

1 Introduction
Expansion of uses and applications of polymers is an unequivocal reality in our today's world, as they are considered convenient substitutes for the common materials, namely metals and wood. As trivially noticed, our world has been incredibly invaded by polymer-made items which our daily living unavoidably deals with which an unabated rate, academic and industrial researches have made an enormous polymer development in response to man's demands. Exciting uses of polymeric materials started from the synthetic silk of Carothers in 1930s to the polyurethane-based artificial heart of Robert Jarvik in 1980s. Chronic problems of today's world, either health-related or others may call, in one way or another, for a polymeric matter. In many cases, crude homopolymers rarely meet the sought-for properties and the needs of their users. Also, the alternatives, encompassing the desired properties, can be either copolymers, blends, or modified homopolymers. Of the several means of modifying polymers, the chemical transformation remains the leading process. It consists in functionalizing a pre-made polymer, that is, to anchor functional groups onto it, or to react the already-existing functional groups of polymer with a molecular reactant. Polymers with redox units, being either integral parts of the polymer matrices or attached ones, are distinguished by oxydo-reduction chemistry features. Of the redox entities are organometallics such as metallocenes and organics such as pyridinium salts and hydroquinone/catechol [1,2]. Not only do the latter molecules show redox properties but also metallic adsorption as well, via their chelating ability. The other feature of these molecules is their phenolic nature, implying a unique chemistry. Indeed, the phenolic OH, being a strong electron-donating group, entails a great reactivity of the phenyl group, hence, the reaction may be out-of-control. Yet, some chemical reactions on some hydroquinone and quinine derivatives were reported [3]. A myriad of natural products have dihydroxybenzene, dimethoxybenzene, or methylene-dioxybenzene-containing structures [4,5]. To cite but a few examples, there are urushiol, catechins and papaverine. Moreover, some dihydroxybenzene-bearing molecules such as 3,4-dihydroxyphenyl-l-alanine (l-DOPA) and nitecapone (a catechol-o-methyl transferase inhibitor) are distinguished by their specific activity against Parkinson's disease [6,7]. Tricatechol-containing macrocyclic compounds were synthesized to study their iron solubilization (iron transport) via the chelating ability, mimicking the natural siderophores (for example Enterobactin) [8]. Owing to their powerful inhibition of free radical polymerization, to their plausible reaction with organometallic reagents in ionic polymerization, and to their ready solubility in the polar media, the hydroxyl functionality in dihydroxybenzene-containing monomers is always blocked before polymerization; the deblocking is an obvious step in the dihydroxybenzene-containing polymers' making process. Besides their blocking role, dialkoxy substituents have been used to adjust the redox potential of a conjugated polymer, its absorption, its emission and its solubility. In fact, long alkyl and/or alkoxy groups efficiently promote the solubility of PPV materials in polar aprotic solvents such as chloroform, dichloromethane, and THF, by lowering the interchain interactions. Yet, the removal of the protecting groups is not always straightforward [9]; different Bronstëd acids and Lewis acids, and different Arrhenius bases and Lewis bases have been employed. Some alkoxyl groups defied removal towards some reagents, while others were readily displaced.
Again, it is enlightening to recall the main properties of the dihydroxybenzene-containing molecules and their corresponding benzoquinones, which are the redox abilities (Scheme 1a) [1,2], the metallic chelation and sequestration (Scheme 1b) [10,11], the adhesive properties to substrates of different natures (metallic, plastics, human organs) (Scheme 1c) [12], and the properties of dialkoxybenzene-bearing molecules, which are electroluminescence, electrical conductivity, and optoelectronic property (Scheme 1d) [13,14]. In making the dihydroxybenzene/dialkoxybenzene-containing materials, these properties have been highly coveted. Needless is to remind the sound application of dihydroxybenzene-containing materials in water treatment, that is, the removal of heavy metals from water effluents and wastewater.
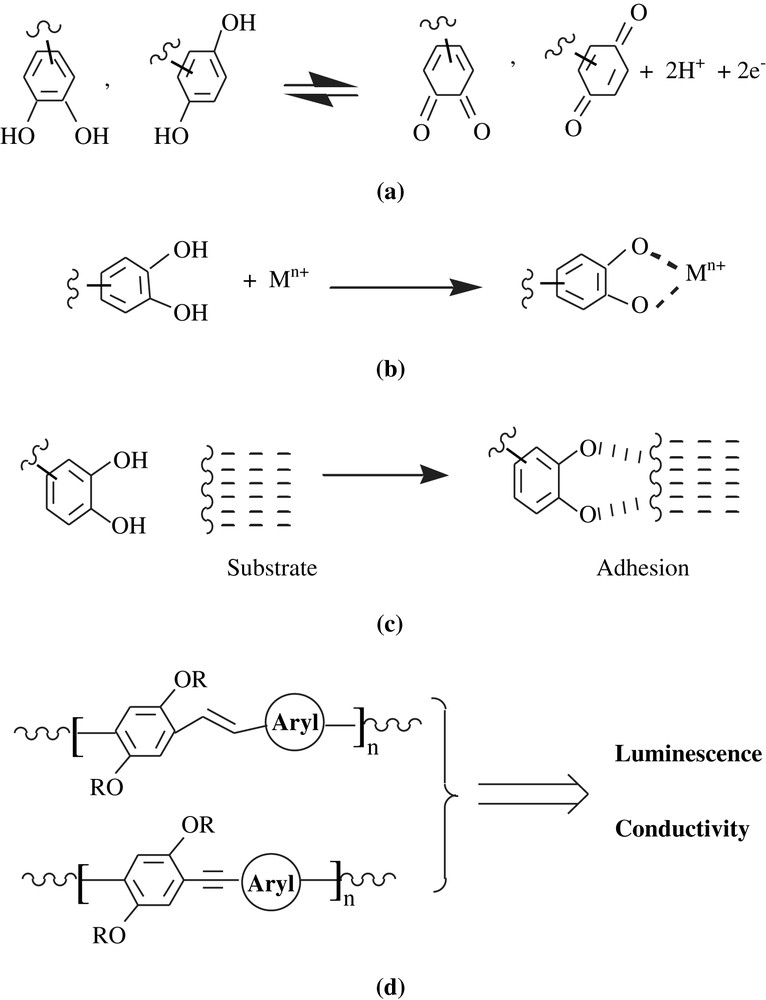
In this mini-review, dihydroxy/dialkoxybenzene-bearing polymers developed in the last decade, their synthetic pathways and their properties are highlighted. A synthesis layout is as follows: addition and metathesis polymers, condensation and coupling polymers, electrosynthesis polymers, attachment polymers, and enzymolysis polymers.
2 Addition and metathesis polymers
Chain-growth polymerization has been always the method of predilection for polymerizing the vinyl monomers, including those having dialkoxybenzene units. Yet, the naked dihydroxybenzene-containing vinyl monomers are not prone to this type of polymerization because of the great affinity of phenol functionality towards free radical and organometallic initiators. Consequently, the OH groups must be blocked at the start and may be deblocked after polymerization processing.
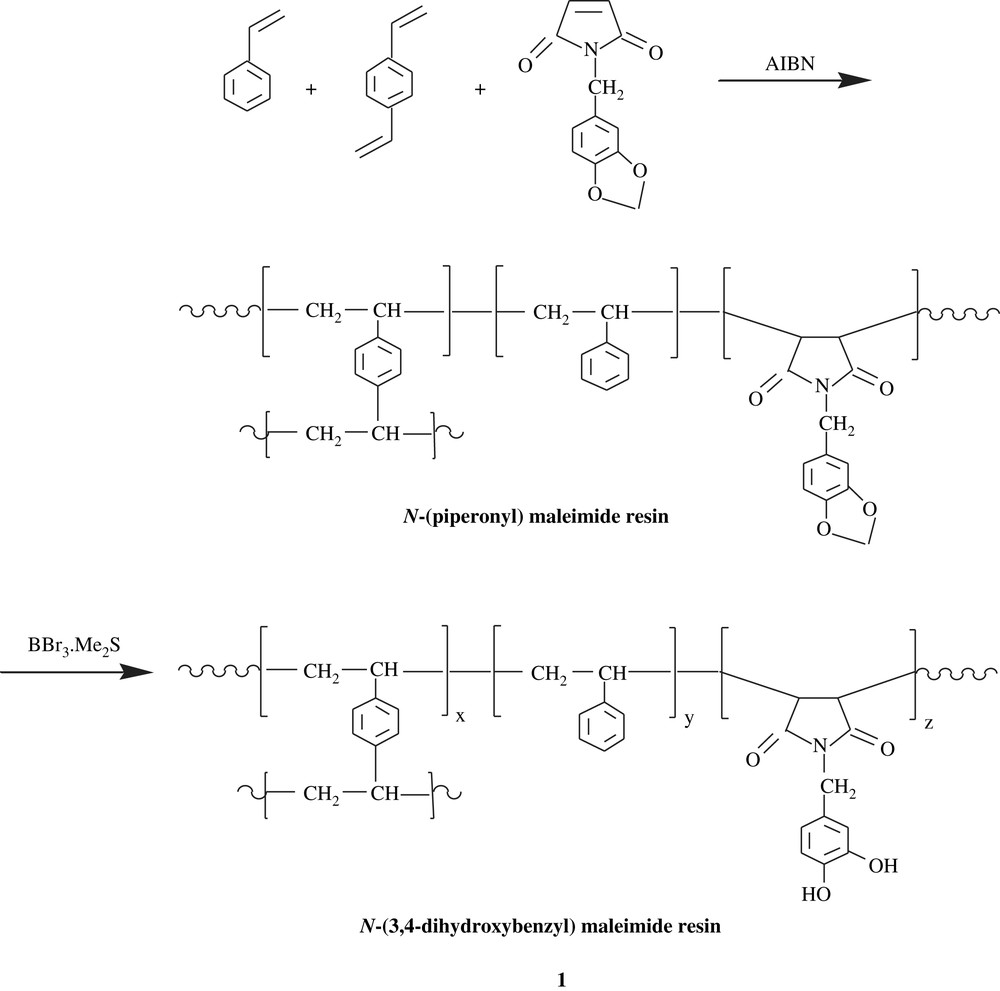
Sherrington et al. [15] prepared a catechol-containing terpolymer, a macroporous resin, via a radical route and a suspension technique, as traced in Scheme 2. The cleavage of formal linkage was effected by BCl3·Me2S to yield N-(3,4-dihydroxybenzyl) maleimide resin, 1. This material was employed as titanate-based catalyst support for transesterification and epoxidation reactions. The efficiency of these polymer-support catalysts was significant for both reactions studied.
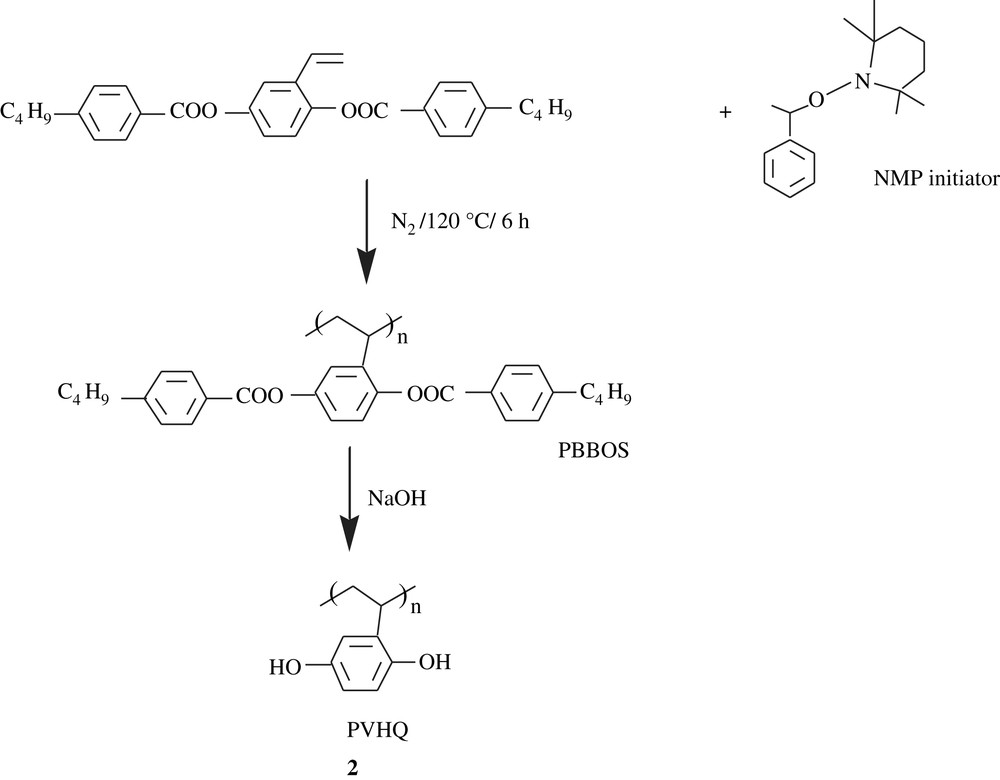
By means of stable free radical polymerization (SFRP), a living/controlling radical polymerization (LCRP), Ober et al. [16,17] were able to make poly(vinylhydroquinone) (PVHQ), 2. The strategy was first to conceive the macromonomer by blocking the hydroxyl function of vinylhydroquinone with 4-n-butylbenzoyl group, followed by reaction with NMP initiator (Scheme 3). A molecular weight of the obtained poly(2,5-bis[(4-butylbenzoyl)oxy]styrene) (PBBOS), of as high as 108 000 and a polydispersity index (PDI) of as low as 1.06, respectively, were determined. Alkaline saponification of PBBOS induced the removal of the protecting group to afford PVHQ.
On the one hand, PBBOS exhibited thermotropic liquid crystallinity, having mesogenic units affixed on the polymer backbone. On the other hand, the electrochemical properties of the deblocked PBBOS were investigated by cyclovoltammetry (CV) in aqueous acetic acid. It was found that the voltammogram (Fig. 1) showed a single redox couple centered at + 290 mV versus Ag/Ag+ electrode.

Cyclic voltammograms of PVHQ (2) modified GC electrode in a 0.10 M acetic acid aqueous solution at various potential scan rates. Reprinted with permission from Chem. Mater., 2001, 13, 2928–2932. © 2001 American Chemical Society [17].
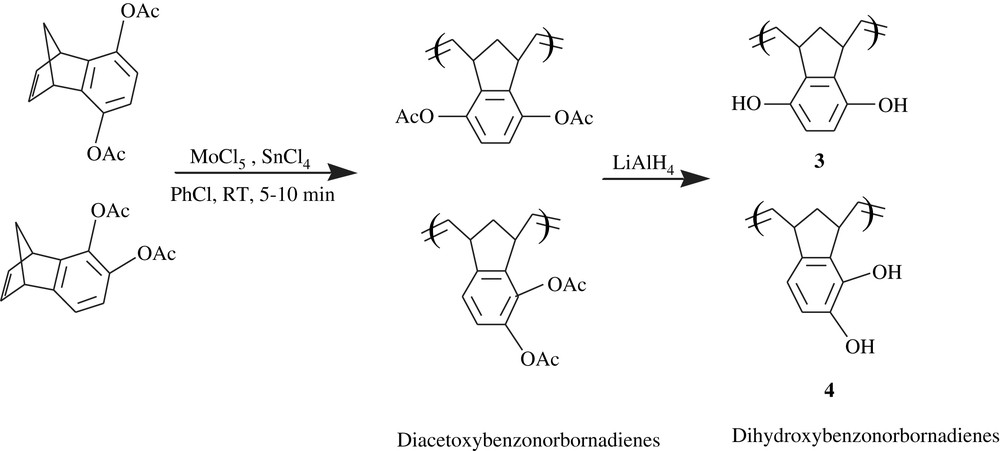
Chow et al. [18] applied the ring-opening metathesis polymerization method (ROMP) to the hydroquinone- and catechol-fused norbornadiene derivatives. The system MoCl5/SnCl4 induced the polymerization of p- and o-diacetoxybenzonorbornadiene in chlorobenzene within short times (5–10 min) (Scheme 4), giving the corresponding polymers in substantial yields (91–99%), with molecular weights ranging from 200 000 to 900 000. The polymers were featured by their good solubility in the common solvents. In this work, worthy of note is the dependence of the molecular weight on the polymerization time and dilution; a high dilution (10–30 mL) coupled with a long reaction time (1–2 h) yielded relatively lower molecular weights (3 × 104–5 × 104), whereas a fifth- to 15th-fold dilution coupled to a shorter time (5–10 min) afforded higher molecular weights in the range of 2 × 105–9 × 105. The removal of the blocking acetoxyl group was quantitatively provoked by LiAlH4 (93%) to give the corresponding dihydroxybenzonorbornadiene polymers, 3 and 4. The latter polymers could be realized in 90% yields by applying the same procedure to the naked dihydroxybenzonorbornadienes; these polymers were found insoluble in common solvents and sparingly soluble in DMF. The subsequent oxidation of these dihydroxy-norbornadienes was successfully accomplished with dichlorodicyanoquinone (DDQ).
Chow [19] also used the ROMP technique to polymerize some derivatives of 2,3-(1,4-dialkoxyaceno)norbornadiene employing a ruthenium carbene complex Cl2(PCy3)2Ru = CHPh as a catalyst to afford the corresponding polymers. These polymeric materials exhibited luminescence both in solution and in films, and were used successfully for the fabrication of light-emitting diodes (LED).
Ring-opening metathesis polymerization (ROMP) approach was successfully employed by Turner [20] in the conversion of tetraoctyloxy-substituted[2.2]para-cyclophanediene, using a second-generation Grubbs catalyst, into monodisperse and soluble p-phenylenevinylene homopolymer (PPV), 5, (Scheme 5). This polymer was faceted with a well-defined molecular weight and an alternating cis–trans microstructure. In fact, the molecular weight ranged from 7200 to 18 000 and the PDI 1.2, suggesting an overall monodispersity. As shown in Fig. 2, the optical absorptions (UV–visible, in dichloromethane) λmax were 482–490 nm; λmax increased with the polymer chain length, thus with a conjugation length. The photoluminescence emissions PLmax (Fig. 2) were all close to 556 nm and were, however, found to be independent of the conjugation length.
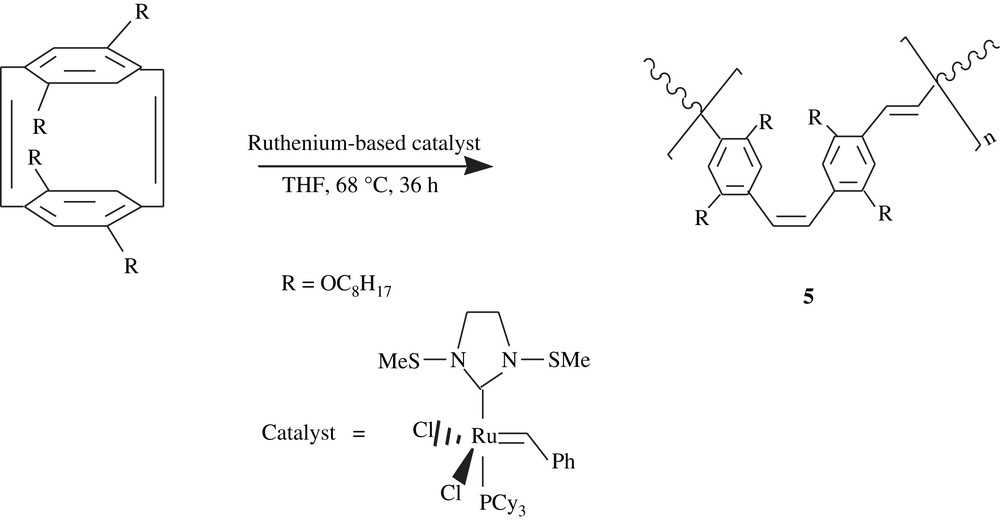

UV–visible (left) and photoluminescence PL (right) spectra for 5. C.-Y. Yu and M.L. Turner, Soluble poly(p-phenylenevinylene)s through ring-opening metathesis polymerization, Angew Chem., Int Ed, 2006, 45, 7797–7800. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
3 Condensation and coupling polymers
Yamamoto and Kimura [21] synthesized poly(2,5-dihydroxy-p-phenylene) or poly(p-hydroquinone) (PPHQ), 6, by dehalogenative organometallic polycondensation of 1,4-diacetoxy-2,5-dibromobenzene using a zerovalent nickel complex Ni(0)Lm as a polymerization promoter (Scheme 6). The deacetoxylation of the polymer by treatment with lithium aluminum hydride followed by treatment with HCl gave PPHQ with a low molecular weight, 8500. The latter polymer had a limited solubility: soluble in DMF, partially soluble in DMSO, and insoluble in acetonitrile, chloroform, toluene, and THF. Surprisingly, they were also insoluble in methanol and water. The electrochemical behavior of 6 was examined by CV in DMF; the voltammograms exhibited two oxidation peaks at 0.4 and 0.7 V, and two reduction peaks at −0.8 and −1.4 V versus Ag/Ag+ electrode. Also, it was observed that the electrochemical behavior of PPHQ strongly depended on the state at which the measurement was taken, i.e., solution or solid state. The pH effected the oxidation potential: the lower the pH, the higher the oxidation potential.
The same author, Yamamoto, made poly(p-phenyleneethynylene) type polymer, 7, by polycondensation between 4,7-di(4-bromophenyl)-2,1,3-benzothiadiazole and 1,4-didodecyloxy-2,5-diethynylbenzene using Pd(PPh3)4 and CuI as a catalyst system (Scheme 7) [22]; the polymer architecture consists of an electron-accepting unit, the 2,1,3-benzothiadiazole, and an electron-donating group, the 1,4-didodecyloxy-2,5-diethynylbenzene. The polymer was soluble in chloroform, THF, and toluene, and its molecular weight was around 40 000. The polymer showed photoluminescence in solutions and in the solid state: PLmax = 548 nm (CHCl3 solution), 560 nm (spin-coated film), and 559 nm (cast film). This material possessed an electrochemical property; the cyclovoltammogram showed two peaks at −1.78 and −2.15 V versus Ag/Ag+ electrode, and the color of the experimented film changes from yellow to green.

Moulay and Mzyène [23,24] took advantage of the side reaction of the chloromethylation reaction of 1,2- and 1,4-dimethoxybenzenes, affording poly(3,4-dimethoxy-o-tolylene), 8, and poly(2,5-dimethoxy-p-tolylene), 9, respectively, via a series of Friedel–Crafts reaction in a stepwise fashion (Scheme 8). Their intrinsic viscosities [η] were very low, 0.034 and 0.019 dL g−1, hinting at low molecular weights as with polybenzyls and poly(p-xylylene)s. Poly(3,4-dihydroxy-o-tolylene) and poly(2,5-dihydroxy-p-tolylene) were produced by treating the corresponding dimethoxylated polymers with 48% HBr in acetic acid. These dihydroxylated resins were insoluble in common organic solvents but soluble in methanol. Their redox activity was ascertained by potentiometric titration with ceric ammonium nitrate (CAN); their mid-potentials Em's were found to be 1017 and 1080 mV at 25 °C, respectively.
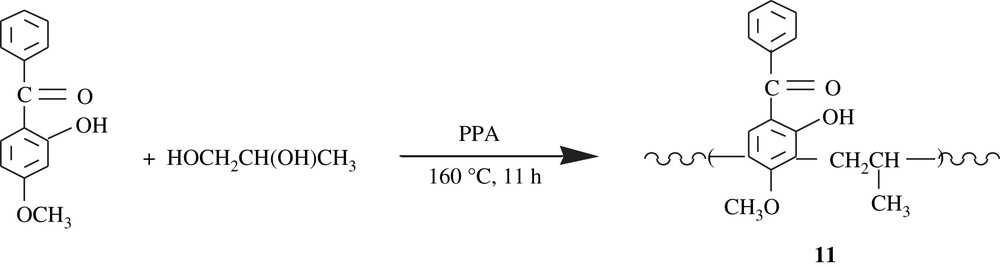
Poly[(2,4-dihydroxybenzophenone)1,4-butylene], 10,poly[(2-hydroxy-4-methoxybenzophenone)1,4-butylene], and poly[(2-hydroxy-4-methoxybenzophenone) propylene], 11, three polychelates and ion-exchange resins, were developed by polycondensation route [25–27]. The reactions involved are illustrated in Schemes 9 and 10. The molecular weights of these polymers were also low, 1236 and 1095 g/mol, respectively. They were successfully tested for the chelation of transition metals, particularly the lanthanides. Worthy of note is that the chelation occurs via one of the hydroxyl groups and the carbonyl group of benzophenone.
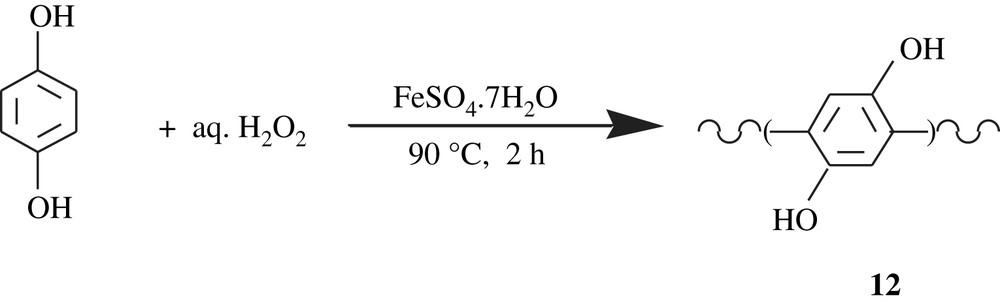
Zagorsky and Toropov [28] patented a work on the synthesis of poly(1,4-dihydroxyphenylene), poly(hydroquinone), 12. The synthesis involved the reaction of hydroquinone with aqueous hydrogen peroxide in the presence of hydrated salts of Fe(II), Mn(II), Co(II), Cr(II), Ni(II), Zn(II), Cu(II). An example of such a reaction is outlined in Scheme 11. The poly(hydroquinones) obtained were soluble in water, DMF, DMSO, poorly soluble in acetone, and insoluble in diethyl ether and ethanol. Their molecular weights did not exceed 1665 Da. The hydroquinone moiety present in the structure was claimed to reduce the toxicity of polyphenylene and promote its antihypoxic, antioxidative, and antiradical properties.
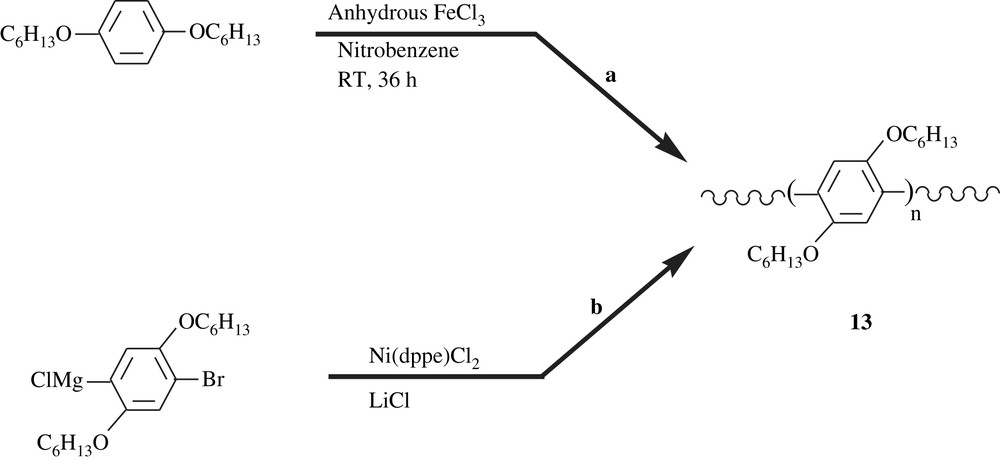
Poly(para-phenylene) (PPP) and poly(para-phenylenevinylene) (PPV) and their derivatives have attracted the attention of many researchers for more than a decade, because they exhibit good electrical conductivity and electroluminescence properties, and thus, their potential applications in light-emitting devices, biosensors, non-linear optics, and others. Various synthetic routes to dialkoxybenzene-bearing PPVs were described in the literature [13,14], including Wittig reaction, Horner reaction, Gilch reaction, Heck coupling, Suzuki coupling, and ROMP metathesis as described in Schemes 4 and 5. Results of the ongoing research have revealed that the dialkoxyl substituents of the phenyl group of PPP and PPV led to an increase in the electric properties of the materials, the conductivity and the luminescence. Herein, for a matter of conciseness, a few of these materials are outlined to shed light on the importance of the dialkoxybenzene-containing polymers. Agarwal et al. [29] have recently reported a facile synthesis of poly(2,5-dihexyloxyl-1,4-phenylene), 13, a dialkoxylated PPP, involving the FeCl3-catalyzed polycondensation of 1,4-dihexyloxybenzene at room temperature as illustrated in Scheme 12a. The dihexyloxylated PPP obtained was featured with a molecular weight of 37 360, a polydispersity index of 1.34, and a good solubility in a spectrum of organic solvents (toluene, chloroform, THF). CdS nanoparticles were admixed with this polymer and the composite thus fashioned has shown enhanced conducting and thermal properties. On the other hand, Yokozawa et al. [30] realized the synthesis of the same dihexyloxylated PPP but via a catalyst-transfer polycondensation, a method well-applied for polythiophenes. The latter authors employed Ni(dppe)Cl2 in the presence of equimolar LiCl to induce polymerization of the 1-bromo-4-chloromagnesio-2,5-dihexyloxybenzene, affording polymer with a molecular weight of 30 000 and a polymolecularity of 1.17–1.27 (Scheme 12b).
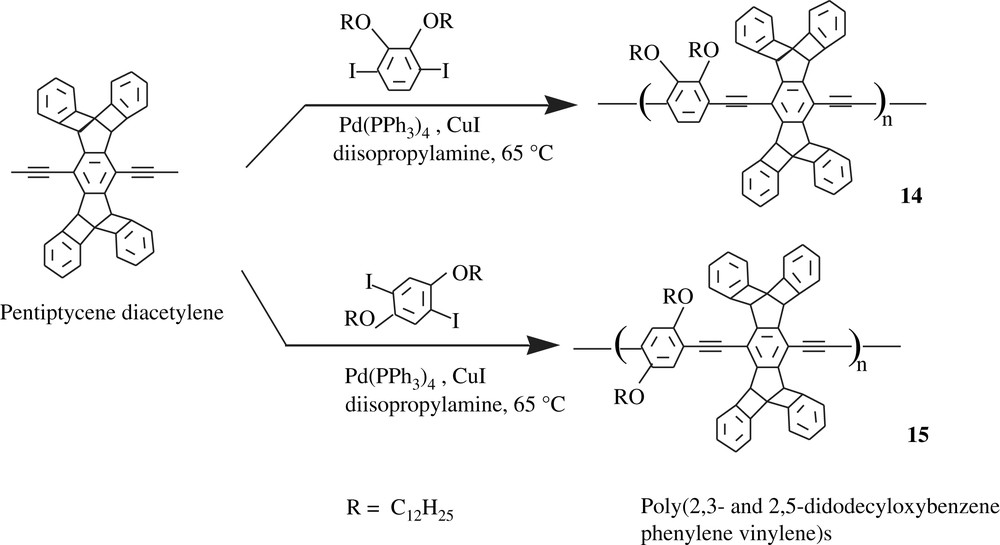
Swager and Zhu [31] synthesized some exotic dialkoxbenzene-bearing poly(phenylenevinylene)s (PPVs) (14 and 15) and poly(phenylene ethynylene)s (PPEs) (16 and 17) that possessed light absorbing and emitting characteristics. The synthetic procedure recalled the Sonogashira conditions in the coupling process as traced in Schemes 13 and 14. In Scheme 14, the coupling polymerization of the pentiptycene diacetylene with 1,4-diiodo-2,3-dodecyloxybenzene occurred in the presence of the Pd(PPh3)4/CuI catalyst system, giving the corresponding polymers of molecular weights of 72 kDa and in yields of 65–75%.

A modification of the synthetic protocol of PPEs was applied for preparing the didodecyloxybenzene-containing PPVs; the vinyldioxaborolane was involved in the coupling reaction with the above-mentioned diiodobenzenes, resulting in polymers in 71–79% yields and of molecular weights of 12 kDa [31]. This work revealed the neat effect of the ortho- and para-positions of the didodecyloxyl group on the light absorption and fluorescence. Indeed, the 2,3-didodecyloxybenzene-bearing PPVs and PPEs showed lower absorption and emission wavelengths than the 2,5-isomers, that is, a shift to a blue light which the authors attributed to an electronic origin. These observations were almost identical in both solutions and thin films.
Ozawa and his collaborators [32,33] developed stereocontrolled synthesis of dialkoxybenzene-containing PPVs with (E)- and (Z)-vinylene units, that is, the all-trans-PPV, and the all-cis-PPV. By means of Hiyama cross-coupling and by the use of Ru(HCl(CO)PPh3)3 as catalyst (Scheme 15), the all-trans-PPV, 18, was obtained as an orange powder in a quantitative yield (99%). Its average molecular weight was in the range of 5700 and 6300 with a molecular weight distribution of 1.40–2.30. It was highly soluble in THF, toluene, chloroform, and insoluble in methanol and hexane.
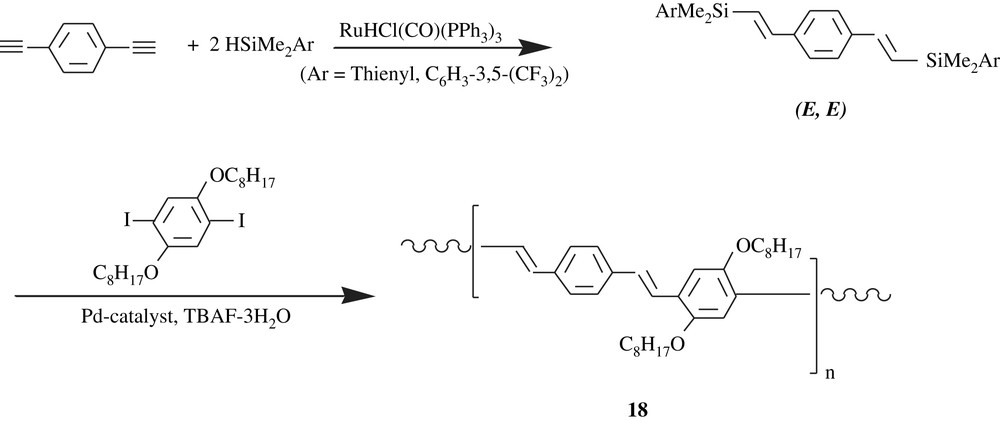
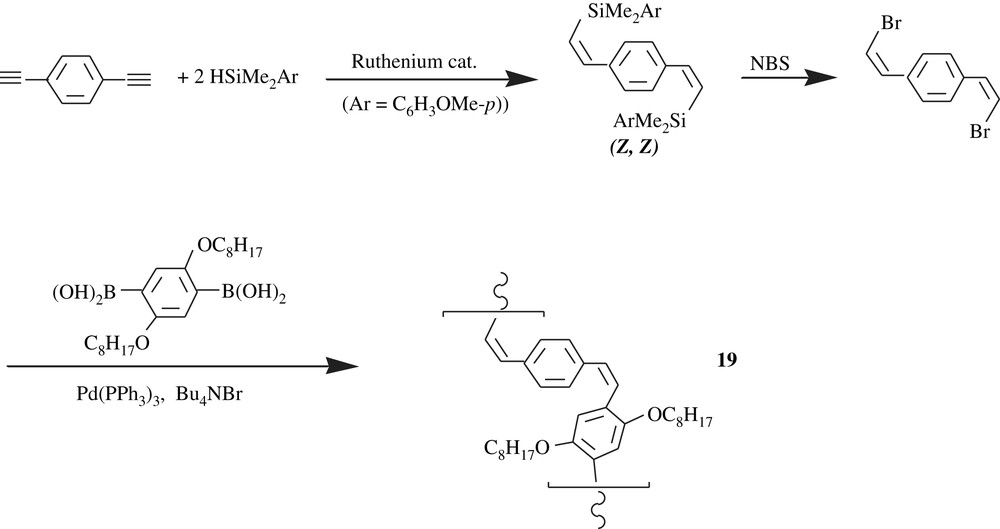
The all-cis-PPV, 19, however, was made through a Suzuki–Miyaura-type polycondensation as illustrated in Scheme 16. The product was an orange solid and was obtained in a high yield, 95%, with a molecular weight of 4700 and a PDI of 1.92. The peculiar facet of the all-cis-PPV was its ready photoisomerization into the all-trans-PPV.
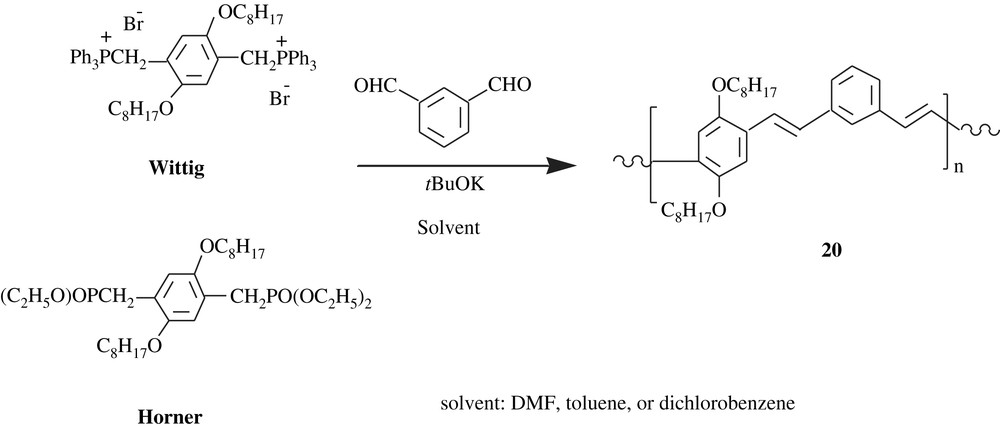
Drury et al. [34] prepared poly(m-phenylenevinylene-co-2,5-dioctyloxy-p-phenylenevinylene), PmVP (20) via two pathways (Scheme 17): one involving a Horner phosphonate ester and the other a Wittig phosphonium bromide. Interestingly, the properties of the produced polymeric material depended not only on the synthetic route, but also on the solvent employed. The average weight molar mass, , of the PmVP was in the range of 8600–46 000 with a PDI of 1.3–3.5. The transition temperature and the melting point of the polymer were 9.5–50 and 86–120 °C, respectively. While the Wittig route yielded PmVP with a low cis-olefin content (14–20%), the Horner pathway afforded the polymer with a high trans-olefin content (99%). Optical properties of the polymer were found to be related to the mode of preparation. There was a blue-shift for the polymer made via Wittig route, which was attributed to the cis-content present in the Wittig product. The optical absorptions (UV–visible in DMF) λmax were 320 and 391 nm for the cis-rich polymer and 330 and 405 nm for the trans-rich one. Their corresponding photoluminescence maxima PLmax in DMF were 449, 482, and 520 nm for the former polymer and 454, 479, and 520 nm for the latter one.
Earlier, Pang et al. [35] also applied the Wittig and the Horner synthetic methods to make a soluble poly(m-phenylenevinylene-alt-2,5-dibutoxy-p-phenylenevinylene), PmVP. The Wittig procedure provided a cis-rich polymer (75%), while the Horner one gave a trans-rich polymer (88%), in a yield of about 60%. The molecular weights of the polymers and their PDIs were 10 000 and 11 600, and 2.5 and 2.1, respectively. The products were yellow solids and soluble in THF, chloroform, and toluene. Their optical absorptions (UV–visible) λmax were 404 and 385 nm for the cis-rich PmVP and the trans-rich one, respectively, hinting at a slightly longer conjugation in the former. However, the emission maxima λem were found nearly identical and equal to 445 and 471 nm.
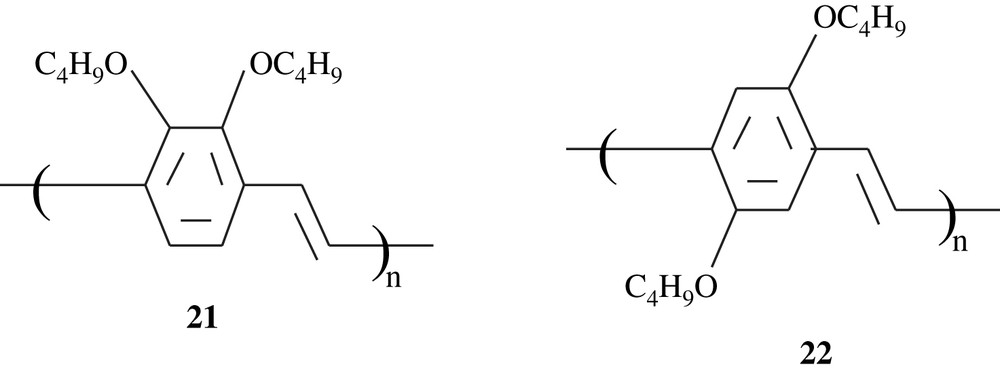
Holmes et al. [36,37] prepared poly(2,3-dibutoxy-1,4-phenylenevinylene), 21, and its copolymer with a silylated analogue monomer by means of a Gilch dehydrohalogenation polycondensation of the corresponding bishalomethyl-substituted benzene monomers. The polymer 21 was found to emit a blue-green light at a frequency higher (λmax = 454 nm, and PLmax = 519 nm) than that of 2,5-polymer analogue 22, and this finding was assigned to steric interactions between the two ortho-dibutoxy groups with the adjacent vinyl units. The photoluminescence (PL) efficiencies of the former polymer were nearly 40%. A double layer emitting device fabricated with the copolymer exhibited high electroluminescence efficiency.
Poly(2,5-methoxy-1,4-phenylenevinylene) grafted organophosphazene copolymers were made by Leung et al. [38]. The copolymers were readily soluble in common organic solvents and fluoresced in the blue color range. They were used to make a series of organic light-emitting diodes (OLED).
Hiroshige et al. [39] were able to make copolymers of PPP and dialkoxylated PPP, 23, via sulfonium salt polymers as depicted in Scheme 18. The pyrolysis of the slightly green sulfonium salts led to reddish materials, which were insoluble in all solvents. However, and because of a solubility matter, it was possible to determine the average molecular weights of only the sulfonium salt precursors, which were found to be in the range of 8.0 × 102–6.5 × 106 amu with a polydispersity index of 1.5. Upon doping with iodine, the reddish materials became black-colored and endowed with electrical conductivity. Their latter parameter reached values as high as 2.9 S cm−1, stating that the dialkoxyl groups would lower the ionization potential of the conjugated polymer and would therefore enhance its affinity to a dopant. The electrical conductivity of these copolymers was found to increase tremendously upon stretching; indeed, the stretching ratio of 4–6 implied a conductivity range of 180–354 S cm−1.
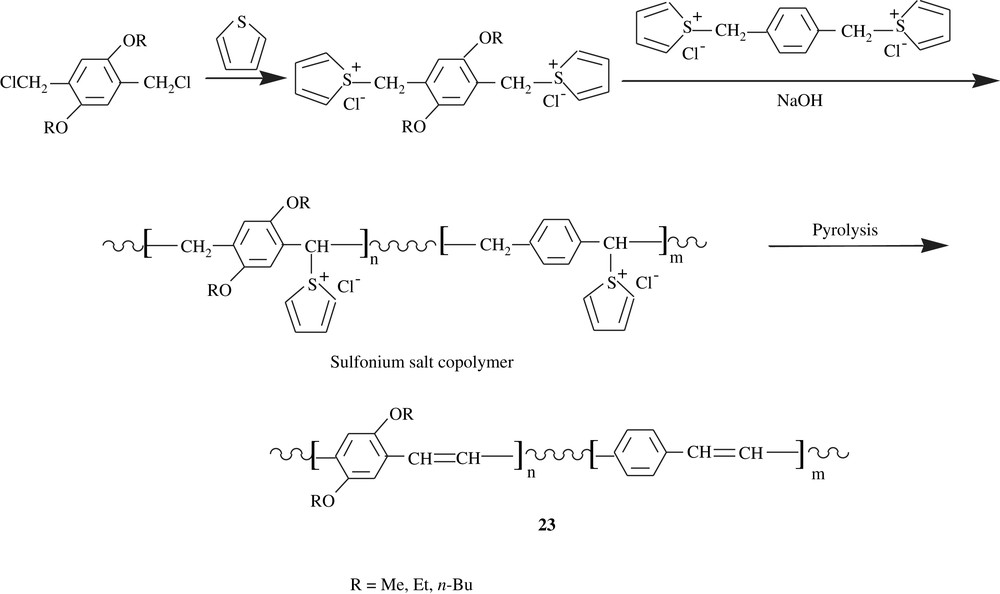
Copolymers of 2,5-dialkoxybenzenes and 3,3′-dialkoxy-2,2′-bithiophenes, 24, were prepared via a polycondensation by means of Stille coupling either of distannylated dialkoxybenzenes with dihalogenated dialkoxybithiophenes or of distannylated dialkoxybithiophenes with dihalogenated dialkoxybenzenes (Scheme 19) [40]. These different monomers were made through multistep syntheses. The polymers were obtained in low to moderate yields, 16–56%, and were of moderate molecular weights, . Their glass transition temperatures were in the range of 184–210 °C, and they decomposed beyond 264 °C. Study of their chiro-optical properties revealed that the matrices were of higher conjugation lengths, accounting for their fluorescence, electroluminescence, and electrical conductivity properties.
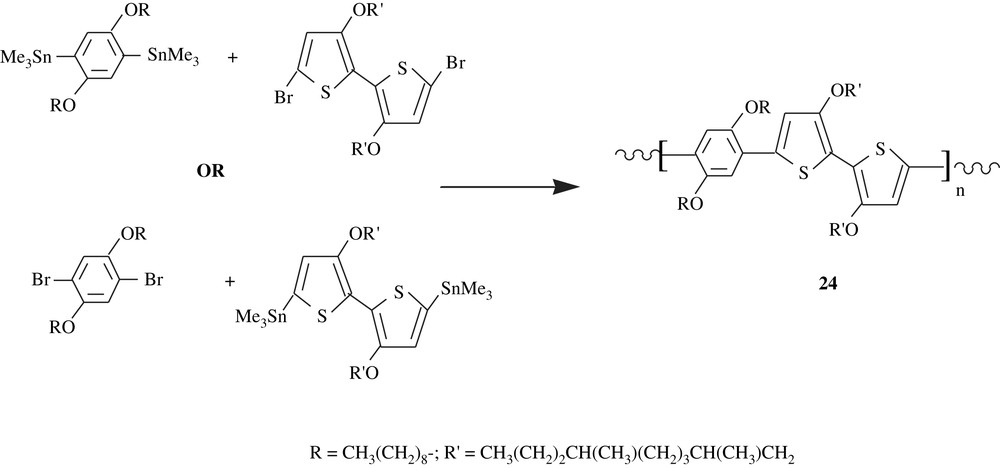
Thompson et al. [41] employed the Sonogashira coupling to prepare, via a multistep synthesis, the polymeric substance C60-OPE/OPV, 25, consisting of didecyloxybenzene and styryl units end-capped with [60] fullerene groups. Both redox and fluorescence properties of this material were assessed. Cyclovoltammetry study showed that only one quasi-reversible wave appeared at the midpotential E1/2 = −1.10 V versus Ag/Ag+ electrode. The results of the fluorescence measurements emphasized the significance of the electronic interactions between the cruciform π-oligomer and C60. Overall, the observed phenomena may make these fullerene-oligomer species potential candidates for advanced optoelectronic devices. The styryl units present in the polymer matrix would certainly have played a major role in inducing the optoelectronic properties of the material as Schubert et al. [42] reported for DO–ST–PPE2–PPV2, 26, a dioctyloxybenzene and distyryl-bearing material that was made via Sonogashira coupling. The distyryl groups acted as electron-donors to the main chain conjugation system and formed a separate bis(styryl)phenylene chromophore system.
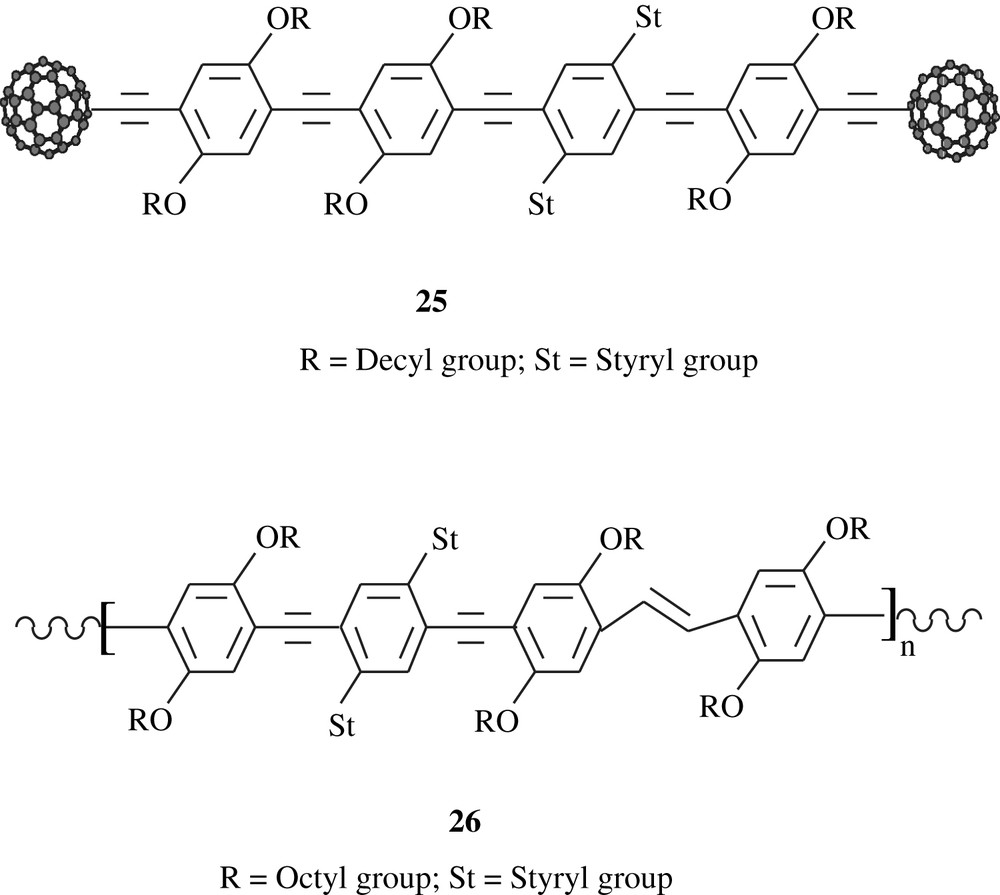
Gross and his coworkers [43] disclosed the preparation of plastic solar cells from C60-oligophenylene–ethynylene material, C60-OPE. The material showed some activity in a photovoltaic device. Cravino [44] recently reviewed the tethered-fullerene conjugated polymers, including the dialkoxybenzene-containing ones, and their (opto) electronic and photovoltaic properties were highlighted. These properties can be promoted by a material having electron-donor and electron-acceptor; fullerene derivatives stand as the most electron-acceptor materials. It is interesting that the alkoxyl groups contributed to the solubility of the fullerene units. It is strongly believed that the conversion of solar energy into electric one by these materials is very promising.
Ramakrishnan and his coworkers [45,46] were able to make an emitting-light material precursor, featured by a tunable two-color patterning (Scheme 20). The material precursor, MEHPPVD, was readily soluble in solvents such as THF and toluene, which facilitated its spin-casting. As can be noted, the substance bears dithiocarbamate (DTC) and methoxy groups on the α carbons, which were prone to elimination, affording MEHPPV. Once the polymer was treated with triphenylsulfonium triflate, a photoacid generator, it was spin-cast, subsequently irradiated by Hg-vapor lamp, and finally heated at 180 °C for 30 min. The results were that, for the acid generated regions, a complete elimination of DTC and methoxy groups yielded MEHPPV-100 (100% elimination, high conjugation, HC); MEHPPV-100, 27, emitted an orange-red color. However, for the regions of the material devoid of acid, a selective elimination of DTC only gave MEHPPV-x, 28 (low conjugation, LC), which emitted a green color, and this color shifted to red upon increasing the elimination.
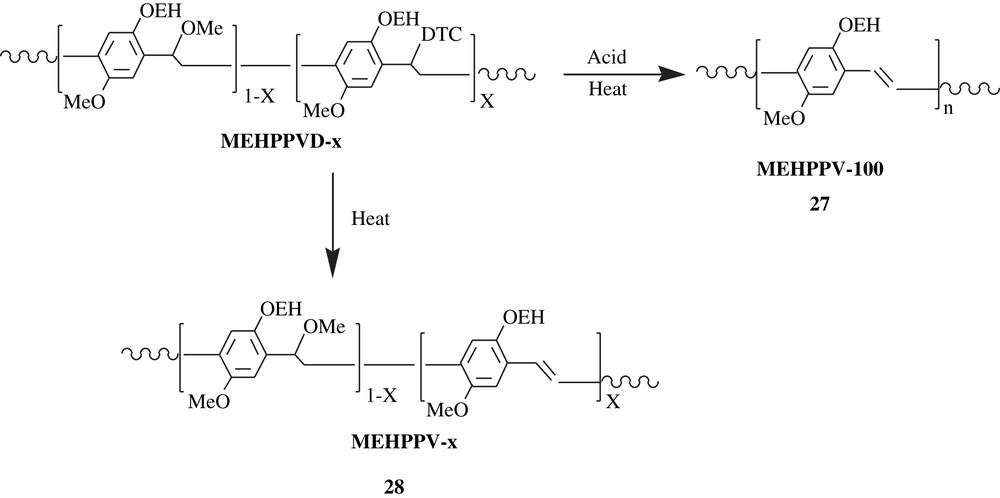
Similar work on the π-conjugated polymers of MEHPPV-type was reported, stressing the role of π-stacking on solution and solid-state luminescent properties [47,48]; the use of suitable bulky substituents has shown to tune the emission property of these materials in solution as well as in the solid state.
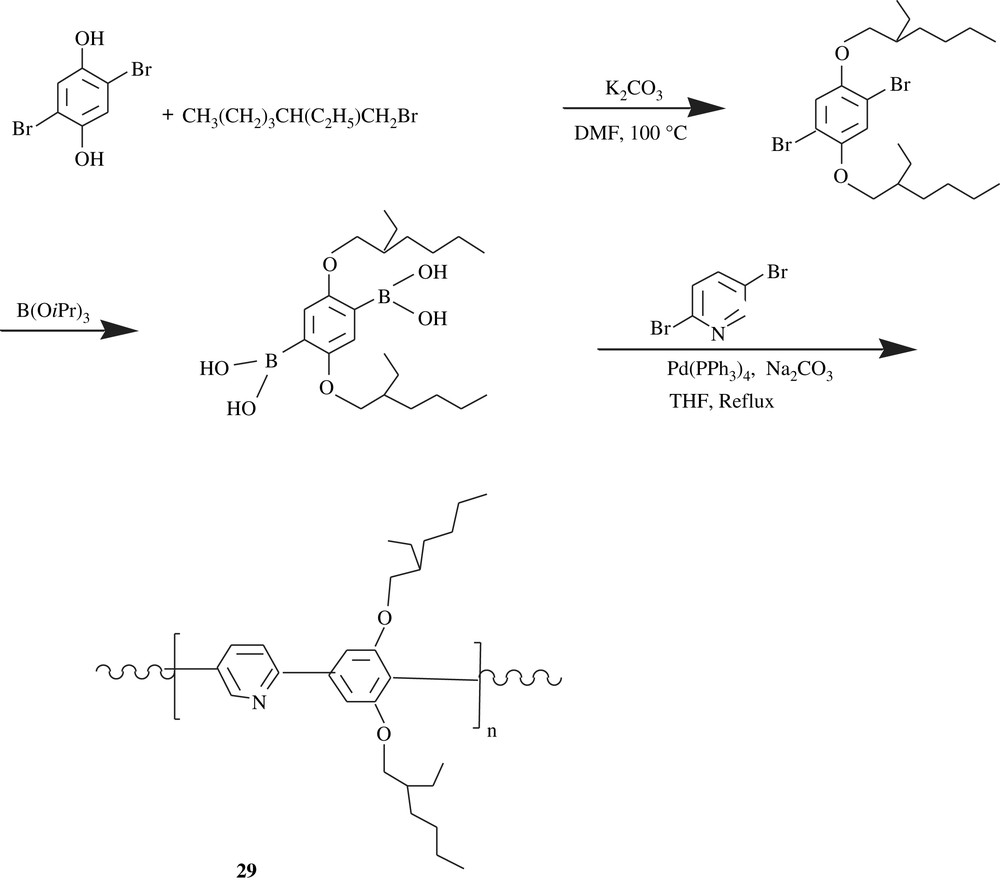
Special dialkoxybenzene-carrying polyphenylenes are those inserting pyridine moieties within their backbones. Indeed, the pyridine-bearing conjugated polymers are highly luminescent. To cite but one example is the poly(2,5-pyridinylene-co-1,4-(2,5-bis(ethylhexyloxy)phenylene)), 29, which was realized according to Scheme 21 [49]. What is fascinating about this polymer was the effect of the protonation of lone pair of pyridine unit by strong acid on the fluorescence and electroluminescence emissions; the protonation caused a red shift emission. Furthermore, a planarization phenomenon of the repeating unit of the polymer could have been arisen from the intramolecular hydrogen bonding (the protonated nitrogen of the pyridine moiety) to the adjacent oxygen of the solubilizing 2-ethylhexyloxy chain, and this planarization would have extended the conjugation and thus tuned the emission profiles.
One of the beautiful and exotic molecules that chemistry has brought forth in the last two decades is dendrimers. In fact, their reality has enriched the polymeric materials field and, as a result, new chemistry has been effectively founded. Yamakawa et al. [50,51] prepared a dialkoxy/dihydroxybenzene-containing dendrimer by means of a divergent method, one of the two techniques for making dendrimers. As traced in Scheme 22, the 2,8,14,20-tetrakis (3,5-dihydroxyphenyl)calix[4]resorcinarene was made by a one-step reaction involving resorcinol and 3,5-dihydroxybenzaldehyde in water; this calix[4]resorcinarene was conceived as a special core molecule for building the first-generation dendrimer, by treating it first with K2CO3/18-crown-6, then with 3,5-bis(allyloxy)benzyl bromide as a dendron. The molecular weight of this first-generation dendrimer was 2997 with a polydispersity index of 1.05. This dendrimer was soluble in DMSO and DEE. Cleavage of the allyloxy groups or deallylation was effected by a reduction using bis(triphenylphosphine) dichloride (Ph3P)2PdCl2 and ammonium formate HCO2NH4 in THF, to afford the corresponding dihydroxy compound for a second-generation process, 30. This strategy was applied for making the third, fourth, and fifth generations. An interesting remark was that the diallyloxy dendrimers were viscous oils, whereas the corresponding dihydroxylated ones were powdery. The poly(aryl ether) Fréchet-type dendrimers thus formed functioned as negative-working and alkaline-developable photoresists.

Lee and his coworkers [52] applied the convergent pathway to produce dendrimers made of the 1,3-dialkoxybenzene-made periphery and of trans-1,2-bis(4-pyridyl)ethylene as a photo-responsible and redox-active core.
4 Electrosynthesis polymers
Electrochemistry has gained a foothold in the realm of organic synthesis, even on the industrial scale. Indeed, numerous organic compounds have been electrochemically prepared. As far as the polymers are concerned, adiponitrile, a monomer precursor for adipic acid employed in polyamide making, has been industrially manufactured by means of electrosynthesis for years. Nowadays, the electrosynthesis is being extended to polymer synthesis. Öktem and his coworkers [53] reported the electrochemical polymerization of 4-ally1-1,2-dimethoxybenzene which cannot be polymerized by a free radical route due to the reactivity of allylic hydrogens (Scheme 23). The reaction was realized in acetonitrile/Bu4NBF4 (TBAFB) and yielded two polymers: an insoluble polymeric film on the electrode surface and a soluble polymer on the anolyte. Spectral characterization of the soluble one, 31, revealed two monomeric units, suggesting that the second unit stemmed from an isomerization of the initial monomer: isomerization of 4-allyl-1,2-dimethoxybenzene to 1,2-dimethoxy-4-propenylbenzene. Besides, the presence of amino group in the polymer structure may hint at the interaction of acetonitrile, the reaction medium, with in situ generated carbonium ion. The molar mass of the soluble polymer was found to be 1350, a range of values for allyl polymers. However, the work has not been continued to the demethylation of the products, ensuing the polyallylcatechols.
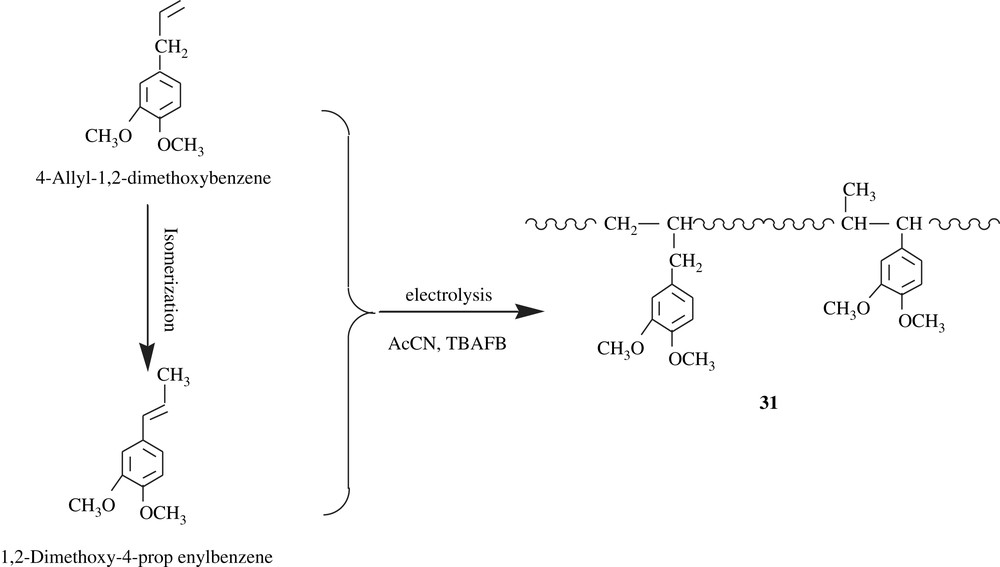
Polyarylenedioxybenzenes, PMDOB (32) and PEDOB (33), were electrochemically synthesized (Scheme 24) by Liu et al. [54] in order to valorize them as conducting polymers. Indeed, they possess a polyacetylenic chain, which is responsible for their conducting properties. Their electrical conductivities were measured and found to be 0.1 and 0.17 S cm−1, respectively. They were soluble in polar solvents such as DMSO, and insoluble in common organic solvents such as THF, DCM, chloroform, and DMF. Unfortunately, neither their average molecular weights were determined nor the cleavage of the alkylenedioxyl group was performed to yield the corresponding dihydroxylated compounds.
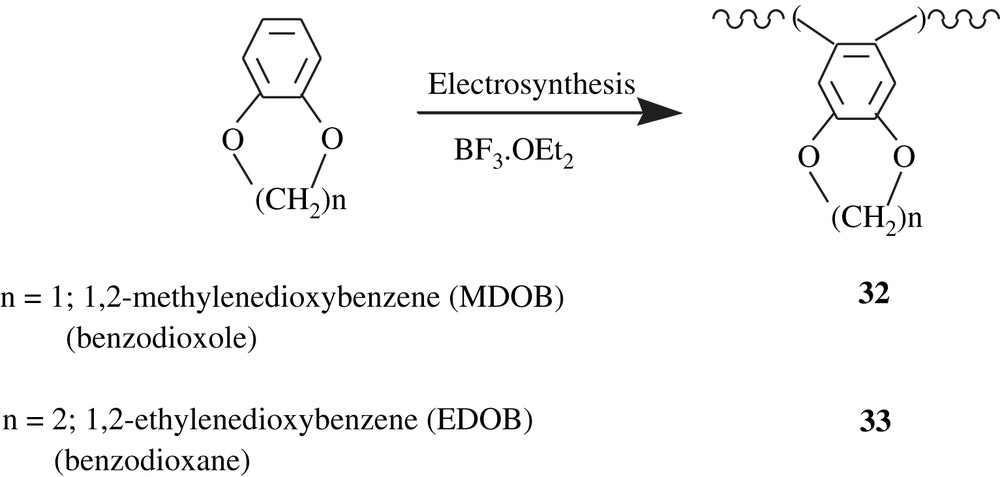
Using acetonitrile/TBAFB system, Kalaji et al. [55] electrochemically polymerized 1,3-dimethoxybenzene (1,3-DMB) into poly(1,3-dimethoxybenzene), poly(1,3-DMB) (34) (Scheme 25). The latter was insoluble in common organic solvents and, thus, its molecular weight could not have been measured. However, the thoroughly undertaken analyses by IR and solid-state 13C NMR enabled to propose the 1,3,4,6-tetrasubstituted structure as shown in the scheme. The polymer was thermally stable up to 400 °C, and has an electrical conductivity of 2.5 × 10−4 S cm−1, a value for semi-conductors.
Earlier, the anodic oxidation of 1,4-dimethoxybenzene, performed in acetonitrile, gave a substituted poly(para-phenylene) with a conductivity of 3.5 × 10−4 S cm−1 [56]. However, the electrochemically synthesized olymers of 1-methoxy-4-ethoxybenzene (PMEP), 1-methoxy-4-n-hexoxybenzene (PMHP) and 1-methoxy-4-[2-(1-adamantyl)-2-oxo-ethoxy]benzene (PMAP), whose molecular weights were in the range of 2200–2900, and, with the exception of PMEP, were soluble in organic solvents [57]. Their conductivities were in the spectrum of 2 × 10−5–2 × 10−2 S cm−1.
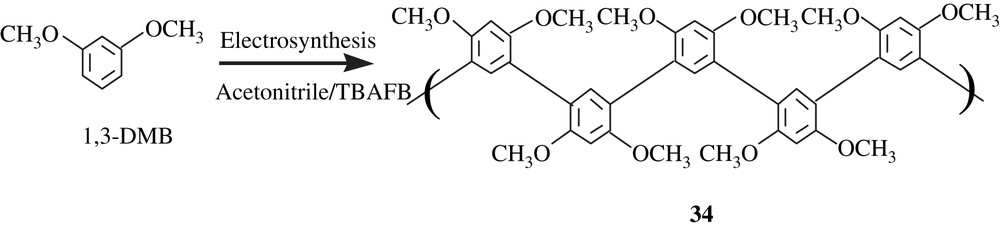
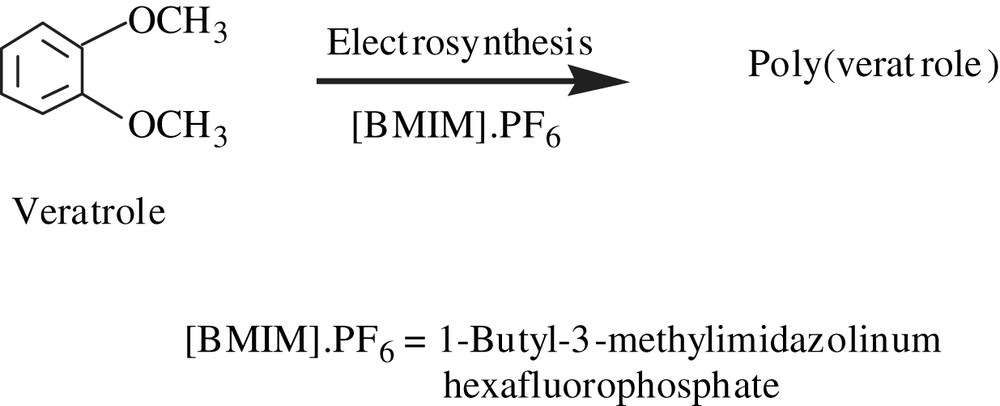
Jouikov and his coworkers [58] reported the formation of polyveratrole through an oxidative self-coupling mechanism when veratrole (1,2-dimethoxybenzene) was submitted to electrolysis in ionic liquids (ILs). In ionic liquids such as those of imidazolinium ([BMIM]·PF6, [BMIM]·BF4, [BMIM]·NTf2), the poly(veratrole) obtained was distinguished by its deep blue color that lasted for several months with no degradation (Scheme 26). Yet, neither the structure of the polymer nor its molecular weight and its solubility have been discussed. However, the polymer was isolated either in an amorphous form or a fibrous morphology, depending on the ionic liquid employed.
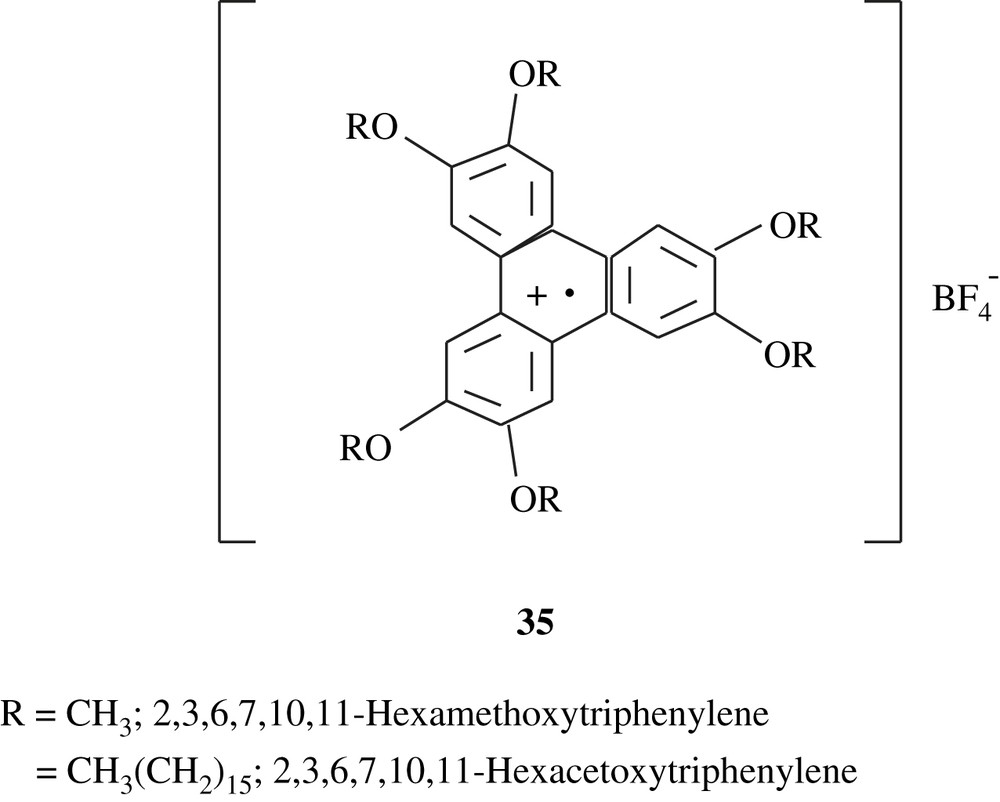
However, the electropolymerization of veratrole in dichloromethane and in the presence of TBAFB gave a green polymeric conducting solid [59]; the structure suggested for this material is shown below (35), reflecting the presence of the phenylene radical cation. Under the same electrosynthesis conditions, Ortiz et al. [60] reported the production of the polymeric substance, 2,3,6,7,10,11-hexaacetoxytriphenylene, from its corresponding monomer which was prepared by a Williamson reaction. The polymeric compound obtained was also green, soluble in acetonitrile, insoluble in dichloromethane, and electrochemically behaved in the same fashion as the one stemming from veratrole.
5 Attachment polymers
One fascinating and revolutionary alternative to reach a material with structure and/or properties that cannot be realized by direct means such as the homopolymerization, the blending of polymers, the copolymerization, the interpenetration of polymers (IPN), or others, is the chemical transformation of an appropriate polymer. The functionalization of a polymer via an attachment of specific molecular units usually ends up with useful polymeric materials.
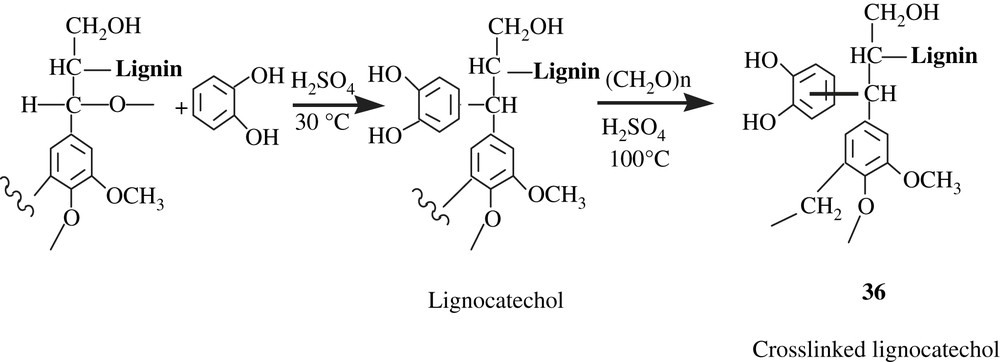
Inoue and his collaborators [61] successfully immobilized the catechol molecule onto a lignin matrix by the Funaoka's phase separation technique, according to the reaction Scheme 27. The lignocatechol obtained underwent a crosslinking by reaction with formaldehyde, affording the gel 36 that was employed for the adsorption of heavy metals (Al(III), Cd(II), Co(II), Fe(III), La(III), Ni(II), Pb(II), Zn(II)). Also, a chromatographic separation of metal ion pair (Pb/Zn) using a lignocatechol gel-packed column was performed; the results showed a neat separation as Pb(II) was selectively adsorbed.
Moulay and Mehdaoui [62,63] successfully applied the conditions of Minisci to poly(acrylic acid) (PAA) and poly(methacrylic acid) (PMA), where the tethered moieties were hydroquinone and catechol as shown in Scheme 28. A homolytic mechanism is involved in such a chemical attachment, quinones (Q) being radical scavengers. The extents of functionalization were 35% and 52%, respectively. The thus-modified polyacrylics (37), PAA-HQ and PMA-HQ, underwent a severe degradation. The redox properties of these materials were estimated by potentiometric titration; the mid-potentials, Em's, were 836 and 898 mV at 20 °C, respectively. Interesting was the fact that the two stage-oxidations were observed at 35 °C for both polymers (Fig. 3), suggesting two-oxidation processes and involving semi-quinone intermediates. The mid-potentials of the polymers were higher than those of monomeric analogues, catechol and hydroquinone. This study stressed the effect of the methyl group on both the degree of substitution and the redox property of the polyacrylic.


Plots of potentiometric titration at 35 °C with 0.05 M ceric ammonium nitrate in 90% acetic acid: A, 1:1 hydroquinone/catechol; B, 37 with R = H; C, 37 with R = Me.
It is worth mentioning the polymer effect that manifested during the potentiometric titration process. While stable values of redox potentials E for hydroquinone/catechol could be read within times shorter than 5 min, a significant delay in the establishment of those for polymeric analogues 37 was noted; times as long as 30–60 min were required to attain an equilibrium for the polymers.
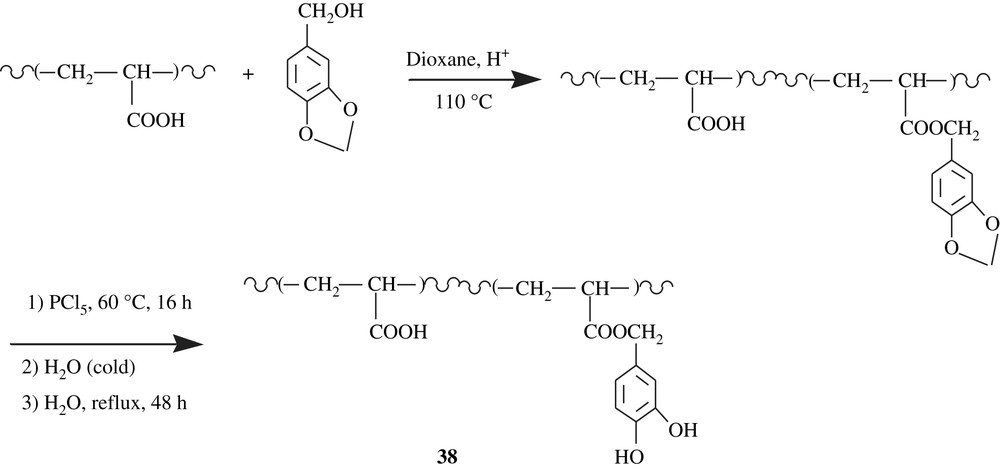
On the other hand, poly(acrylic acid) was functionalized with a piperonyl group through an esterification as depicted in Scheme 29 [64]. An optimal extent of substitution of 72% was attained. The deblocking process was realized with PCl5 followed by hydrolysis, and the spectral characterization clearly showed a quantitative deprotection, leading to catechol-containing poly(acrylic acid), 38. This polychelate was tested for Cu(II) and Cd(II) and the results showed a better chelation for Cd(II).
Also, a pH-metric study was undertaken and the results indicated that the acidity of catechol-containing PAA, 38, was lower than that of PAA. The titration curves in Fig. 4 show that the pHs of 38 were slightly lower than those of PAA. A general and interesting observation was that the pHs were more acidic in the presence of a metallic ion. For example, the pHs for PAA and 38 matched at 12–12.5 for volumes of titrant ranging from 5 to 12 mL, and for the same titrant volumes these pHs decreased to 7.5 and 5–5.5 in the presence of Cd(II) and Cu(II), respectively. This would suggest that the metallic adsorption enabled the dissociation of the hydroxyl groups of both the catechol and carboxylic acid units, making the materials more acidic. The chelation of copper by these materials may have afforded complexes more stable than those with cadmium; that is, the copper-complexed materials became more acidic than those with cadmium.

Plots of the potentiometric titrations, pH = f(VNaOH), with 0.1 N NaOH.
Cellulose was also chosen as an immobilizing support for pyrocatechol through an –NH–CH2–CH2–NH–SO2–C6H4–NN– linker, resulting in a macromolecular chelator 39 (Scheme 30) [65]. Quantitative chelation (98.0–99.4%) for Cu(II), Zn(II), Fe(III), Ni(II), Co(II), Cd(II), and Pb(II) was pH-dependent, that is, in the range of 4–7. The sorption capacity of the catechol-containing cellulose for these heavy metals was 85.3 and 186.2 mmol/g. This resin was also tested for river and tap waters and the results were promising. The degree of modification of the starting cellulosic material by pyrocatechol was lacking.
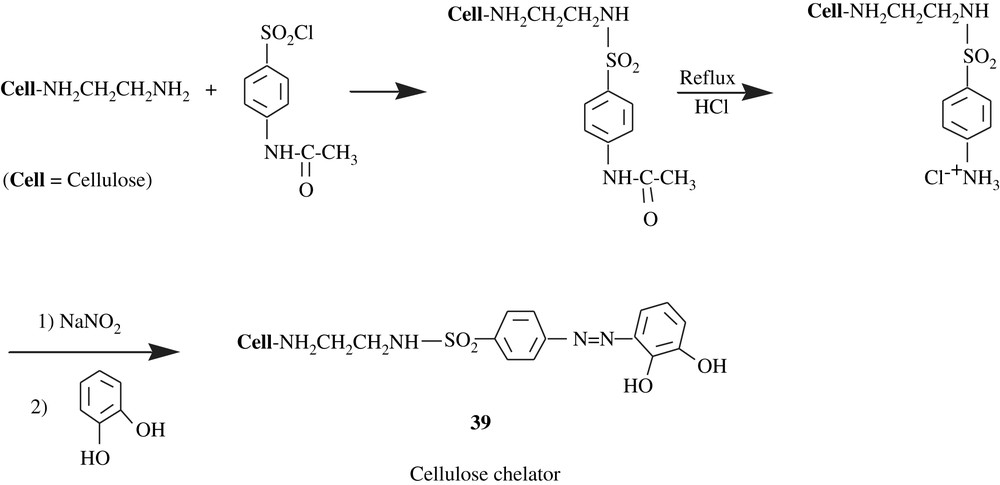
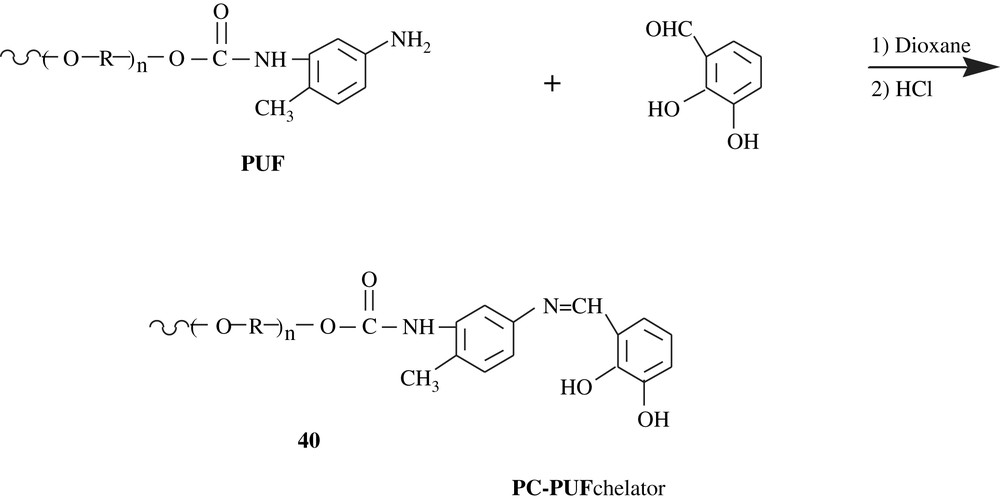
Lemos et al. [66] functionalized polyurethane foam by pyrocatechol for their use in online pre-concentration system for cobalt, nickel, and copper in food samples (spinach, rice flour, corn flour and black tea). Flame atomic absorption spectrometry (FAAS) was employed to determine the levels of these metals. The modification reaction involved the reaction of the aldehyde function of 2,3-dihydroxybenzaldehyde with the amino group of the terminal toluidine unit of the polyurethane chain 40 (Scheme 31).
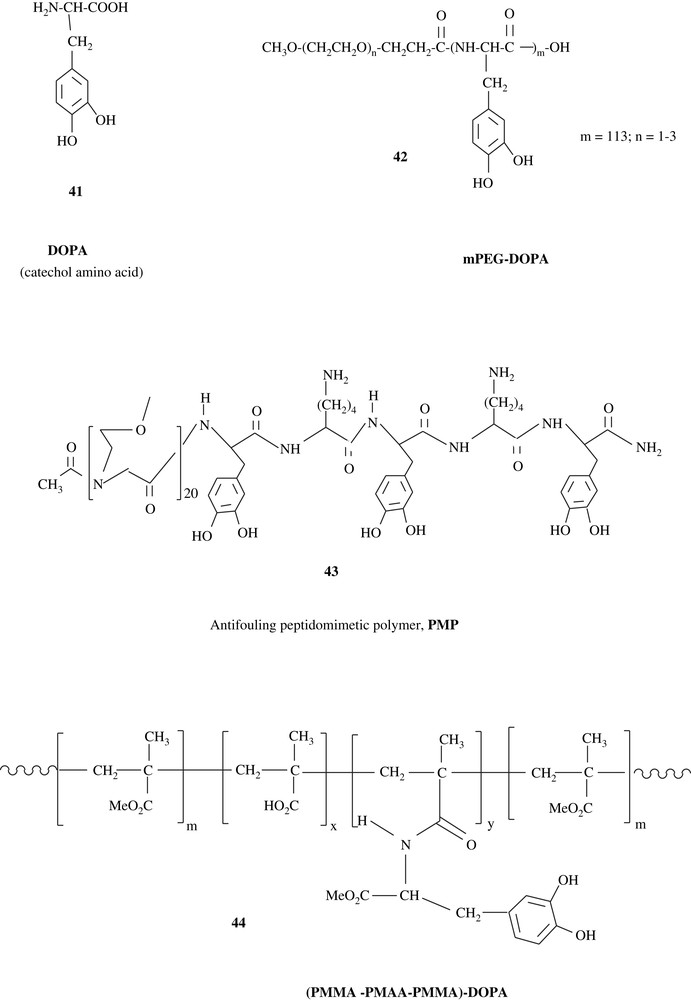
Intensive research is taking place on the synthesis of mussel adhesive proteins (MAP), for the prevention of mussels, geckos, and barnacles from attaching to surfaces such as ship hulls and piers, and for the use in medical implants and diagnostics [12,67–70]. Owing to its adhesive properties, 3,4-dihydroxyphenyl-l-alanine (l-DOPA) (41) was linked to pre-made synthetic polymers such as polyethylene glycol (PEG), affording antifouling substances, which make the coated surfaces very resistant to cell adherence. The antifouling polymers mPEG–DOPA (42) and PMP (43), which were built up by a solid-state synthesis pathway, are illustrative. DOPA units of the materials were believed to form strong charge–transfer complexes to metal oxide surfaces. Indeed, 43 showed a potential capacity of maintaining fouling resistance for several months. Furthermore, l-DOPA was anchored on poly(methyl methacrylate)–poly(methacrylic acid)–poly(methyl methacrylate) (PMMA–PMAA–PMMA) triblock copolymers 44 [67]. The hydrogel prepared from this triblock showed strong adhesive properties towards TiO2 surfaces.
Wilker et al. [71] modeled the bioadhesives, which marine mussels and barnacles deposit on surfaces, by making a copolymer of styrene and 3,4-dihydroxystyrene. The styrenic units of the copolymer mimicked the protein part of bioadhesive and the 3,4-dihydroxystyrene (a catechol functionality) ones acted as DOPA. In addition, the authors claimed that poly (styrene-co-3,4-dimethoxystyrene)s, the precursors of the poly(styrene-co-3,4-dihydroxystyrene)s, also displayed adhesive properties. These precursors were made in 83–96% yield by anionic polymerization (addition polymerization) resulting in molar masses of 9500–14 600. Their Tg's were intermediate between that of polystyrene and that of poly(3,4-dimethoxystyrene), that is, 106.3 and 53.1 °C, respectively. The demethylated copolymers were then crosslinked using ions such as Fe3+, IO4−, and Cr2O72− before the adhesive test.
6 Enzymolysis polymers
Nowadays, enzymes are not only confined to the biosynthesis but are also extended to in vitro work. The enzymatic synthesis of polymers is an integral facet of the worldwide ongoing research [72–75]. For example, lipase was employed to produce polyesters [76,77]. Best of all, the enzymatic polymerization has become an efficient alternative to treat the wastewater from aromatic compounds such as phenol and its derivatives, catechol, hydroquinone, and the dihydroxybenzene-containing compounds [78]. Indeed, the enzymes able to induce this type of polymerization are the following: laccase, lipase, peroxidase, bilirubin oxidase, tyrosinase, soybean peroxidase (SBP) and horseradish peroxidase (HRP). Moreover, a sound account on the phenolic polymers syntheses by enzymatic routes has been elegantly penned by Ritter [79].
The synthesis of poly(quinones) via chemical and electrochemical transformations of hydroquinones has proved unsuited owing to the formation of a large amount of by-products and to the complex polymer structures. However, enzymatic syntheses were found to yield poly(hydroquinones), thus poly(quinones), with definite structures and good solubility. For example, Dordick et al. [80] were able to make poly(hydroquinone) through a multi-enzymic pathway as depicted in Scheme 32. The first step involved the transfer of β-d-glucose to hydroquinone, a reaction catalyzed by β-glucuronidase to afford arbutin. Then, the polymerization of the latter compound by HRP/H2O2 gave poly(arbutin), a water-soluble phenol polymer with a molecular weight of 1600–3200, which, upon deglycosylation by an acidic hydrolysis, is converted into poly(hydroquinone) 45. This poly(hydroquinone) was indeed soluble in polar solvents such as DMF, DMSO, THF, MeOH, and acetone.

Laurinavicius et al. [81], however, polymerized arbutin by laccase affording polyarbutin with a degree of polymerization ranging from 23 to 29. The partially hydrolyzed polyarbutin was used as a mediator for the oxidation of the quinone-dependent glucose dehydrogenase. Sensor activity of this polymeric material was assessed by cyclovoltammetry. A carbon electrode coated with this polymer responded positively without use of a reagent, indicating that the polymer was acting as a mediator and as an immobilizing matrix.
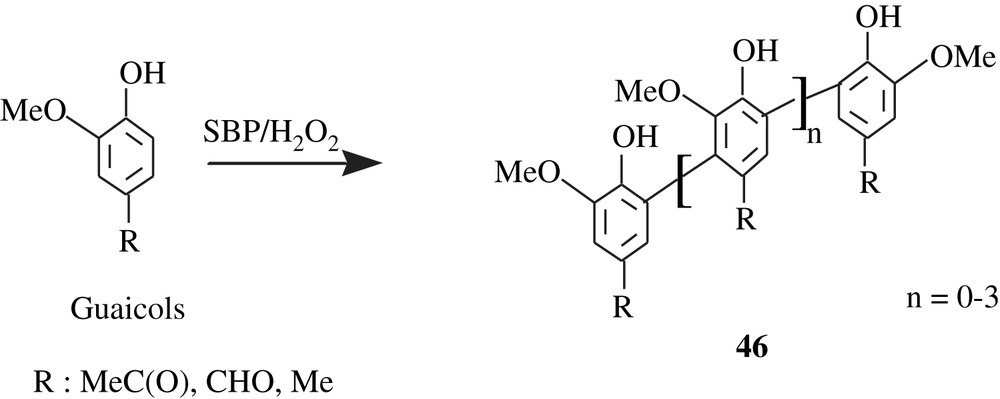
Soybean peroxidase (SBP) was employed to catalyze the oligomerization of some guaiacols (o-methoxyphenols) leading to the products 46 (Scheme 33) [82].
Kobayashi et al. [83,84] polymerized syringic acid with HRP/H2O2 to provide a polyphenylene oxide derivative (PPO), 47, with a molecular weight of 15 000 (Scheme 34). It is worth recalling that PPOs have been commonly made via an oxidative coupling using a copper/amine catalyst system [85]. In this reaction, a mechanism has been proposed involving a decarboxylation followed by a coupling. The structure of the polymer, exclusively of 1,4-oxyphenylene unit with a carboxylic acid and a phenolic hydroxyl group as the chain termini, was ascertained by IR, 1H and 13C NMR spectroscopies and by the MALDI-TOF mass spectrometry.

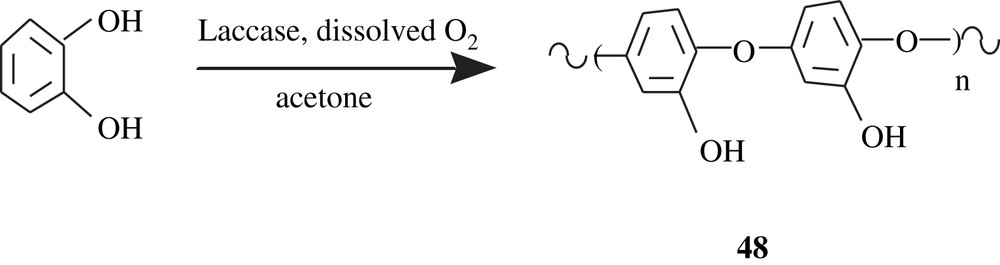
Tanyolaç et al. [86–88] disclosed the results of their work on the laccase polymerization of catechol, affording poly(catechol) 48 with a defied water-solubility; its molecular weight was not given but, based on its FTIR spectrum, its structure was elucidated, as shown in Scheme 35.
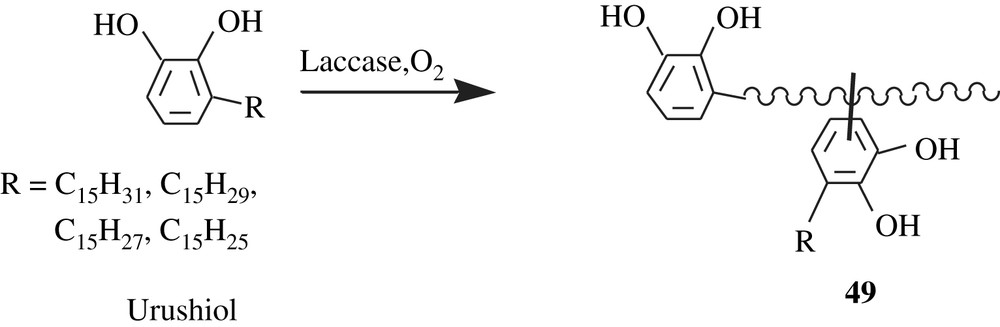
Urushiols, laccols, and thitsiol are natural catechol-bearing molecules found in saps that are exuded from the far-east Asian countries' trees: Rhus vernicefera, Rhus succedanea, and Melanorrhoea usitata, respectively. These molecules are components of oriental or Japan lacquers that were employed in ancient coatings. The composition of each of these components is a mixture of alkyl- and alkenylcatechols; a triunsaturated R group is mostly predominant. A feature of these molecules is that they can undergo polymerization via a laccase-catalyzed oxidative coupling, affording glossy films [89]. One of the possible structures of the polymer obtained is 49 depicted in Scheme 36; this structure was the result of a coupling dimerization, stemming from a Friedel–Crafts alkylation of one urushiol molecule with a generated carbocation within the R group of another urushiol molecule.
Urushiol still attracts researchers particularly for its coating performance. Interesting is that artificial urushiol has been successfully made by a multistep synthesis and results have elucidated a tight structure/property relation [90,91]. Recently, Hu et al. [92] reported the UV-induced polymerization of a naturally occurring urushiol in the absence of a photoinitiator. The film obtained was thoroughly characterized by FTIR and ESR, revealing the formation of o-quinone moiety within the structure and the generation of the phenoxy radical that is responsible for the film-forming. A feature of this study was that the UV-curing of urushiol followed a different mechanism (a free radical one) from the well-known laccase-catalyzed oxidation, therefore produced different films. In fact, the phenoxy radicals produced were believed to attack the unsaturations of the R to promote the curing process.
Mueller et al. [93] designed a combination of two different polymeric materials via an enzymatic pathway, for heart valve coating. Of the two kinds of polymers, poly(hydroquinone) and poly(resorcinol) were fashioned in the form of thin-film materials to assure adhesion to the heart valve. The second series of materials are of another chemical nature, and are able to hinder blood coagulation. Hyperbranched poly(resorcinol) and linear poly(hydroquinone) were prepared by polymerizing resorcinol and hydroquinone with horseradish peroxidase (HRP) in the presence of H2O2 or oxygen. The polymers' structures were ill-resolved and their yields were low; yet, the resorcinol and hydroquinone units in their corresponding could be linked via ether and carbon–carbon bonds, as that illustrated in Scheme 35.
7 Conclusion
This minireview should have thrown some light on the diversity of the dialkoxy/dihydroxybenzene-bearing polymers that were successfully and cleanly prepared, and fully characterized. Not only the synthetic pathways of these particular materials were various, but also their properties were peculiar and not limited. Modern synthetic approaches, coupled with the classical routes, have been applied to bring forth these valuable substances. Their displayed properties, described in this paper, are due in part to the presence of the dialkoxy/dihydroxybenzene units. Indubitably, the intensive ongoing research in different laboratories would come up with other dialkoxy/dihydroxybenzene-containing resins, which may be characterized by novel and interesting properties, and hence, new applications.