

1 Introduction
[FeFe]-hydrogenases are enzymes that are able to catalyze the reversible oxidation of molecular hydrogen: H2 ⇌ 2H+ + 2e−. Such a very simple reaction could have fundamental importance for the possible future development of a hydrogen-based economy [1]. However, the current approaches for molecular hydrogen oxidation imply the use of very expensive platinum-containing catalysts, while H2 production at industrial level still depends on hydrocarbons. In this framework, [FeFe]-hydrogenases represent a promising model for the development of new-generation catalysts; in fact, these iron-containing enzymes are generally very efficient in H2 production, and they are able to evolve molecular hydrogen directly from acidic aqueous solutions supplied with a convenient source of electrons [2].
The [FeFe]-hydrogenase active site is generally referred to as the “H-cluster” (see Fig. 1); this is a peculiar Fe6S6 cluster which can be ideally subdivided in two distinct portions: a classical Fe4S4 moiety and an Fe2S2 subcluster (commonly termed “[2Fe]H”) bearing CO and CN− ligands [3]; these subclusters are linked to each other through the sulphur atom of a cysteine residue. The two iron atoms of the binuclear subsite are termed “proximal” (Fep) or “distal” (Fed), depending on their positions with respect to the Fe4S4 moiety. Notably, one of the carbonyl groups included in the [2Fe]H subsite bridges the Fep and Fed centers and it moves to a semibridging position when the enzyme is in its completely reduced form. The coordination environment of the iron ions included in the binuclear cluster is completed by a bidentate ligand which has been proposed to correspond either to a di(thiomethyl)amine (DTMA) or to a propanedithiolate (PDT) residue [3b,4].
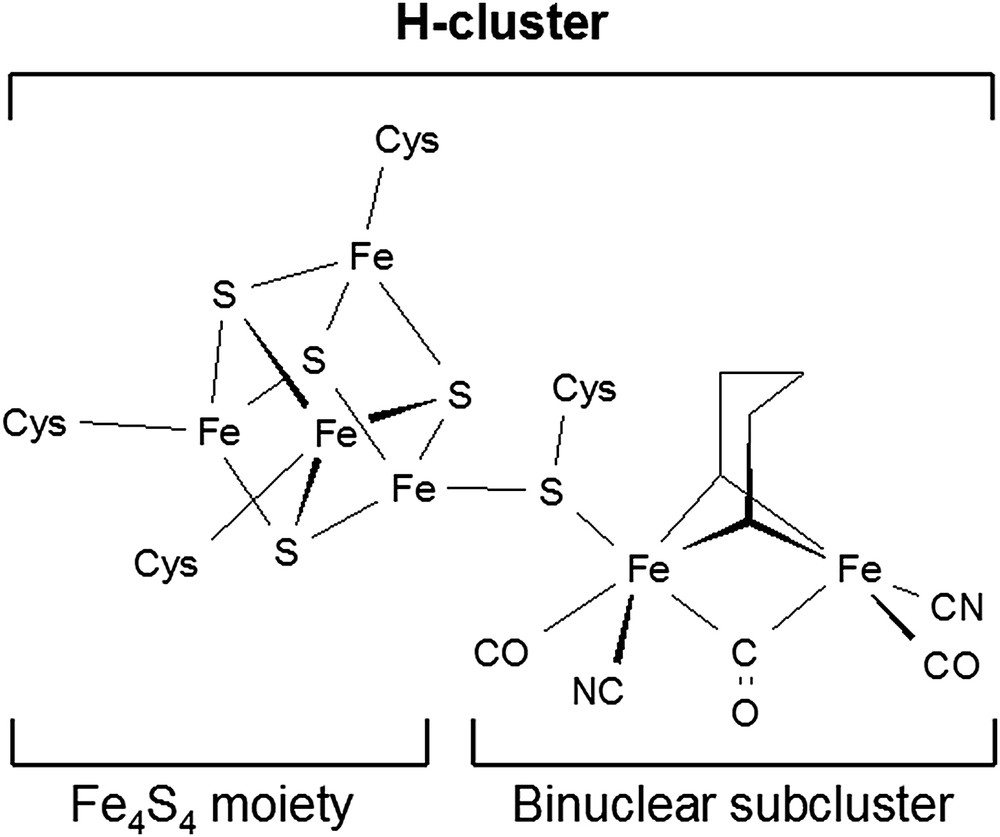
Schematic structure of the H-cluster.
Different redox states of the [2Fe]H cluster have been characterized spectroscopically [5]. The fully oxidized and fully reduced forms of the enzyme are EPR silent and have been proposed to correspond, on the basis of similarities between the FT-IR spectra of the enzyme and of model compounds, to Fe(II)Fe(II) and Fe(I)Fe(I) species, respectively. The partially oxidized form is paramagnetic and should correspond to the Fe(I)Fe(II) redox state [6]. Moreover, spectroscopic studies of [FeFe]-hydrogenases are consistent with a [2Fe(II)2Fe(III)] oxidation state for Fe4S4 moiety included in the H-cluster, both in the oxidized and reduced forms of the enzyme.
As for the catalytic mechanism of [FeFe]-hydrogenases, the most plausible picture [4b–d,7] involves binding of a proton to the Fe(I)Fe(I) [2Fe]H subsite, which would lead to the formation of an Fed-bound terminal hydride ligand; a second protonation reaction and two mono-electron reduction steps would then precede H2 evolution, thus closing the catalytic cycle (Fig. 2). Alternative mechanisms have been proposed involving the formation of a μ-H ligand [8]; however, recent works [7,9] reported evidences that a terminal hydride group – which can be formed also by protonation of biomimetic assemblies [10] – is more reactive than a hydride ligand in bridging position between Fep and Fed.
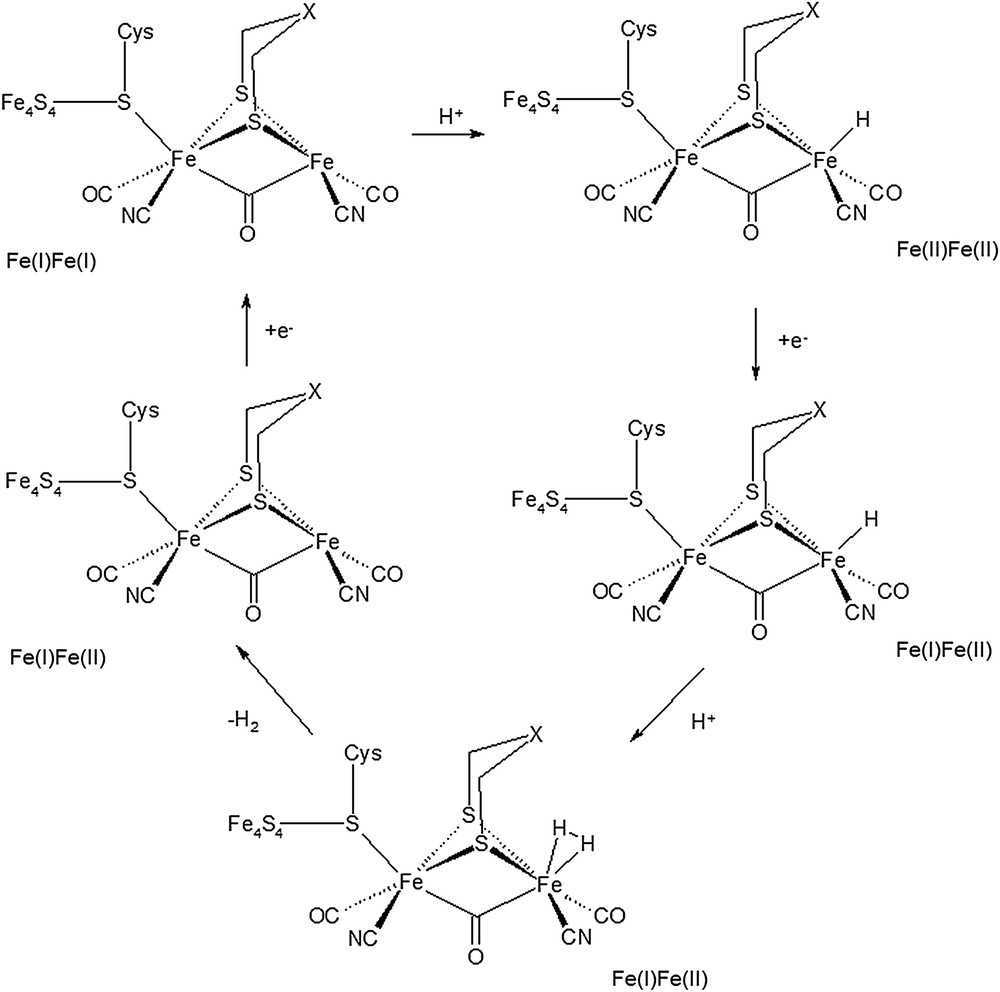
A plausible catalytic mechanism for molecular hydrogen evolution in [FeFe]-hydrogenases.
The apparent structural simplicity of the Fe2S2 site has stimulated several research groups towards the synthesis of biomimetic diiron clusters that could reproduce the geometric and functional features of the [2Fe]H moiety [11]; however, the reproduction of the μ-CO structure (the so-called “rotated” conformation, see Fig. 3) typical of the [2Fe]H subsite has not been achieved until recently [12]; in fact, most synthetic biomimetic assemblies are characterized by an all-terminal disposition of ligands (i.e., they show an “eclipsed” conformation, see Fig. 3).
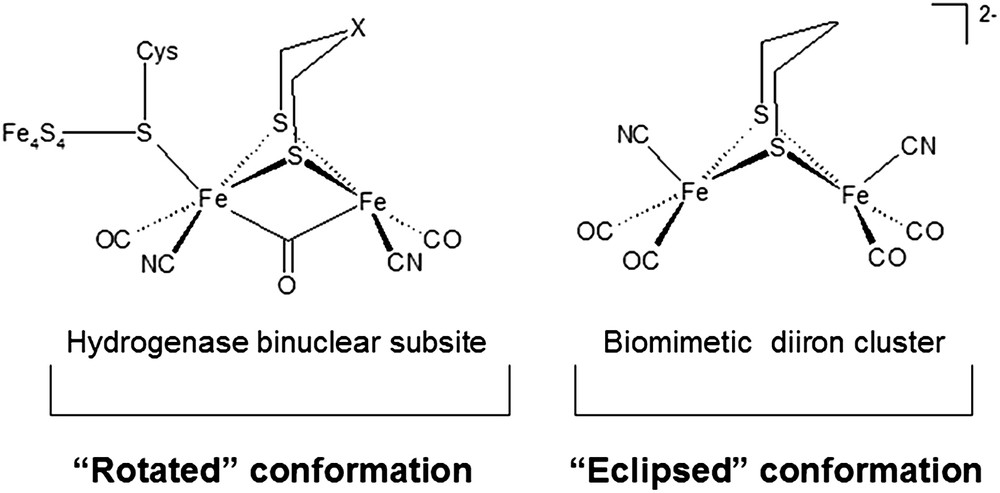
Structure of the [FeFe]-hydrogenase binuclear subsite and geometry of the synthetic complex [(μ-pdt)(Fe(CN)(CO)2)2]2− [13].
Another challenge in this research area is represented by the reconstitution of the whole Fe6S6 framework in a synthetic complex; such a goal has been recently achieved by Pickett and coworkers [14], who obtained and characterized the complex [Fe4S4(L)3{Fe2(CH3C(CH2S)3)(CO)5}]2− (L = 1,3,5-tris (4,6-dimethyl-3-mercaptophenylthio)-2,4,6-tris-(p-tolyl-thio)benzene; see Fig. 4). Differently from the case of the H-cluster in [FeFe]-hydrogenases, the binuclear portion of this organometallic complex does not include cyanide ligands. In fact, the only diatomic ligands included are CO groups, which are disposed in an eclipsed conformation around the iron centers of the binuclear subsite. As far as the interaction between the biomimetic complex and protons is concerned, the eclipsed disposition of CO ligands is expected to favour the formation of slowly reacting μ-H adducts; in fact, the biomimetic Fe6S6 complex is able to electrocatalyze H+ reduction, but it is less efficient than the enzyme in favouring H2 evolution from protons and electrons. A conformational rearrangement of the Fe6S6 cluster towards the rotated geometry is expected to facilitate the formation of highly reactive terminal hydride groups, but there are no experimental evidences of the formation of rotated adducts during the electrocatalytic process.
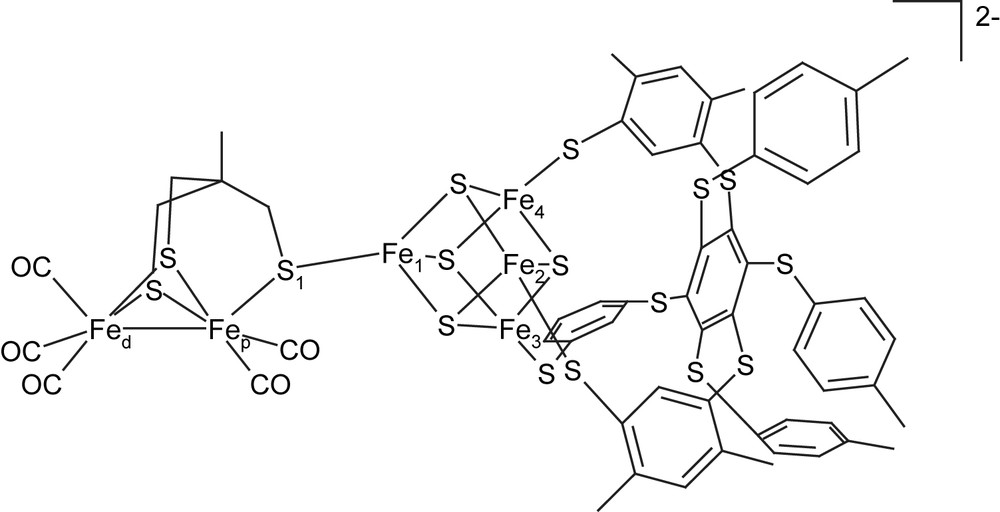
Schematic structure of the biomimetic Fe6S6 complex recently synthesized by Pickett and coworkers [14].
Prompted by the above observations, we performed a density functional theory (DFT) investigation on the structural properties of the biomimetic Fe6S6 assembly in two different redox states: the dianionic state, in which the pentacarbonyl binuclear subcluster should attain the Fe(I)Fe(I) state, and the monoanionic form, which should represent a model of the partially oxidized H-cluster. For these two states, both the rotated and the eclipsed conformations were optimized, in order to evaluate the relative stabilities of the isomers, and the effects that mono-electron oxidation can have in terms of ligand rearrangement around the metal centers of the binuclear subsite. Comparisons between the binuclear subcluster included in the biomimetic Fe6S6 assembly and simpler synthetic diiron models like Fe2CO6(μ-PDT) and Fe2(CO)6(S2C6H4) are also discussed, in order to give further insights into the effects of the introduction of a cubane moiety in a biomimetic assembly.
2 Methods
DFT calculations have been carried out using the pure functional BP86 [15] and a valence triple-ζ basis set with polarization on all atoms (TZVP) [16]. Calculations have been carried out with the TURBOMOLE 5.7 suite [17] applying the resolution-of-the-identity technique [18].
Stationary points of the energy hypersurface have been located by means of energy gradient techniques. The effect of the solvent (acetonitrile, ε = 36.64) has been evaluated according to the COSMO approach [19]. For all models, geometry optimizations have also been carried out in a vacuum; however, if not otherwise stated the discussion is based on the results obtained by including the COSMO representation of the solvent in the model.
The theoretical investigation on the models of the H-cluster is a non-trivial task because the Fe4S4 subunit is composed of two Fe2S2 layers with high-spin (HS) Fe atoms coupled antiferromagnetically to give an overall low-spin ground state. The ground state wave function of such spin-coupled systems corresponds to a linear combination of determinants that would require a multi-configurational treatment, not possible within the DFT scheme. However, in the single determinant approximation, the antiferromagnetic interactions can be modelled according to the broken symmetry (BS) approach, introduced by Noodleman [20,21]. The BS approach consists of the localization of opposite spins of the mono-determinant wave function in different parts of the molecule. The BS wave function, being of spin-unrestricted type, does not represent a pure-spin state, but a weighted average of pure-spin states (and energies).
As noted by Flieder and Brunold [22], the Fe atoms of the Fe4S4 cluster are not equivalent in Fe6S6 complexes, and up to three different schemes of spin localization could be considered, each of them being characterized by a different definition of the Fe2S2 layers. For each of the three coupling schemes, two BS wave functions can be generated by interchanging α and β spin orbitals between the two Fe2S2 layers. Therefore, a total of six BS solutions could be computed for each complex. However, previous theoretical studies of H-cluster models showed that the structural differences among models characterized by different BS configurations are very small [4d]; moreover, the computed relative stabilities never differ by more than 10 kJ mol−1, when rotated and eclipsed models of the partially oxidized and reduced forms of the H-cluster in different BS configurations are considered [4d]. In view of these considerations, we only considered the coupling scheme in which the two Fe2S2 layers are defined by the Fe1–Fe2 and Fe3–Fe4 metal sites (see Fig. 4 for atom labels). According to this scheme, the BS solutions in which majority of the α and β spin orbitals are localized on the Fe1–Fe2 layer will be labelled as BS1 and BS2, respectively.
Geometry optimization was performed on models of the synthetic Fe6S6 assembly in which the bulky L ligands coordinated to the cubane irons (see Section 1) were replaced by CH3S− groups. Thus, the Fe6S6 models here discussed are complexes of the general formula [Fe4S4(SCH3)3{Fe2(CH3C(CH2S)3)(CO)5}]n−, where n = 1 or 2.
As for the calculation of the overall charges of the cubane moiety and of the binuclear subcluster included in the Fe6S6 models, we applied the following approach: Mulliken atomic charges of the Fe4S4(SCH3)3 portion of the model were calculated and a summation extended to all these charge values was carried out. Then, the overall charge of the Fe2S2 subsite could be obtained by simply subtracting the charge value of the cubane (i.e., of the Fe4S4(SCH3)3 moiety) from the total charge of the Fe6S6 complex.
3 Results and discussion
Before discussing the results of our work, it is worth illustrating the details of the nomenclature that will be used in this paper for the various models here investigated. All the models of the synthetic Fe6S6 cluster have the “Fe6S6” tag in their names in square brackets. The presence of a bridging carbonyl ligand in the rotated conformers is evidenced by a “b”, added as a subscript; as for eclipsed models – i.e., models which show an all-terminal disposition of ligands – a “t” subscript is present in their names. The charge of the Fe6S6 complex is also always included as a superscript.
3.1 DFT characterization of dianionic hexanuclear model compounds
In a previous study, the structure of the eclipsed, dianionic form of the synthetic Fe6S6 assembly shown in Fig. 4 was described [14]. It turned out that the distance between iron atoms in the binuclear subcluster (2.6 Å) is compatible with the presence of an Fe–Fe bond. Moreover, the authors concluded that the redox state of the binuclear subcluster is Fe(I)Fe(I), thus implying that the cubane moiety attains the [2Fe(II)2Fe(III)] state.
In the present contribution, we carried out the geometry optimization of both the eclipsed and the rotated conformation of the dianionic hexanuclear cluster; a comparison between the two isomers is presented in terms of their structural properties, spin density values and relative stabilities.
For the [Fe6S6]b2− and [Fe6S6]t2− complexes the two broken symmetry solutions BS1 and BS2 (see Section 2) converge to the same energy and geometry.
Model [Fe6S6]t2−, when soaked in a polarizable continuum medium at ε = 36.64, shows an Fep–Fed distance of 2.55 Å (from now on, Fep will refer to the iron atom of the binuclear cluster closer to the cubane moiety, while Fed will indicate the other iron center included in the diiron subsite). The Fep and Fed spin density values are negligible (Table 1) and the overall charges of the Fe2S2 and Fe4S4 subsites are −0.54 and −1.46, respectively; these observations are compatible with an Fe(I)Fe(I) redox state for the diiron subcluster.
Atomic spin densities of the Fe atoms in the Fe6S6 clusters computed at BP86/def-TZVP level of theory
| 〈S2〉 | Fe1,2 | Fe3,4 | Fep | Fed | |
| [Fe6S6]b2− | 6.46 | 3.07, 3.08 | −3.06, −3.07 | 0.01 | 0.03 |
| [Fe6S6]t2− | 6.48 | 3.07, 3.08 | −3.07, −3.08 | 0.04 | 0.01 |
| [Fe4S4(SCH3)4]2− | 6.43 | 3.05, 3.05 | −3.05, −3.05 | – | – |
| [Fe6S6]b1− | 5.73 | 2.92, 2.88 | −2.40, −2.46 | 0.002 | 0.02 |
| [Fe6S6]b1− | 5.71 | 2.91, 2.88 | −2.43, −2.40 | 0.04 | 0.02 |
| [Fe4S4(SCH3)4]1− | 5.64 | 2.85, 2.85 | −2.39, −2.39 | – | – |
Relevant insights regarding the chemistry of the synthetic hexanuclear cluster are given by the analysis of the structural properties of the rotated conformer [Fe6S6]b2− (see Fig. 5) and by the computation of its stability with respect to the corresponding eclipsed conformer. In [Fe6S6]b2−, the Fep–Fed bond distance (2.57 Å) is only slightly larger than the corresponding distance in [Fe6S6]t2−. Such a value of the Fe–Fe interatomic distance well reproduced the Fep–Fed bond length in the reduced enzyme, which has been reported to be 2.55–2.61 Å [4a]. The CO ligand localized in the region of space between Fep and Fed is in semibridging position; in fact the Fep–C(μ-CO) and Fed–C(μ-CO) distances are 2.19 Å and 1.80 Å, respectively. These bond length values poorly reproduce the corresponding values in the reduced enzyme, in which the Fep–C(μ-CO) distance can reach the very large value of 2.56 Å, while the Fed–C(μ-CO) bond length was found to be as short as 1.69 Å [4a]. However, it should be noted that computational data regarding the position of this CO group have to be discussed with caution, since previous DFT results showed that the energy landscape associated with the movement of the semibridging CO is very flat [4d].
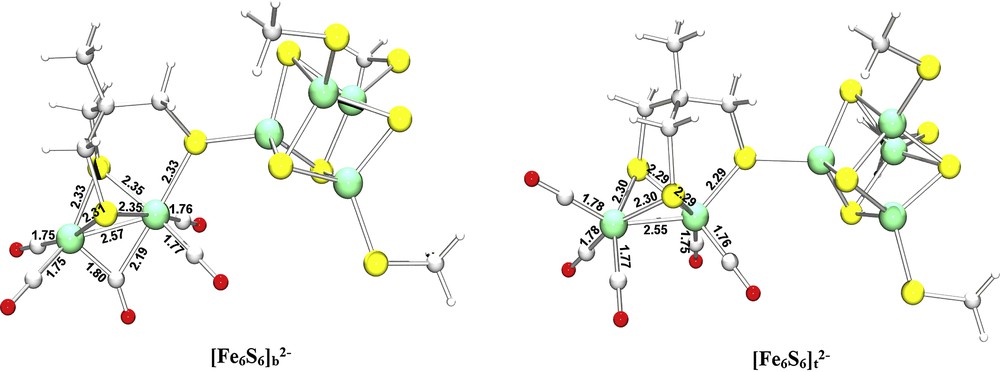
Optimized geometries of model complexes [Fe6S6]b2− and [Fe6S6]t2−. Selected distances are given in Å.
As far as Fep and Fed spin densities are concerned, they are again very close to zero (Table 1), while the comparison of [Fe6S6]b2− and [Fe6S6]t2− total energy values evidences that the μ-CO adduct is less stable than the eclipsed conformer by 11.8 kJ mol−1 (the energy difference reduces to 7.4 kJ mol−1 when geometry optimizations are carried out in a vacuum). Notably, the energy gap for an analogous isomerization reaction is significantly larger in the Fe(I)Fe(I) form of the hexacarbonyl diiron complex Fe2(CO)6(μ-PDT) [23]. In fact, the rotated conformer of this neutral assembly is 33.9 kJ mol−1 less stable than the eclipsed adduct (this energy difference refers to geometry optimizations carried out in a vacuum); if one considers that the difference between the first coordination spheres of Fe2(CO)6(μ-PDT) and of the binuclear subcluster in the synthetic Fe6S6 adduct is limited to the substitution of a CO group with a cubane moiety, one can argue that an Fe4S4 assembly in its [2Fe(II)2Fe(III)] is better than a carbonyl ligand in stabilizing the rotated form of the binuclear cluster.
3.2 DFT characterization of monoanionic hexanuclear model compounds
In a very recent paper, Tye et al. and Felton et al. noted that the one-electron oxidation of the hexacarbonyl Fe(I)Fe(I) diiron complex Fe2(CO)6(S2C6H4) facilitates the rotation of CO groups bound around one of the iron centers, thus leading to the stabilization of the rotated conformer with respect to the corresponding eclipsed isomer [24]. Such a structural reorganization is expected to be correlated with the desymmetrization of the cluster in terms of redox states of the iron centers, one of which formally becomes bivalent after the mono-electron oxidation of the Fe(I)Fe(I) adduct. This observation prompted us to investigate the structural and redox properties of the monoanionic hexanuclear compounds [Fe6S6]b1− and [Fe6S6]t1− (see Fig. 6) that would arise from the mono-electron oxidation of the Fe(I)Fe(I)–[2Fe(II)2Fe(III)] complexes [Fe6S6]b2− and [Fe6S6]t2−.
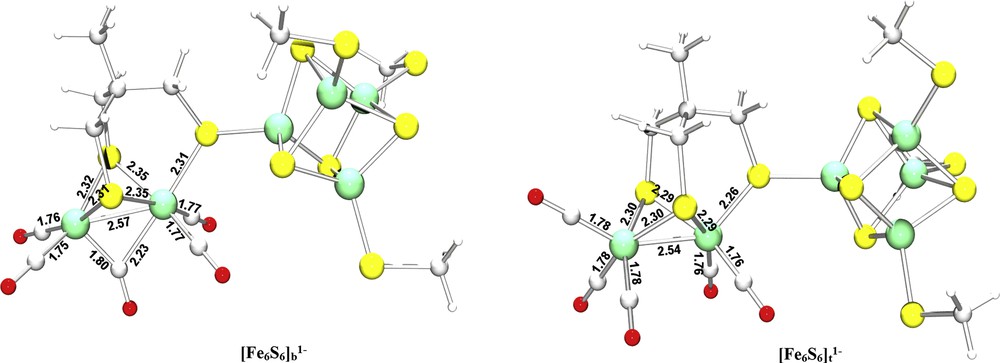
Optimized geometries of model complexes [Fe6S6]b1− and [Fe6S6]t1−. Selected distances are given in Å.
For [Fe6S6]b1− and [Fe6S6]t1− the BS1 and BS2 solutions converge to a very similar geometry, where the only appreciable difference is the Fe1–S1 bond length, which in BS1 is about 0.02 Å longer than that of BS2. The energy difference between the BS1 and BS2 solutions is also very small (6.5 kJ mol−1 and 2.1 kJ mol−1 for [Fe6S6]b1− and [Fe6S6]t1−, respectively), with BS1, which is the only form discussed in the following, being the lowest energy solution.
First of all, let us discuss the main features of the rotated adduct [Fe6S6]b1−; computation of Fep and Fed spin density values (0.02 in both cases) clearly evidences that the unpaired electron of this open shell species does not reside in the binuclear subcluster. This suggests that the oxidation involves exclusively the Fe4S4 cluster included in the Fe6S6 assembly; such a conclusion is supported by the calculated spin densities of Fe atoms in the Fe4S4 cluster, which are very similar to those computed for the [Fe4S4(SCH3)4]1− complex (Table 1), and by the variation of the computed charge of the Fe4S4 moiety; in fact, the charge of the cubane in [Fe6S6]b2− is −1.42, but its absolute value lowers by 0.85 charge units after the one-electron oxidation step.
The above-reported considerations indicate that the redox state of the iron centers in [Fe6S6]b1− is Fe(I)Fe(I)–[3Fe(III)Fe(II)]. Notably, the [3Fe(III)Fe(II)] state has never been observed in the cubane moiety of the enzyme active site, which should normally maintain the [2Fe(III)2Fe(II)] state (see Section 1 of the present paper). The fact that the binuclear subcluster included in [Fe6S6]b1− remains in the Fe(I)Fe(I) redox state has relevant bearings on the structural features of the diiron subsite, the geometry of which preserves most of the features that characterize the diiron subsite in [Fe6S6]b2−. In particular, the Fep–Fed distance is still as large as 2.57 Å, and the μ-CO group unsymmetrically bridges Fep and Fed (the Fep–C(μ-CO) and Fed–C(μ-CO) distances are 1.80 Å and 2.23 Å, respectively).
In [Fe6S6]b1− the two layers of the Fe4S4 cluster are defined by an all-ferric pair of Fe3+ ions and a mixed valence pair of Fe2+ and Fe3+ ions, with the unpaired electron fully delocalized between the two Fe atoms of the mixed valence pair (Table 1). The different localizations of the all-ferric pair, which are adjacent or distant to the binuclear cluster in BS1 and BS2 solutions, respectively, are correlated to the difference in the Fe1–Sb bond length discussed above and with the small energy difference between BS1 and BS2.
As a next step in the present study, we evaluated the possibility that the oxidation of the hexanuclear dianionic cluster could lead to a stabilization of its rotated conformation, analogously to what has recently been proposed for the diiron synthetic assembly Fe2(CO)6(S2C6H4) (see above). To this end, the [Fe6S6]t1− structure was optimized (see Fig. 6) and its stability with respect to [Fe6S6]b1− was calculated. It turned out that [Fe6S6]b1− is 22.3 kJ mol−1 less stable than [Fe6S6]t1−, an energy difference which is surprisingly larger than the corresponding stability difference for the dianionic compounds (11.8 kJ mol−1, see Section 3.1). In other words, the inclusion of the cubane moiety in the synthetic cluster disfavours the rotation of the ligands around Fed upon mono-electron oxidation of the H-cluster.
Finally, the unpaired electron in [Fe6S6]t1− still resides in the cubane moiety, as witnessed by Fep and Fed spin density values, which are very small (see Table 1). This observation, together with the computation of the overall charge of the tetranuclear subcluster in [Fe6S6]t2− and [Fe6S6]t1− (−1.46 and −0.61, respectively), clearly evidences that the mono-electron oxidation leading to the monoanionic eclipsed derivative does not involve the binuclear subcluster, similarly to the case of the rotated conformer.
4 Conclusions
In the present contribution, an investigation was presented regarding computational models of a recently synthesized Fe6S6 complex structurally related to [FeFe]-hydrogenases active site. All the DFT models here presented share the same atomic composition, but differ in terms of charge and ligand dispositions around the metal centers of their binuclear subsite (their general formula is [Fe4S4(SCH3)3{Fe2(CH3C(CH2S)3)(CO)5}]n−, where n = 1 or 2).
Two main issues were investigated for each redox state of the complex: (i) the relative stabilities of rotated and eclipsed conformations and (ii) the redox state of the iron centers in the Fe2S2 and Fe4S4 subsites.
Results can be summarized as follows: the dianionic complex shows an Fe(I)Fe(I)–[2Fe(II)2Fe(III)] redox state, both in its rotated and eclipsed conformation. However, the rotated conformation turned out to be significantly less stable than the eclipsed adduct (ΔE = 11.8 kJ mol−1 and 7.4 kJ mol−1 at ε = 36.64 and 1, respectively). The energy difference for the corresponding rearrangement in the Fe(I)Fe(I) binuclear complex Fe2(CO)6(μ-PDT) is much larger (33.9 kJ mol−1, considering models optimized in a vacuum), indicating that a cubane moiety in trans position with respect to the CO bridging Fep and Fed can favour the rotation of ligands on the distal iron atom.
As far as the mono-electron oxidation of the dianionic complex is concerned, computation of spin density values and atomic charges clearly indicates that the Fe4S4 moiety is directly involved in the redox step, while the binuclear subcluster maintains an Fe(I)Fe(I) redox state.
It is known that a one-electron oxidation of simple Fe(I)Fe(I) models of [FeFe]-hydrogenases binuclear subsite can lead the rotated geometry to be more stable than the corresponding eclipsed adduct [24]. However, such a stabilization of the rotated isomer was not observed in the monoanionic Fe6S6 synthetic model, probably because the binuclear subsite remains in its Fe(I)Fe(I) redox state after a one-electron oxidation of the dianionic, Fe(I)Fe(I)–[2Fe(II)2Fe(III)] assembly. This point represents a major difference between the synthetic assembly and the enzyme active site; in fact, the completely reduced H-cluster attains the Fe(I)Fe(I)–[2Fe(II)2Fe(III)] state, but its binuclear subsite switches to the Fe(I)Fe(II) state upon partial oxidation of the enzyme. Future studies will be devoted to the elucidation of the structural and electronic determinants underlying the difference in redox properties between the H-cluster and the biomimetic Fe6S6 model.