm−1; 28 715 reflections were collected, 7946 unique [R(int) = 0.0186] of which 7336 were observed [I > 2.0σ(I)]. At convergence, R1 = 0.0253, wR2 = 0.0641 [I > 2.0σ(I)] and R1 = 0.0279, wR2 = 0.0655 (all data), for 542 parameters. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 673897.
Acknowledgements
Part of this work was carried out by S.E.K. at University College London. He gratefully acknowledges the Royal Society (London) for a fellowship to spend time at UCL. We also thank Professor Derek Tocher and Dr. Abil Aliev for help with aspects of the crystallography and NMR spectroscopy, respectively.