

1 Introduction
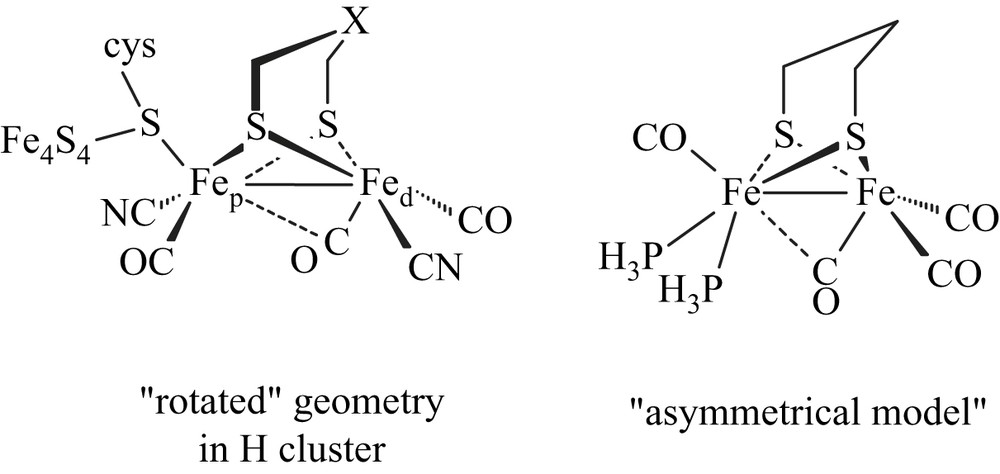
Synthetic organometallic diiron molecules featuring structural key characteristics of the active site of the Fe-only hydrogenase are intensively developed in order to have a better understanding of the activity of this natural system and to elaborate efficient electrocatalysts for the production of H2 [1]. Theoretical studies have pointed out that asymmetry of an electron-rich diiron site could be one of the key structural features to get diiron molecules able to stabilize a “rotated” geometry, and thus to afford better mimic of the natural active site (Scheme 1) [2].
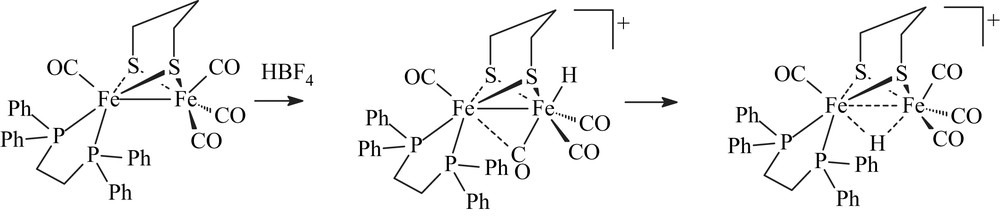
Recent use of bidentate ligands, such as diphosphines [3–6], bis(N-heterocyclic carbene) [7], phenanthroline [8] and carbene-pyridine [9], has allowed the synthesis of numerous electron-rich unsymmetrical systems [Fe2(CO)4(κ2-L2)(μ-pdt)]. NMR studies of the protonation of some of them gave first evidence of the formation of terminal hydride species, as being the major key intermediate in the natural process [10]. Indeed, low-temperature protonation experiments with the unsymmetrical dithiolato diiron complex [Fe2(CO)4(κ2-dppe)(μ-pdt)] revealed the formation of terminal hydride intermediates in a process leading to the μ-hydride derivative [Fe2(CO)4(κ2-dppe)(μ-H)(μ-pdt)](BF4) [6a] (Scheme 2).
These results gave the first chemical precedent of a terminal hydride species resulting from protonation at a single metal atom in the dithiolato diiron model. They strongly suggest that unsymmetrical disubstitution combined with sufficient basicity favours an adequate geometrical arrangement at the diiron centre to lead to terminal hydride upon protonation. It is worth noting that the sole example of structurally characterized terminal hydride species has been obtained by hydridation at a bimetallic iron(II) centre, while no stable dithiolate diiron complexes (at temperature close to 298 K), having a terminal hydride, has been reported [11]. This prompted us to replace the dppe (Ph2PCH2CH2PPh2) ligand by dmpe (Me2PCH2CH2PMe2) in [Fe2(CO)4(κ2-dppe)(μ-pdt)], i.e. a diphosphine having a similar bite angle but more electron-donating than dppe.
In the continuity with our previous works [6–8] exploring the combinations of chelate ligands with the hexacarbonyl precursor [Fe2(CO)6(μ-pdt)] (pdt = S(CH2)3S), we describe here the synthesis, characterization and behaviour in acidic media of the unsymmetrically disubstituted diiron complex [Fe2(CO)4(κ2-dmpe)(μ-pdt)] (1). Furthermore, our attempts to get new systems with the unsymmetrical bidentate ligand Ph2AsCH2CH2PPh2 are also reported.
2 Results and discussion
2.1 Synthesis and characterization of [Fe2(CO)4(κ2-dmpe)(μ-pdt)] (1)
Treatment of [Fe2(CO)6(μ-pdt)] with dmpe in refluxing THF for 1 h resulted in the formation of the unsymmetrically disubstituted complex [Fe2(CO)4(κ2-dmpe)(μ-pdt)] (1) with moderate yields (47%) (Scheme 3).
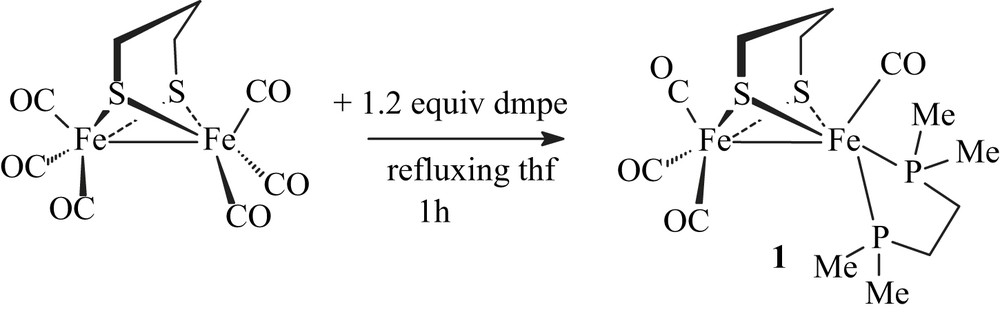
Reaction conditions to yield 1 are milder than those used for synthesizing its dppe analogue [6a]. The replacement of two carbonyl ligands by dmpe does not require either the presence of Me3NO nor warming in refluxing toluene overnight. This reflects the expected behaviour of the dmpe ligand that is a better electron-donor than dppe. Compound 1 was characterized by elemental analyses, IR and NMR spectroscopies. Typical ν(CO) bands are observed at 2008(s), 1936(s), 1893(w) cm−1 in the infrared spectrum of a CH2Cl2 solution of 1. These absorptions are as expected shifted to lower frequencies relative to those of the dppe analogue [ν(CO) = 2019(s), 1949(s), 1906(w) cm−1] [6a], and they are very close to those of [Fe2(CO)4(κ2-phen)(μ-pdt)] (2008(s), 1939(s) and 1897(s) cm−1) [8]. The 1H NMR spectrum of 1 displays a set of poorly resolved resonances for the pdt and dmpe groups. The 13C–{1H} NMR spectrum at 298 K of 1 shows a carbonyl pattern characteristic of an unsymmetrically disubstituted complex, i.e. a singlet and a triplet (J = 12.0 Hz) at 218.4 and 221.2 ppm. These signals are assigned unambiguously to the {Fe(CO)3} and {Fe(CO)P2} moieties, respectively. The 31P–{1H} NMR spectrum of 1 in CD2Cl2 exhibits two singlets at 64.2 and 62.7 ppm in an approximate 1:6 ratio. Upon cooling, the more deshielded resonance signal splits into two sets of signals appearing as ill-resolved AB quartets, and the peak at 62.7 ppm splits into two singlets (Fig. 1 and extended Fig. a in Supplementary data). The presence in solution of four isomers is hence observed at low temperature. These data agree with previous studies of analogous compounds, which indicate that fluxional processes related to the diphosphine group and to the dithiolate bridge are operative in solution [4a,6]. Accordingly, the former peak was assigned to a basal–apical isomer and the latter to a basal–basal form, with two possible orientations of the pdt bridge for each isomer (Fig. 1). It should be noted that unlike the dppe compound [Fe2(CO)4(κ2-dppe)(μ-pdt)], the basal–basal isomer of 1 is here the major species. In our previous work, we found that the ratios between the basal–basal and basal–apical isomers of [Fe2(CO)4(κ2-dppe)(μ-pdt)] depended on the solvent [6a], suggesting a slow equilibrium between the two forms. This was supported by the following experiment: when [Fe2(CO)4(κ2-dppe)(μ-pdt)] was completely dissolved in CD2Cl2 or toluene-d8 at 190 K, the 31P–{1H} NMR spectrum at 190 K of the resulting solution showed only the presence of the basal–apical isomer. The basal–basal form of the dppe complex was detected when the temperature reached 223 K, indicating that the equilibrium becomes operative.
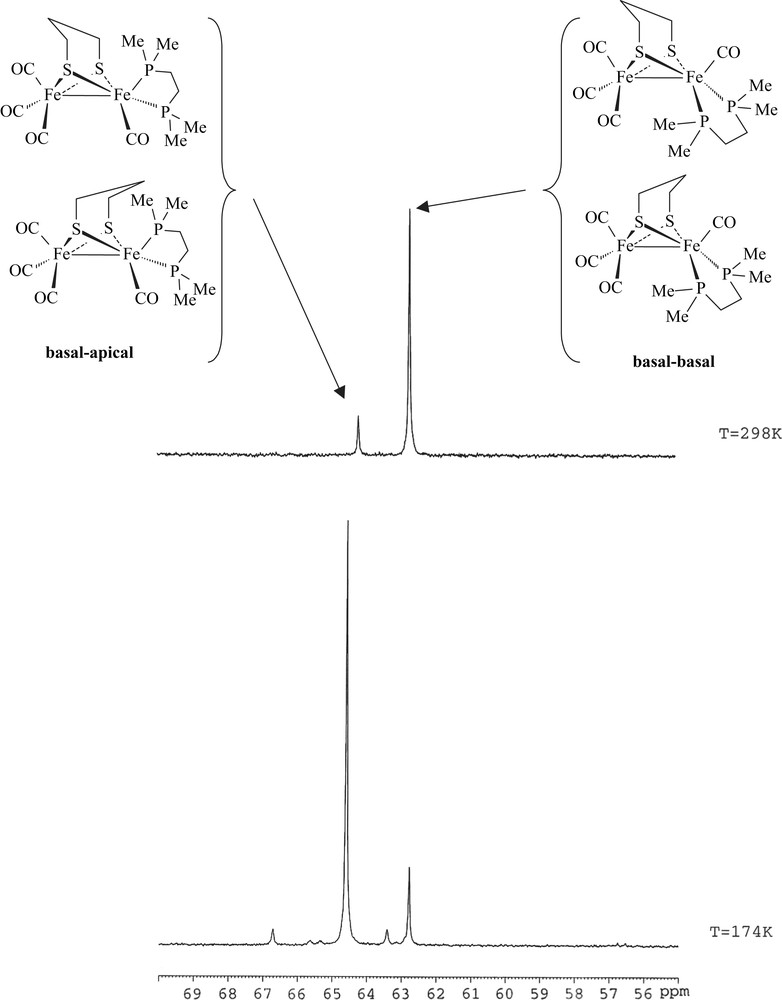
31P–{1H} NMR spectra (in CD2Cl2) of [Fe2(CO)4(κ2-dmpe)(μ-pdt)] (1) at 174 and 298 K.
X-ray analysis of single crystals of the complex 1 obtained from a diethyl ether solution established without any ambiguity the binding mode of the bidendate diphosphine to a single iron atom in a basal–basal position (Fig. 2). One of the more striking features of the structure of 1 is the Fe–Fe distance (2.6038(5) Å), which is longer than that observed in [Fe2(CO)4(κ2-dppe)(μ-pdt)] (2.547(7) Å) [6a] and is very close to that determined in other related compounds containing a basal–basal diphosphine, such as [Fe2(CO)4{κ2-(PPh2)2NR}(μ-pdt)] (R = iPr, 2.6236(4) Å; allyl, 2.6042(4) Å) and [Fe2(CO)4(κ2-dppm)(μ-pdt)] (2.5879(7) Å) [3]. In addition, the basal–basal chelation of the diphosphine induces a noticeable dissymmetry of the Fe2S2 core, which is revealed by a shortening of the Fe–S bonds trans to the Fe–P bonds [S(1)–Fe(1), 2.2182(7); S(1)–Fe(2), 2.2658(7); S(2)–Fe(1), 2.2369(6); S(2)–Fe(2), 2.2755(7) Å]. A similar effect has been also observed in [Fe2(CO)4(κ2-phen)(μ-pdt)] [8]. As expected, complexes [Fe2(CO)4(κ2-dppe)(μ-pdt)] and [Fe2(CO)4(κ2-dmpe)(μ-pdt)] possess very near bite angles (P–Fe–P), 87.7(4)° and 86.05(3)°, respectively.
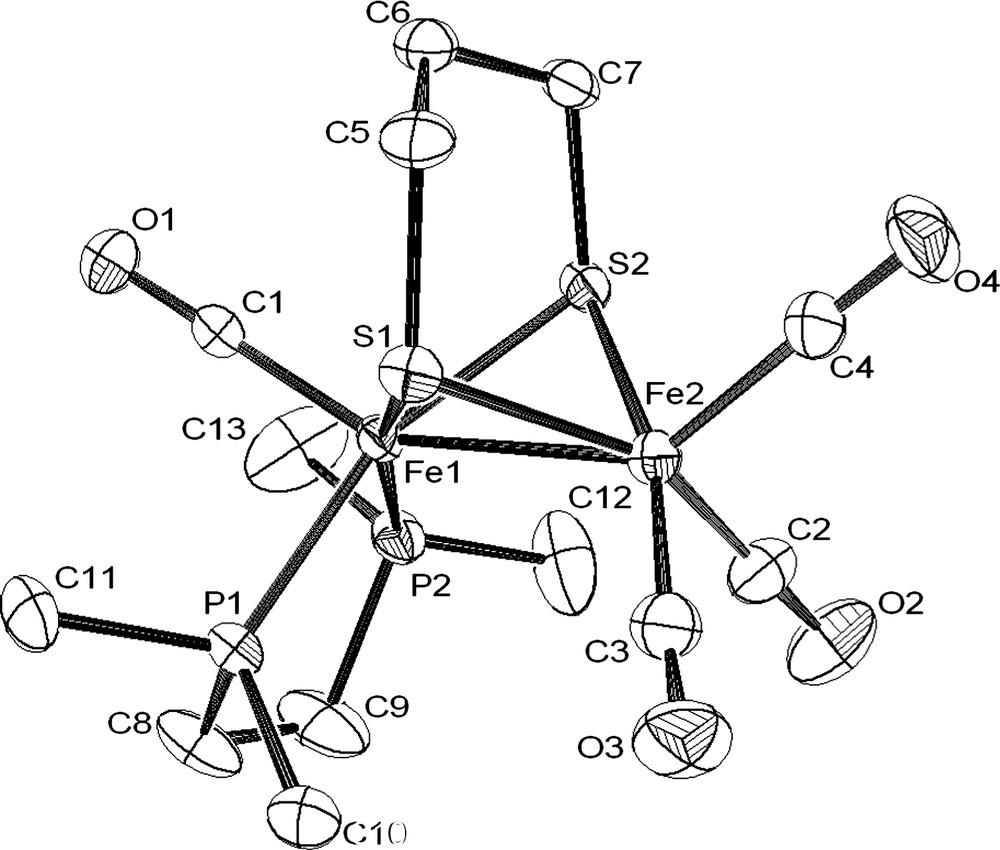
Molecular structure of [Fe2(CO)4(κ2-dmpe)(μ-pdt)] (1) with selected bond lengths (Å) and angles (°): Fe(1)–Fe(2), 2.6038(5); Fe(1)–P(1), 2.1989(7); Fe(1)–P(2), 2.2099(7); S(1)–Fe(1), 2.2182(7); S(1)–Fe(2), 2.2658(7); S(2)–Fe(1), 2.2369(6); S(2)–Fe(2), 2.2755(7); P(1)–Fe(1)–P(2), 86.05(3); Fe(1)–S(1)–Fe(2), 70.99(2); Fe(1)–S(2)–Fe(2), 70.48(2). P(1)–Fe(1)–Fe(2), 107.94(2); P(2)–Fe(1)–Fe(2), 111.74(2).
2.2 Protonation study of 2
Protonation of 1 with HBF4·Et2O was carried out in CH2Cl2 at 298 K, giving immediately a red solution of the μ-hydride derivative [Fe2(CO)4(κ2-dmpe)(μ-H)(μ-pdt)](BF4) (2) (Scheme 4).
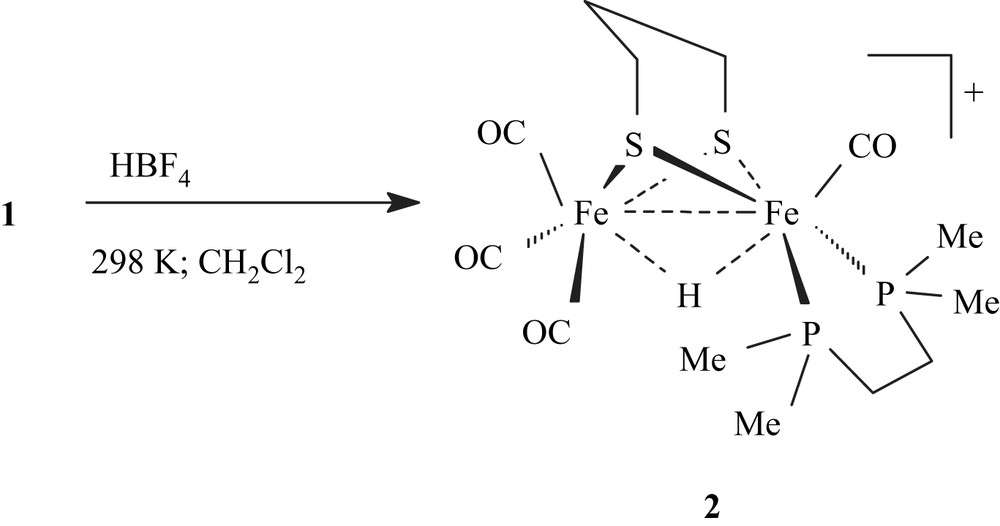
IR carbonyl absorptions of 2 are as expected shifted to higher energy (2093(s), 2036(s), 1972(s) cm−1) relative to those of 1. The 1H NMR spectrum of 2 in CD2Cl2 confirms that protonation occurs at the Fe–Fe site. Accordingly, a characteristic high-field signal assigned to an Fe2(μ-H) group appears as a triplet at −13.92 ppm with a coupling constant, 2JPH, of 22.0 Hz, in agreement with a basal–basal position of the diphosphine in 2. The 31P–{1H} NMR spectrum displays a singlet at 68.0 ppm. Addition of Et3N (6 equiv) to a solution of 2 in dichloromethane resulted in the slow deprotonation of 2 in 1 within 2 h.
Protonation experiments of 1 at low temperature confirmed previously reported results concerning unsymmetrically disubstituted derivatives [Fe2(CO)4(κ2-L2)(μ-pdt)] [L2 = dppe, phen, bis(N-heterocyclic carbene)] [6–8].
Treatment of 1 with excess of HBF4·OEt2 in CD2Cl2 at 188 K resulted in the immediate appearance, in the 1H NMR spectrum, of a signal at −4.68 ppm, corresponding to the formation of a species 3 featuring a terminal hydride at the unsubstituted iron atom (Fig. 3). The 31P–{1H} NMR spectrum displayed a singlet at 65.5 ppm. The stability of this species up to 243 K allowed us to record a 13C–{1H} NMR spectrum, which shows in the carbonyl region three ill-resolved signals at, 236.2, 212.0 and 203.0 ppm, with relative intensities of 1, 1 and 2 (Fig. b in the Supplementary data). The more deshielded chemical shift at 236.2 ppm suggests either the presence of a semi-bridging carbonyl or a strong electron-donor effect of the hydride in a trans position relative to this CO group. An 1H–13C HMBC 2D NMR experiment shows a correlation between the hydride signal and the carbonyl peaks at 236.2 and 203.0 ppm (Fig. c in the Supplementary data). This strongly suggests, as it has been proposed in previous works [6–8], the presence in 3 of an {FeH(CO)3} moiety with a terminal hydride, two equivalent carbonyl groups (basal position) and a third semi-bridging carbonyl trans to the hydride. Until now, neither reliable low-temperature IR data nor suitable single crystals of 3 can be obtained. The transformation of intermediate 3 upon increasing the temperature was monitored by 1H NMR and 31P–{1H} NMR. No noticeable change was observed up to 243 K. Above this temperature, a triplet at −14.46 ppm (JPH = 20 Hz) and a doublet at −14.34 ppm (JPH = 25 Hz) appeared in the 1H NMR spectrum, while the 31P–{1H} NMR spectrum displayed a singlet at 68.0 ppm and an AB pattern at 67.2 (d, JPP = 31.2 Hz) and 65.9 (d, JPP = 31.2 Hz) (Fig. 3). These observations accord with the formation of μ-hydride species with basal–basal (2ba–ba) and basal–apical (2ba–ap) coordination of the diphosphine ligand. Compound 2ba–ap appears to be an intermediate and its complete transformation into 2ba–ba was observed when the temperature raised to 298 K. Surprisingly, unlike the related dppe compound, the 1H NMR spectrum of the dmpe derivative 2, in acidic solution at low temperature, did not exhibit any signal that could be assigned to a terminal hydride attached to the iron atom supporting the dmpe ligand.
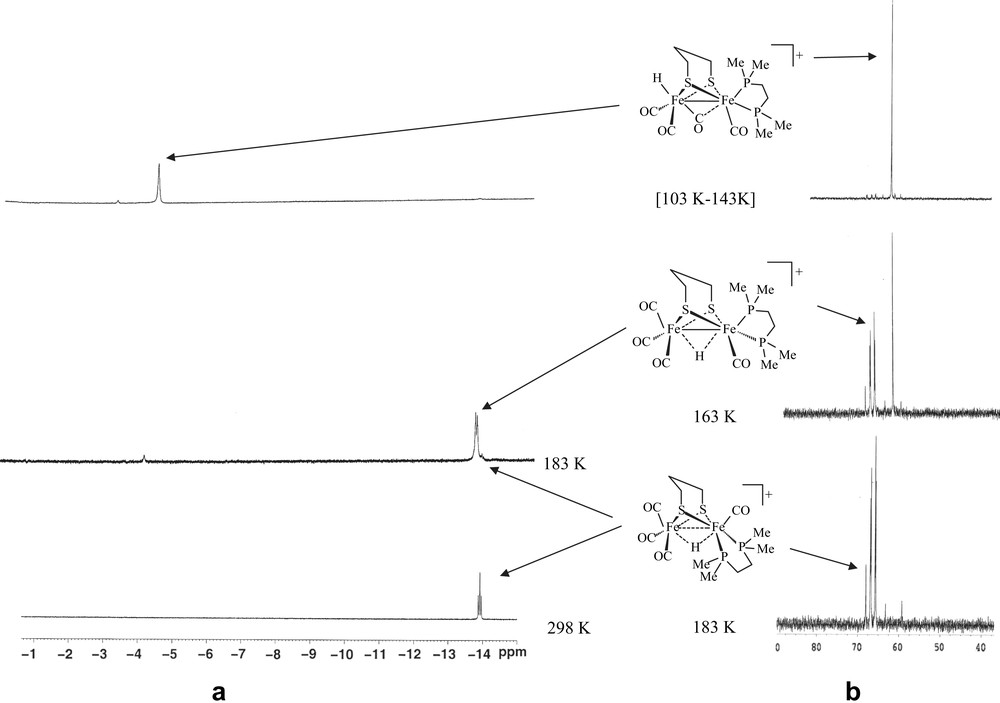
1H and 31P–{1H} NMR spectra of [Fe2(CO)4(κ2-dmpe)(μ-pdt)] (1) in the presence of HBF4·OEt2 in CD2Cl2 at various temperatures.
In order to check the influence of the dissymmetry on the protonation of the disubstituted diiron centre, we decided to study the protonation of the symmetrical complex [Fe2(CO)4(PMe3)2(μ-pdt)] [12] under conditions similar to those above. Its treatment with excess (3 equiv) of HBF4·OEt2 in CD2Cl2 solution at 198 K resulted in the immediate appearance of two signals at −14.73 ppm (singlet) and −14.86 ppm (doublet, J = 22 Hz). These resonances are assigned to two species having a bridging hydride and the phosphine groups in apical–apical and basal–apical position. Couplings between bridging hydride and phosphorus atom lying in apical position are not observed. This result suggests that both the basicity of the diiron site and the unsymmetrical substitution are required to get a terminal hydride in disubstituted tetracarbonyl dithiolato systems [Fe2(CO)4L2(μ-pdt)]. Finally, on warming the solution to 298 K, the two previous hydride signals disappeared, while a triplet appeared at −15.2 ppm, which was assigned to the already known basal–basal product [Fe2(CO)4(PMe3)2(μ-pdt)(μ-H)]+ reported by Darensbourg et al. [12]. Such an assignment was corroborated by recording the 31P–{1H} NMR spectrum. Moreover, similar investigation realized with the symmetrically substituted derivative [Fe2(CO)4{P(OMe)3}2(μ-pdt)] [13] gave results close to those reported above. The 1H NMR spectrum in CD2Cl2 at 178 K indicated the formation of three bridging hydride species upon protonation by HBF4·OEt2 at this temperature (see Fig. d in the Supplementary data). When the temperature reached 298 K, the conversion of two of these isomers into the third one was observed and was achieved after four days.
2.3 Reaction of 1 with arphos (arphos = Ph2AsPCH2CH2PPh2)
Heating [Fe2(CO)6(μ-pdt)] with the mixed phosphine–arsine Ph2PCH2CH2AsPh2 (arphos) and Me3NO·2H2O in refluxing THF for 4 h gave the disubstituted complex [Fe2(CO)4(arphos)2(μ-pdt)] (4b). The formation of the monosubstituted derivative [Fe2(CO)5(arphos)(μ-pdt)] (4a) was observed in some experiments, but only small amounts of this species are isolated after purification by chromatography (Scheme 5).
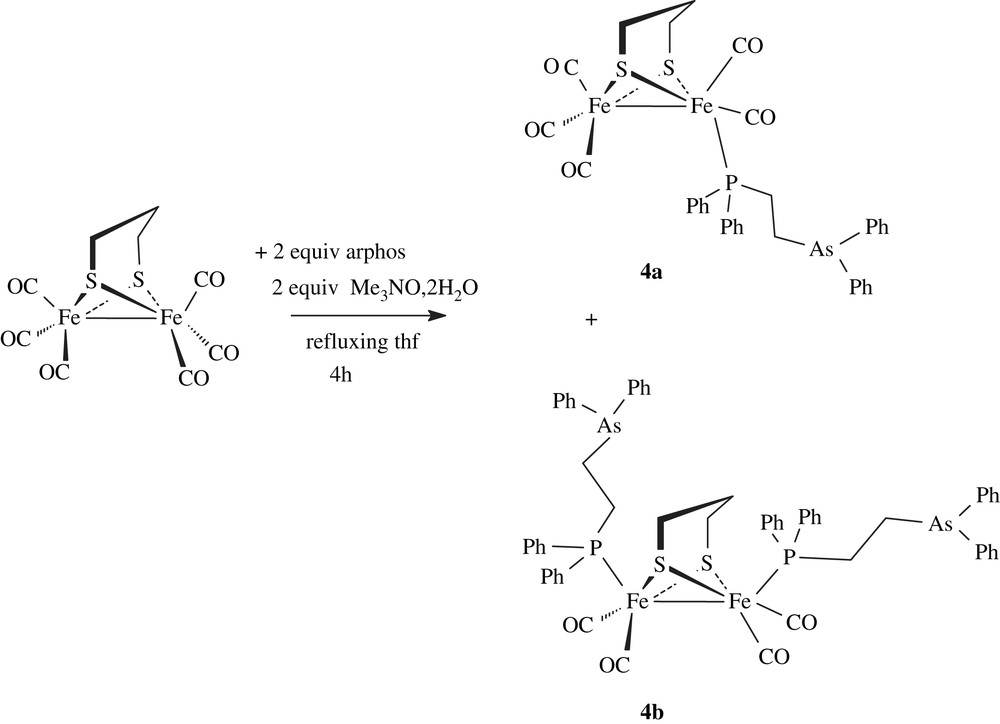
The coordination of the phosphine–arsine ligand was revealed by the deshielding of chemical shift of the phosphorus atom in the 31P–{1H} spectra (CD2Cl2) of 4a and 4b. The related signal is observed at −12.1 ppm in the free molecule Ph2PCH2CH2AsPh2 and at 60.4 and 58.3 ppm in 4a and 4b, respectively. IR spectra in the carbonyl region of 4a and 4b in CH2Cl2 solution were typical of monosusbtituted (ν(CO): 2044(s), 1981(vs), 1959(sh) and 1928(w) cm−1) and disubstituted (ν(CO): 1989(s), 1951(s), 1919(s) cm−1) phosphine derivatives [13]. 1H NMR spectra of 4a and 4b show the expected signals for arphos and pdt groups. The structure of complex 4b was confirmed by X-ray analysis of single crystals obtained from dichloromethane–ether solutions (Fig. 4). The main feature of this structure is the presence of two dangling monodentate arphos ligands in apical positions. The Fe2S2 core has a typical butterfly structure. Distances and angles are unexceptional and very close to values determined for diiron dithiolate analogues [14].
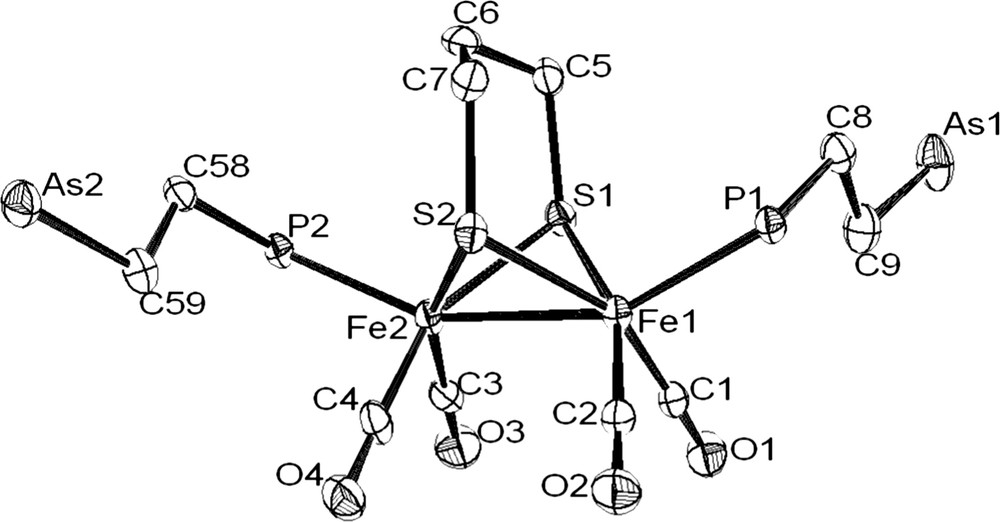
Molecular structure of [Fe2(CO)4(arphos)2(μ-pdt)] (4b) with the phenyl rings and the hydrogen removed for clarity. Selected bond lenghts (Å) and angles (°): Fe(1)–Fe(2), 2.5148(13); Fe(1)–P(1), 2.227(2); Fe(2)–P(2), 2.234(2); S(1)–Fe(1), 2.277(2); S(1)–Fe(2), 2.280(2); S(2)–Fe(1), 2.278(2); S(2)–Fe(2), 2.281(2); Fe(1)–S(1)–Fe(2), 66.98(6); Fe(1)–S(2)–Fe(2), 66.96(6); P(1)–Fe(1)–Fe(2), 152.34(7); P(2)–Fe(2)–Fe(1), 156.18(7).
Given these results and in the absence of a diiron chelate compound, we decided not to pursue the work with the phosphine–arsine ligand.
3 Concluding comments
In summary, we have reported an extension of our previous works concerning the protonation of diiron molecules [Fe2(CO)4(κ2-L2)(μ-pdt)] with chelating dppe [6], bis(N-heterocyclic carbene) [7] and phenanthroline [8] ligands. We have shown that the use of the dmpe ligand instead of dppe allows such a complex to react faster with protons due to the better electron-donating properties of the dmpe ligand. Moreover, its protonation at low temperature gives a more stable terminal hydride intermediate, suggesting the importance of the basicity of the bimetallic site. We have also demonstrated that symmetrically disubstituted derivatives [Fe2(CO)4L2(μ-pdt)] do not yield terminal hydride complexes upon protonation, indicating that asymmetry is required to favour the protonation of disubstituted complexes at a single metal, prior to forming species containing bridging hydride compound.
4 Experimental section
4.1 Methods and materials
All the experiments were carried out under an inert atmosphere, using Schlenk techniques for the syntheses. Solvents were deoxygenated and dried according to standard procedures. [Fe2(CO)6(μ-pdt)] was prepared according to the reported procedure [15]. The NMR spectra (1H, 31P) were recorded at room temperature in CD2Cl2 solution with a Bruker AMX 400, AC300 and DRX 500 spectrometers and were referenced to SiMe4 (1H), H3PO4 (31P). 1H–13C experiments were carried out on a Bruker DRX 500 spectrometer. The infrared spectra were recorded on a Nicolet Nexus Fourier transform spectrometer. Chemical analyses were made by the “Service de microanalyse I.C.S.N.” Gif-sur-Yvette (France), or the “Centre de microanalyse du CNRS”, Vernaison (France). Crystal data for compounds 1a–c and 2a were collected with an Oxford Diffraction X-Calibur-2 CCD diffractometer, equipped with a jet cooler device and graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved and refined by standard procedures [16].
4.2 X-ray structure analysis
Crystal data: 1, C13H22Fe2O4P2S2, F.Wt = 480.07, T = 295(2) K, monoclinic, space group P21/c, a = 12.9610(9), b = 10.5514(7), c = 14.4917(9) Å, β = 93.133(5)°, V = 1978.9(2) Å3, Z = 4. Refinement of 212 parameters gave R1 = 0.0326, wR2 = 0.0824 and |Δρ| < 0.827 e Å−3 for all 6164 unique reflections. Compound 4b, C59H54As2Fe2O4P2S2, F.Wt = 1214.62, T = 170(2) K, monoclinic, space group P21/n, a = 12.2473(6), b = 17.3095(12), c = 26.5034(14) Å, β = 91.856(5)°, V = 5615.6(6) Å3, Z = 4. Refinement of 510 parameters gave R1 = 0.0564, wR2 = 0.1316 and |Δρ| < 0.638 e Å−3 for all 6988 unique reflections. Further crystallographic data for 1 and 4b are provided as supporting information.
4.3 Synthesis of 1
4.3.1 Preparation of [Fe2(CO)4(dmpe)(μ-pdt)] (1)
A solution of [Fe2(CO)6(μ-pdt)] (0.3 g, 0.777 mmol) and dmpe (0.140 g, 0.93 mmol) in THF (100 mL) was heated at reflux for 1 h. The initially red solution became brownish. After evaporation of the solvent, the residue was chromatographed on a silica gel column. Elution with hexane–dichloromethane (50:50) afforded a purple red solution of 1, which was evaporated under vacuum. Compound 1 was washed with pentane and obtained as a red air-stable powder (0.176 g, 47% yield). Crystals of 1, suitable for X-ray analysis, were formed by crystallization at room temperature from an Et2O solution. IR (CH2Cl2, cm−1): ν(CO) 2008(s), 1936(s), 1893(w). 1H NMR (CD2Cl2, 25 °C) major isomer δ: 2.25 (2H), 2.04 (2H), 1.88 (4H), 1.63 (1H), 1.42 (1H) (pdt + dmpe), 1.54 (6H, dmpe), 1.29 (6H, dmpe). 13C–{1H} NMR (CD2Cl2, 25 °C) major isomer δ: 221.2 (t, JCP = 12.0 Hz, CO), 218.4 (s, CO), 34.0 (s, pdt), 33.0 (m, CH2–P), 28.7 (s, pdt), 22.3 (m, PCH3), 18.6 (m, PCH3). 31P–{1H} NMR (CD2Cl2, 25 °C) δ: 64.2(s) (minor isomer, 14%), 62.7(s) (major isomer, 86%). Anal. Calcd (%) for C13H22Fe2O4P2S2: C, 32.5; H, 4.6; P, 12.9. Found: C, 31.7; H, 4.5; P, 12.7.
4.4 Protonation reaction of 1
Protonation of 1. A solution of 1 (0.1 g, 0.208 mmol) in dichloromethane (10 mL) was treated with 2 equiv of HBF4·Et2O. The mixture was stirred for 15 min. The volume was then reduced under vacuum and diethyl ether was added to precipitate a red powder of 2 (0.95 g, 80% yield). IR (CH2Cl2, cm−1): ν(CO) 2093(s), 2036(s), 1972(s). 1H NMR (CD2Cl2, 25 °C) δ: 2.75 (m, 2H, pdt), 2.58 (m, 2H, dmpe), 2.54 (m, 1H, pdt), 2.42 (m, 2H, pdt), 2.18 (m, 2H, dmpe), 1.98 (m, 1H, pdt), 1.62 (d, JPH = 9.5 Hz, 6H, dmpe), 1.46 (d, JPH = 10,0 Hz, 6H, dmpe), −13.92 (t, JPH = 22.0 Hz, 1H, Fe2(μ-H)). 31P–{1H} NMR (25 °C) δ: 68.0(s).
4.5 Reaction of [Fe2(CO)6(μ-pdt)] with arphos (arphos = Ph2AsPCH2CH2PPh2)
A solution of [Fe2(CO)6(μ-pdt)] (0.20 g, 0.52 mmol) and arphos (0.44 g, 1.04 mmol) in THF (100 mL) was heated under reflux in the presence of Me3NO·2H2O (0.185 g, 1.10 mmol) for 4 h. The orange solution became red-brown. After evaporation of the solvent, the residue was chromatographed on a silica gel column. Elution with hexane–dichloromethane mixtures afforded two red–orange bands. The first gave the monosubstituted complex 4a and the second the disubstituted derivative 4b. Compound 4b was isolated as a red powder (0.16 g, 25% yield).
Compound 4a: IR (CH2Cl2, cm−1): ν(CO) 2044(s), 1981(vs), 1959(sh) and 1928(w). 1H NMR (CD2Cl2, 25 °C) δ: 7.6–7.3 (m, 20H, arphos), 2.51 (m, 2H, arphos), 2.14 (m, 2H, arphos), 1.77 (m, 2H, pdt), 1.41 (m, 4H, pdt). 31P–{1H} NMR (25 °C) δ: 60.4(s).
Compound 4b: IR (CH2Cl2, cm−1): ν(CO) 1989(s), 1951(s), 1919(s). 1H NMR (CDCl3, 25 °C) δ: 7.6–7.3 (m, 40H, arphos), 2.53 (m, 2H, arphos), 2.23 (m, 2H, arphos), 1.59 (m, 4H, pdt), 1.08 (m, 2H, pdt). 31P–{1H} NMR (25 °C) δ: 58.3(s). Anal. Calcd (%) for C59H54As2Fe2O4P2S2. CH2Cl2: C, 55.4; H, 4.3. Found: C, 55.1; H, 4.2.
5 Supporting information
Spectroscopic data, crystallographic information files (CIF) for 1 and 4. The crystallographic material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as supplementary material CCDC 678127, 678128 and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number). See http://www.ccdc.cam.ac.uk/deposit for crystallographic files in .cif format.
Acknowledgements
This work was supported by the CNRS (Programme “Énergie”, PRI-4.1) and the ANR (Programme “PhotoBioH2”). The “Ministère de l'Éducation nationale, de l'Enseignement supérieur et de la Recherche” is acknowledged for providing a studentship to S.E., and “Université de Bretagne occidentale” is acknowledged for financial support.