

1 Introduction
The search for cheap and robust electrocatalysts as an alternative to platinum for the reduction of proton to dihydrogen has led to resurgence of the chemistry of diiron dithiolate complexes. This arises from the striking resemblance of the long-known [Fe2(μ-SR)2(CO)6] molecules with the [2Fe]H-subsite of the iron-only hydrogenases [1,2] (Scheme 1) that catalyse the reduction of protons to dihydrogen with a high efficiency. Since they started in 1999 [3], the modeling studies based on complexes with {2Fe2S} or {2Fe3S} frameworks have experienced a huge development [4].
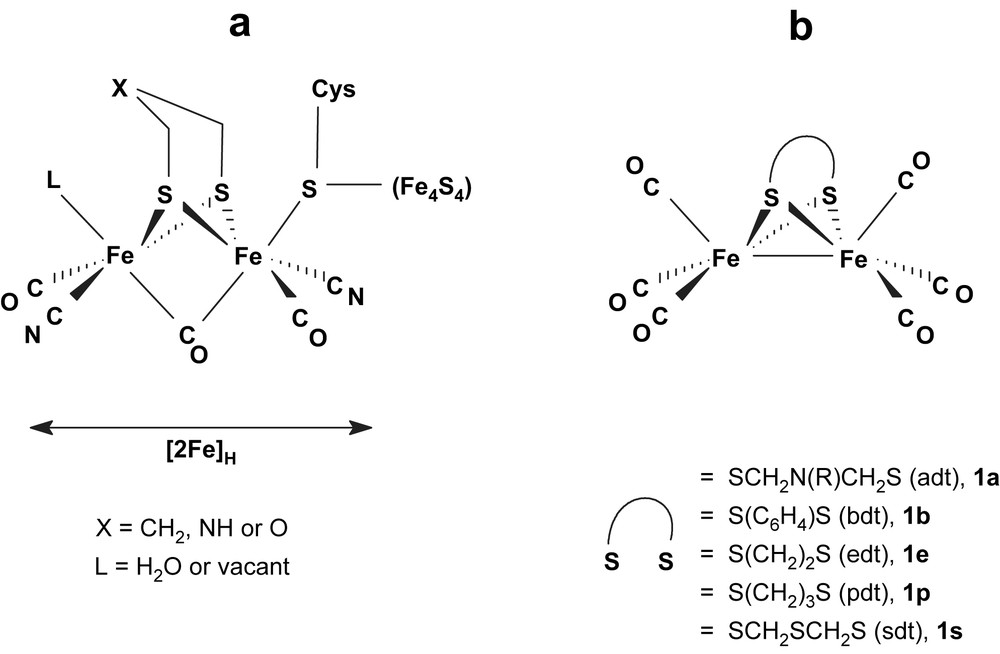
Representations of (a) the active site (H cluster) of [FeFe]H2ases [1,2], and of (b) synthetic model compounds.
These studies demonstrated that simple diiron models are able to electrocatalyse the H+/H2 conversion, but also that they are all much less efficient than the enzymes. In this contribution, we chose to emphasize a few aspects of the electrochemistry of diiron dithiolate complexes that we believe are important for a better understanding of the electron transfer-induced reactivity of these complexes both in the absence and in the presence of protons, and that may also be helpful to drive future synthetic studies towards more efficient catalytic systems.
2 Electrochemistry of [Fe2(μ-dithiolate)(CO)6] complexes in acid-free medium
The perception of different facets of the rich electrochemistry of diiron dithiolate complexes has much improved in the past few years. Detailed CV investigations showed that the reduction of [Fe2(μ-dithiolate)(CO)6] (dithiolate = SC6H4S: bdt, 1b [5a,6]; SCH2SCH2S: sdt, 1s [7]; SCH2N(R)CH2S with R = CH2CH2OMe, i-Pr: adt, 1a [8]) in acetonitrile involves the transfer of two electrons in a single reversible or quasi-reversible step, due to the potential inversion (E1/2red2 − E1/2red1 > 0) of the two individual electron transfers. Whereas the successive steps could not be resolved in the case of 1b or 1s [5a,7], wave-splitting occurred at fast scan rates for the adt analogue 1a so that the two one-electron processes were discernible [8]. The heterogeneous electron transfer rate constant for the two steps, ksred1 and ksred2, are similar for the bdt-bridged complex 1b [5a,6], while ksred1 < ksred2 for 1s [7] and ksred1 ≫ ksred2 for the azadithiolate derivative 1a [8]. As a consequence of the large ΔEpred2, the oxidation of 1a2– occurs at a potential positive of the oxidation of the anion so that the dianion undergoes a two-electron oxidation to the neutral complex (Scheme 2).
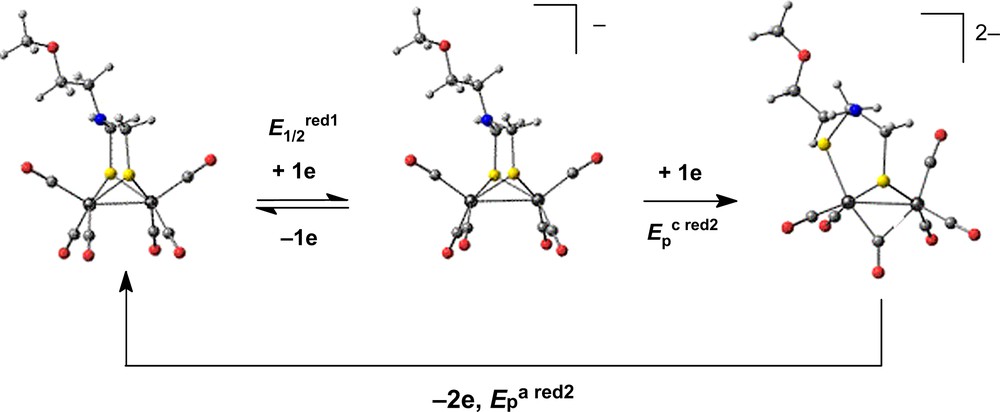
Basic reduction mechanism of [Fe2{μ-SCH2N(CH2CH2OMe)CH2S}(CO)6], 1a, in MeCN showing the calculated structures of the redox partners [8].
Typically, potential inversion occurs when a chemical reaction, in most cases a structural rearragement [9–12], makes the transfer of a second electron thermodynamically more favourable than the first. Density functional theory (DFT) calculations showed that the structure change is distributed over the two one-electron reductions of [Fe2(μ-bdt)(CO)6], with a substantially rearranged anion [6], while the [Fe2(μ-adt)(CO)6]− anion retains essentially the structure of its neutral precursor, the major structural change occurring upon the transfer of the second electron (Scheme 2). This is consistent with the slow electron transfer rate of this step for complex 1a [8].
The electrochemical behaviour of [Fe2(μ-pdt)(CO)6] (pdt = S(CH2)3S, 1p) was also investigated by CV in thf and MeCN electrolytes [8,13–15]. On a timescale sufficiently short to isolate the electron transfers from the following chemistry, the reduction mechanism of 1p is similar to that of the adt analogue (Scheme 2), with the two successive one-electron steps clearly detected by CV at fast scan rates [8]. However, there is a major difference between 1p and 1a in that potential inversion of the reduction steps is observed only for the latter. Therefore, the disproportionation of the anion that is an important reaction for the adt complex is negligible for the pdt analogue whose anion gives rise to a different chemistry [15–17]. Thorough thin-layer spectroelectrochemical (SEC) and theoretical investigations of the reduction of 1p [15] and [Fe2(μ-edt)(CO)6] (edt = S(CH2)2S, 1e) [17] remarkably illustrated the variety of the reduction products arising from the reactivity of the one-electron reduced species. The [Fe2(μ-dithiolate)(CO)6]− anion indeed undergoes the initial loss of a CO ligand (dithiolate = pdt, edt or adt), which leads to the formation of dithiolate-bridged tetrairon dianions for the pdt and edt derivatives [15–17] (Scheme 3). DFT calculations showed that this is impeded by an interaction of the CH2CH2OMe or i-Pr substituent of the N bridgehead atom with the coordination site exposed by the loss of a CO from 1a– [8].
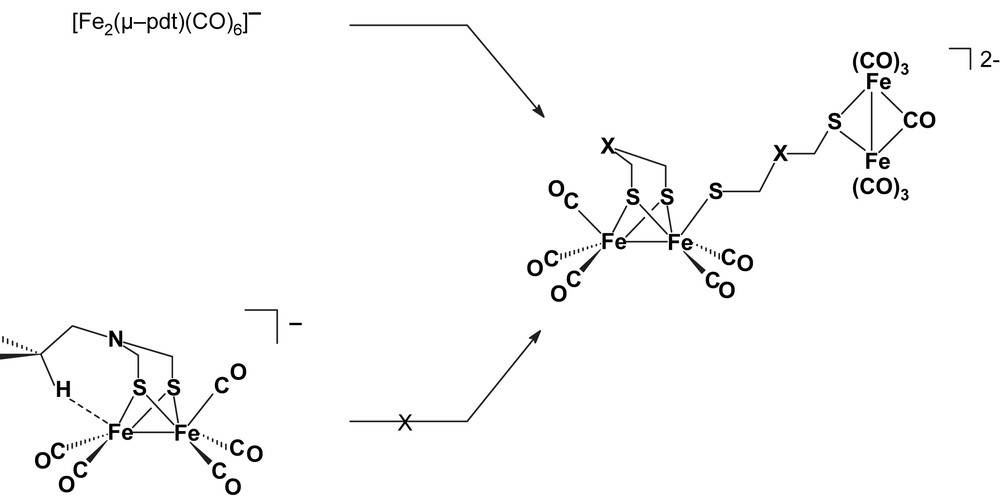
X = CH2 or N(R) (R = CH2CH2OMe, i-Pr) [8,15–17].
Electrochemical, SEC and theoretical studies of various [Fe2(μ-dithiolate)(CO)6] thus revealed that the nature of the S-to-S link has a subtle impact on the reductive behaviour of these compounds, and emphasised the variety of the products that result from the transfer of one and two electrons [5–8,15–17]. In this respect, we proposed previously [4k] that a complex where a CO would be replaced by a X or L ligand (respectively anionic or neutral) would be an interesting synthetic target in that the coordinatively unsaturated anion that may arise from its electrochemical reduction (Scheme 4, A) might generate a terminal hydride under acidic conditions. Such a complex with a labile ligand, namely [Fe2(μ-pdt)(CO)5(L)] (L = MeCN or NH2Pr), is now known [18] and can be used as a precursor to investigate the coordination of various ligands. However, a recent study demonstrated that the reduction of such a derivative would not be a judicious strategy to open a coordination site in an acid-free medium because of the fast formation of the dimeric dianion [19] (Scheme 4, B).
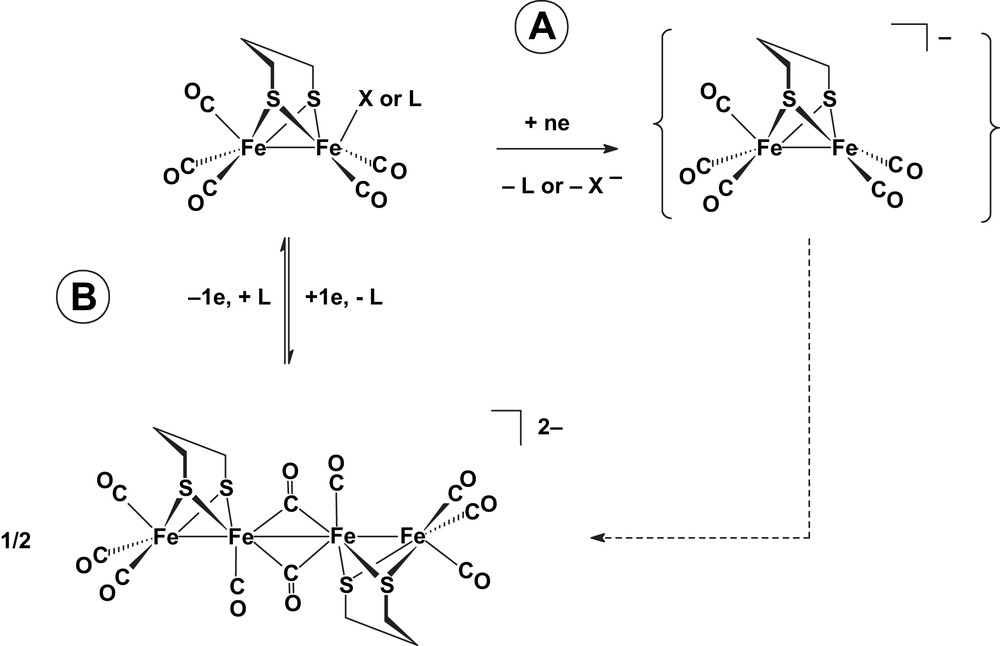
Adapted from Ref. [19].
The electrochemical studies of [Fe2(μ-dithiolate)(CO)6] complexes in acid-free media are an important prerequisite to their use as potential electrocatalysts for proton reduction. This is particularly clear in the case of non-protonatable complexes (dithiolate = bdt, pdt, edt), since the first step of the proton reduction is then an electron transfer. Therefore, reduction products formed in the absence of acid may also take part in electrocatalytic proton reduction processes, since the cleavage of Fe–S bonds, for example, generates new sites for protonation.
3 Electrochemistry in acidic medium – mechanisms of proton reduction processes
While a range of differently substituted dithiolate complexes has now been produced, detailed investigations of the mechanisms of proton reduction by [FeFe]H2ase diiron model compounds are still principally limited to hexacarbonyl species. Several mononuclear [20] and tetranuclear [21] catalysts have also been thoroughly investigated.
3.1 Mechanisms of electrocatalytic proton reduction by diiron hexacarbonyl compounds
The diiron model compounds can be separated into two categories, depending on whether they can be protonated or not, that is, for hexacarbonyls, depending on the nature of the dithiolate bridge. The presence of the N bridgehead atom imparts specific properties to [Fe2{μ-SCH2N(R)CH2S}(CO)6] complexes, since the reduction in acidic medium is shifted towards less negative potentials provided the acid is strong enough to protonate the amine function (pKa = 8.1 for R = Me [22]). Consequently, when the N–H proton is involved, the resulting electrocatalytic process has no counterpart for the pdt, edt or bdt-bridged hexacarbonyl analogues. In contrast, when the acid is too weak to protonate the N atom, the proton reduction processes that are initiated by an electron transfer step are similar to those supported by the propanedithiolate analogue.
A recent study of the reduction of [Fe2{μ-SCH2N(R)CH2S}(CO)6] 1a (R = CH2CH2OMe, i-Pr) in acidic medium showed that different electrocatalytic proton reduction processes may occur depending on the strength and on the concentration of the acid [23]. The process operating at −1.2 V that involves protonation at the N atom of the bridge is observed when HBF4·Et2O or HOTs are used as a proton source. This process, which does not take place with the weaker CF3CO2H acid (pKa = 12.6 in MeCN [24]), was proposed to be limited by the slow release of H2 from the 1a–2H species under pseudo-first-order acidic conditions (Scheme 5).
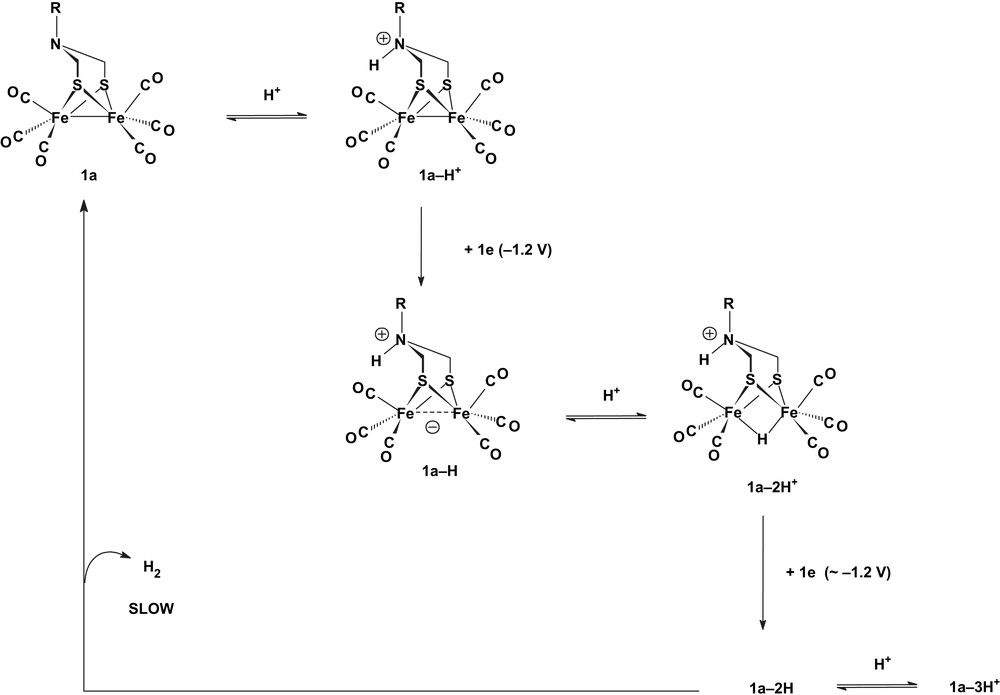
Proposed mechanism of proton reduction specific to hexacarbonyl azadithiolate-bridged complexes in a MeCN electrolyte [23].
In the presence of more than 3–4 molar equivalents HBF4·Et2O (but not HOTs1), the protonation of 1a–2H competes with the loss of H2 from this intermediate. Thus, in the presence of an excess of the strong acid HBF4·Et2O, the mechanism in Scheme 5 becomes superseded by a more negative one (around −1.4 V) based on the reduction of 1a–3H+, in which the N–H proton is probably only a spectator. In this case, the reduction of protons supported by the adt [23] and pdt [15] derivatives should involve similar intermediates of which different isomers (Scheme 6) were considered on the basis of DFT calculations [8,25]. Digital CV simulations indicated a slow release of H2 (k = 2–5 s−1) from 1a–3H+ [23] and 1p–2H [15] (Scheme 6), as well as from the analogue with a PhP(CH2)3PPh bridge (2–2H) [26], while 1a–3H, 1p–2H− and 2–2H− release H2 at a much faster rate, in full agreement with theory [25].
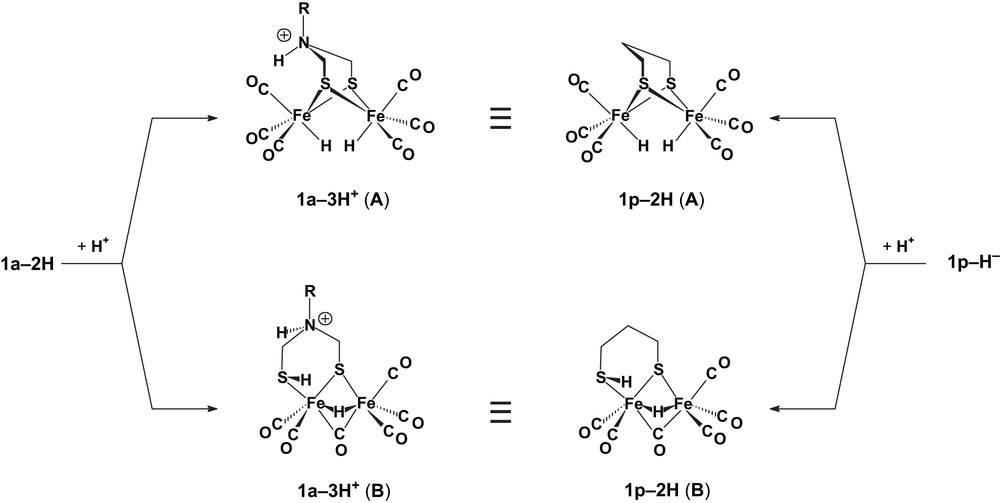
Connection between intermediates possibly involved in proton reduction processes by 1a and 1p complexes [15,23,25].
It should be noted that intermediates with bridging hydride and CO ligands and a cleaved Fe–S bond also take part in the electrocatalytic reduction of the weak acetic acid by 1b [6] (Scheme 7). The latter occurs around −2.1 V instead of ca. −1.3 V for HOTs because, in opposition to HOTs (pKa = 8.3 in MeCN [27]), CH3CO2H (pKa = 22.3 in MeCN [24]) is too weak to protonate 1b–H−. When acetic acid is used as a proton source to the bdt-bridged complex, the catalyst is no more the neutral complex 1b but its one-electron reduced product, [Fe2(μ-bdt)(CO)6]− [6], Scheme 7.
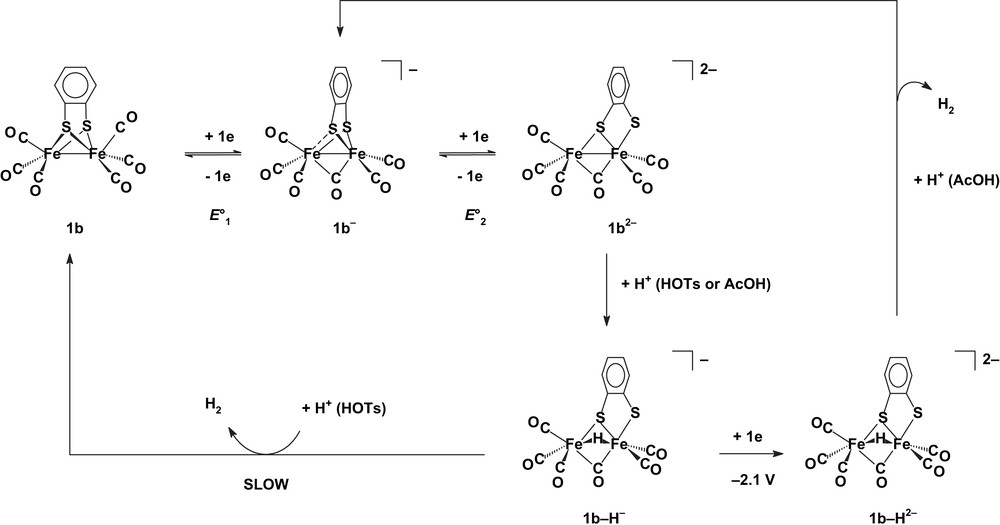
Proton reduction processes catalysed by [Fe2(μ-bdt)(CO)6]0/− 1b0/− [5b,6] (E°2 − E°1 > 0; MeCN electrolyte).
Therefore, the effect of the acid strength on the mechanisms and potentials of proton reduction processes has been clearly illustrated in the case of complexes 1a and 1b [5,6,23]. Rerouting from one mechanism to another depends upon a combination of thermodynamic and kinetic factors such as the relative values of the pKas of the acid and of the protonated metal complexes involved in the proton reduction processes, and the rate of their slowest step.
3.2 Thermodynamic and kinetic aspects of proton reduction processes
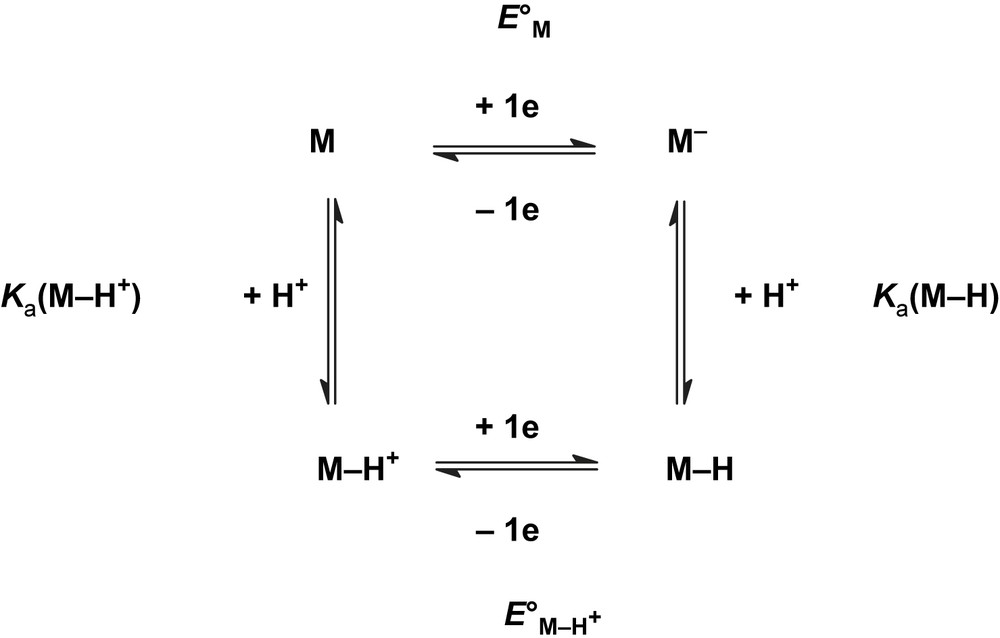
The substitution of donor ligands (L) for CO in non-protonatable complexes increases the basicity of the diiron site so that treatment of [Fe2(μ-dithiolate)(CO)4(L)2] species with acid leads to the formation of products with a bridging hydride [28–32]. The negative shift of the reduction potentials due to the replacement of COs by electron-donor ligands L and/or X− (Table 1, 3rd column) is compensated by a positive one, resulting from the formation of the more easily reducible protonated complex (Table 1, 6th column). The positive shift may be rationalised considering the thermodynamic cycle in Scheme 8 that depicts protonation of diiron dithiolate complexes, M and M−. In case of a reversible one-electron reduction of M and M–H+,2 and when the electron and proton transfers occur in separate steps, the potential shift observed upon protonation is dependent on the pKas of M–H and M–H+, with ΔE° = 0.059 [pKa(M–H) − pKa(M–H+)]. Therefore, the more the basicity of the protonation site is enhanced by the reduction of M, the larger in magnitude ΔE° should be. Since the LUMO of the diiron dithiolate complexes has a predominant σ∗Fe–Fe character, one may anticipate that the basicity of the diiron site will be strongly dependent upon the redox state of the complex.
From the potential shifts observed upon formation of a bridging hydride (Table 1, 6th column), it can be estimated3 that the pKa of the Fe–Fe site increases by 15 ± 2 units upon one-electron reduction of M. This magnitude is similar to the lowering of the pKa of the Ni–H entity by more than 14 units upon one-electron oxidation of the [HNi(PNP)2]+ complex (PNP = Et2PCH2N(Me)CH2PEt2) [20a]. The increase in the electron density at the diiron site upon reduction should also be transmitted to the ligands, but this effect is expected to be diluted so that the reduction should have a lesser impact on the pKa of ligands (terminal or bridging) than on the pKa of the Fe–Fe site. Assuming that no proton migration is concomitant with the reduction of the ligand-protonated M–H+ complex, this should lead to a smaller ΔE° and the values in Table 1 indicate that such is the case.
It is also apparent from Table 1 that the reduction potentials of the singly protonated diiron complexes all fall in a relatively narrow range, from −1.1 to −1.6 V. While infrared spectroscopy shows that the monosubstituted derivatives [Fe2(μ-bdt)(CO)5P(OMe)3] (3b, E1/2red = −1.48 V [34a]) and [Fe2(μ-pdt)(CO)5P(OMe)3] (3p, Epred = −1.98 V [34b]) do not protonate significantly upon addition of HBF4·Et2O, CV of 3b and 3p in the presence of HOTs exhibits a new acid-responsive reduction at a potential less negative than that of their primary reduction in the absence of acid. These observations point to a CE mechanism in which the protonation equilibrium is shifted by the reduction of 3b–H+ (Epred = −1.18 V) or 3p–H+ (Epred = −1.65 V). For complexes that are not protonated by strong acids such as [Fe2(μ-bdt)(CO)6] 1b [5b] and [Fe2(μ-pdt)(CO)6] 1p [15], the electrocatalytic proton reduction also occurs in the same potential range.4 On thermodynamic grounds, one may thus question the validity of the approach consisting in switching from hexacarbonyl to substituted diiron dithiolate complexes.
However, the kinetic aspects of proton reduction processes must also be considered. Different bottlenecks were identified so far. Simulation of the voltammetric responses in the presence of various concentrations of HOTs indicated that the rate-determining step in the H+/H2 catalysis by complexes 3b [34a] and 3p [34b] is the first protonation step. For the [Fe4{Me(CH2S)3}(CO)8] (4Fe6S) complex that has fused {2Fe3S} cores [21], a potential proton reduction process around −1.3 V was also hindered by the slow protonation of the anion, [Fe4{Me(CH2S)3}(CO)8]−, by LutH+ (Lut = 2,6 dimethylpyridine; pKa = 14.05 in MeCN [24]). From the Brönsted relation [35] it may be anticipated that the protonation would be faster if an acid stronger than, respectively, HOTs or LutH+ was used. For 4Fe6S, the kinetic problem was solved at a thermodynamic cost (0.36 V, ∼35 kJ mol−1) by generating a stronger base, namely the [4Fe6S]2– dianion, which resulted in a kinetically very efficient electrocatalysis with a fast elimination of dihydrogen (k = 2000 s−1) [21]. This suggested the possibility that a terminal hydride might be formed upon protonation of [4Fe6S]2–, since a terminal hydride is known to be more reactive than a bridging one towards dihydrogen formation in FeIIFeII complexes [36]. In contrast, the slow step of the less negative proton reduction process for [Fe2{μ-SCH2N(R)CH2S}(CO)6] 1a (R = CH2CH2OMe, i-Pr) consisted in the elimination of H2 from the {2H+/2e} reduced species (1a–2H, Scheme 5) [23]. Such was also the case for catalysis by [Fe2(μ-pdt)(CO)6] 1b [15,25] and [Fe2{μ-PhP(CH2)3PPh}(CO)6] 2 [26]. The slow rate of this reaction (k = 2–5 s−1) that comprises two steps, that is formation and release of a H2 ligand, might be due to an inadequate H+⋯H– positioning, or to a mismatch of the pKas of the protonated site and the dihydrogen ligand [20], in the {2H+/2e} reduced species. The faster H2 elimination upon addition of a further electron might thus result from an electron transfer-induced rearrangement bringing the proton and the hydride in a closer proximity, or from an increased basicity of the hydride.
One of the interests of unsymmetrically disubstituted FeIFeI complexes is precisely that they may favour the “rotated” structure [37] (Scheme 9), more closely related to that of the [2Fe]H subsite of the enzyme (Scheme 1a), and thus facilitate the formation of terminal hydrides. This would circumvent the geometric failure of the usual models that are found in the more stable [4a,4b] eclipsed geometry and protonate at the Fe–Fe bond.
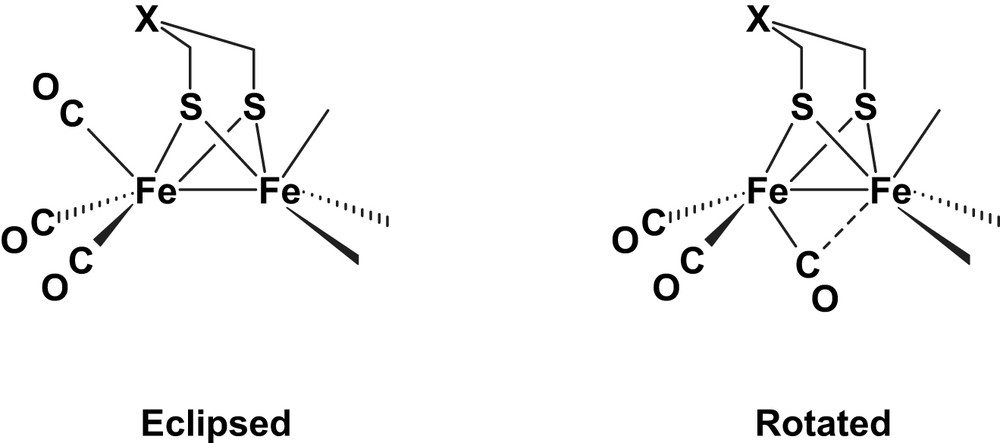
Therefore, [Fe2(μ-dithiolate)(CO)4(κ2-LL′)] complexes might contribute to solve both the kinetic problems mentioned above. Their increased basicity compared to that of their hexacarbonyl precursors should facilitate protonation that may occur at a terminal position, due to the favoured rotated geometry. This, in turn, is expected to speed up dihydrogen elimination at the ultimate stage of the process. This approach is clearly heading to substrate activation at a single metal site, with or without the assistance of the second coordination sphere (Scheme 10).

Putative reduced intermediates with a rotated geometry.
The detection of transient terminal hydrides upon protonation of several [Fe2(μ-pdt)(CO)4(κ2-LL′)] complexes [32], the high kinetic performances of the 4Fe6S cluster [21] and of several mononuclear complexes with precisely positioned internal proton relays [20] towards electrocatalytic H2 production/uptake lends support to this strategy.
4 Conclusion
This contribution attempted to summarize some recent and important advances in the electrochemistry of diiron dithiolate hexacarbonyl complexes that clarified the reduction processes of several such species and unravelled the different mechanisms by which they catalyse the reduction of protons to dihydrogen. The past few years also witnessed very active developments of the synthesis of more sophisticated compounds, such as unevenly substituted species, that are likely to allow further developments of bioinspired catalysis of proton reduction to dihydrogen.
Acknowledgements
This work was supported by ANR (programmes “PhotoBioH2” and “CatH2”), CNRS and Université de Bretagne Occidentale. The Ph.D. students whose names are in the referenced papers from our laboratory are acknowledged for their contribution.
1 In contrast to HBF4·Et2O, HOTs does not protonate the 1a–2H intermediate efficiently. When HOTs is used as a proton source, there is also a second catalytic proton reduction process; it is observed around −1.6 V (instead of −1.4 V for HBF4·Et2O) since it requires that 1a–2H be reduced, which was proposed to occur around −1.6 V [23].
2 The reduction of symmetrically disubstituted diiron complexes with a bridging hydride (represented by M–H+ in Scheme 8) was reported to be a partially reversible one-electron step [28]; the reduction of the N-protonated 1a–H+ (also represented by M–H+ in Scheme 8) was an irreversible one-electron step [23].
| Complex | EpredM (V) | ΔEred (V)a | Protonation site | (V) | ΔEpred (V)b | Ref. |
| [Fe2(μ-adt)(CO)6]c | −1.56d | – | N | ∼−1.2 | ∼0.4 | [23] |
| [Fe2(μ-pdt)(CO)4(PMe3)(CN)]– | −2.56 | −0.96 | Fe–Fe | −1.55 | 1.01 | [28] |
| Fe–Fe and CN− | −1.45 | [28] | ||||
| [Fe2(μ-pdt)(CO)4(PMe3)2] | −2.29 | −0.69 | Fe–Fe | −1.37d | 0.92 | [28] |
| [Fe2(μ-pdt)(CO)4(PMe3)2] | −2.25 | −0.65 | Fe–Fe | −1.5 | 0.75 | [29] |
| [Fe2(μ-pdt)(CO)4(PTA)2]e | −2.02 | −0.42 | PTA | −1.59 | 0.43 | [29] |
| [Fe2(μ-adt)(CO)4(PMe3)2]f | −2.18 | −0.65 | Fe–Fe | −1.10 | 1.08 | [30] |
| N | −1.55 | 0.63 | [30] | |||
| N and Fe–Fe | −1.00 | [30] | ||||
| [Fe2(μ-adt)(CO)4(PMe3)2]g | −2.18 | −0.62 | N | −1.49 | 0.69 | [31] |
| [Fe2(μ-adt)(CO)4(κ2-dppe)]h | −2.05i | −0.47 | N | −1.53 | 0.52 | [33] |
| [Fe2(μ-pdt)(CO)4(κ2-bisNHC)]j | −2.42 | −0.82 | Fe–Fe | −1.48 | 0.94 | [32b] |
a ΔE red = [Epred (complex) − Epred (hexaCO precursor)].
b .
c adt = (SCH2)2N(CH2CH2OMe).
d E1/2red.
e PTA = 1,3,5-triaza-7-phosphaadamantane.
f adt = (SCH2)2N(CH2C6H5).
g adt = (SCH2)2N(CH2C6H4-2-Br).
h adt = (SCH2)2N(i-Pr).
i Under CO.
j BisNHC = IMe–CH2–IMe, IMe = 1-methylimidazol-2-ylidene.
3 The reduction steps are positively shifted from their thermodynamic potentials by the chemistry following the electron transfer event, so that several potentials in Table 1 are peak potentials (Ep) rather than E1/2 or thermodynamic E° values. The ΔpKa worked out from the potentials of chemically irreversible reduction steps are thus approximate.
4 Note that proton reduction catalysed by [Fe2(μ-bdt)(CO)6] occurs at the more negative potential of –2.1 V when the proton source is acetic acid, because this acid cannot protonate [HFe2(μ-bdt)(CO)6]– [5,6], see Scheme 7.