

1 Introduction
Ultrafast molecular photo-switching in the solid state [1] represents challenging issues for the scientific and technological purpose of controlling the macroscopic physical state of a material by an ultra-short laser pulse. The direct observation of such phenomena is key for understanding the mechanisms driving these out-of-equilibrium processes. Spin-crossover materials are acclaimed prototypes of photo-active molecular materials [2], offering various photoswitchable functions (magnetic, optical, dielectric…) for in-depth investigations. They are also of great interest for understanding, for example, the role of binding ligand in models of the heme protein [3,4]. In addition to magnetic, optical or structural analysis developed to understand short-lived, long-lived or photo-steady states [2,5–9], moving the field of molecular photo-magnetism towards the ultrafast time domain is an important issue awaiting material science. Investigations of such ultrafast molecular switching were performed in solution with optical [10,11] or XAS [12–14] techniques. Here we demonstrate, by using optical pump–probe techniques with 100 fs time resolution, that a femtosecond laser pulse applied to a catecholato-Fe(III) spin-crossover solid switches constituting molecules of the crystal to an out-of-equilibrium high-spin state on the 100 fs time-scale.
2 Material and experiment
Spin conversion is the prototype of a bistability and cooperative transformation in molecular solids [1,2,6–19]. For these molecular crystals, the electronic distribution around the metal ion switches under the effect of various stimuli (temperature, pressure, magnetic field or light irradiation) from low-spin (LS) to the high-spin (HS) state.
Strong interest in such materials comes from possible applications (bistable devices or data storage systems) and new potentialities for the development of efficient and easy to handle photosensitive devices. The possibility to trigger the spin-state of a complex by light irradiation is particularly attractive for investigating out-of-equilibrium processes and related mechanisms. The photoinduced spin conversion occurring under continuous or pulsed light irradiation is called the LIESST effect (Light-Induced Excited Spin-State Trapping), as the photo-excited spin-state can be stabilized through a structural relaxation [2]. In femtochemistry, investigations of the dynamics of such Fe(II) molecules in solution provided compelling picture of the coordination core structural reorganization as being associated with the short-lived photoinduced HS state [10–14].
However, studies of solid materials are of fundamental importance with regard to specific mechanisms, time-scales, efficiencies for energy conversion, electronic and atomic relaxation. Unlike in dilute solutions, cooperative response in spin-crossover solids occurring through elastic interactions between molecules [1,2] may result in advantageous effects.
Until now, investigations of the dynamics of the photoinduced spin-state switching in the solid state driven by nanosecond laser pulses have mainly focused on the relaxation processes from the metastable photoinduced HS state to the stable LS state [2,16–18]. Here we present a 100 fs time-resolved optical study of the ultrafast low-spin to high-spin photo-switching transformation process in the spin-crossover solid [(TPA)Fe(TCC)]PF6 (TPA = tris(2-pyridylmethyl)-amine and TCC = 3,4,5,6-tetrachlorocatecholate dianion).
[(TPA)Fe(TCC)]PF6 belongs to the family of Fe(III) molecular compounds switching from a LS state at low temperature (S = 1/2) to a HS state at high temperature (S = 5/2) [15]. There exist two polymorphs of this system: the first one is orthorhombic [15], whereas the second one is monoclinic [19]. The orthorhombic polymorph investigated here undergoes a spin-crossover centred around T1/2 = 210 K as observed in Fig. 1, which depicts the temperature dependence of χMT product (χM – molar magnetic susceptibility, T – temperature). Magnetic susceptibility measurements were carried out with a Quantum Design SQUID Magnetometer (Model MPMS5S) calibrated against a Palladium standard. Measurements were performed on a small collection of single crystals. The fraction of HS molecules XHS was estimated from the χMT product (χM = molar magnetic susceptibility) containing weighted contribution of molecules in HS and in LS state
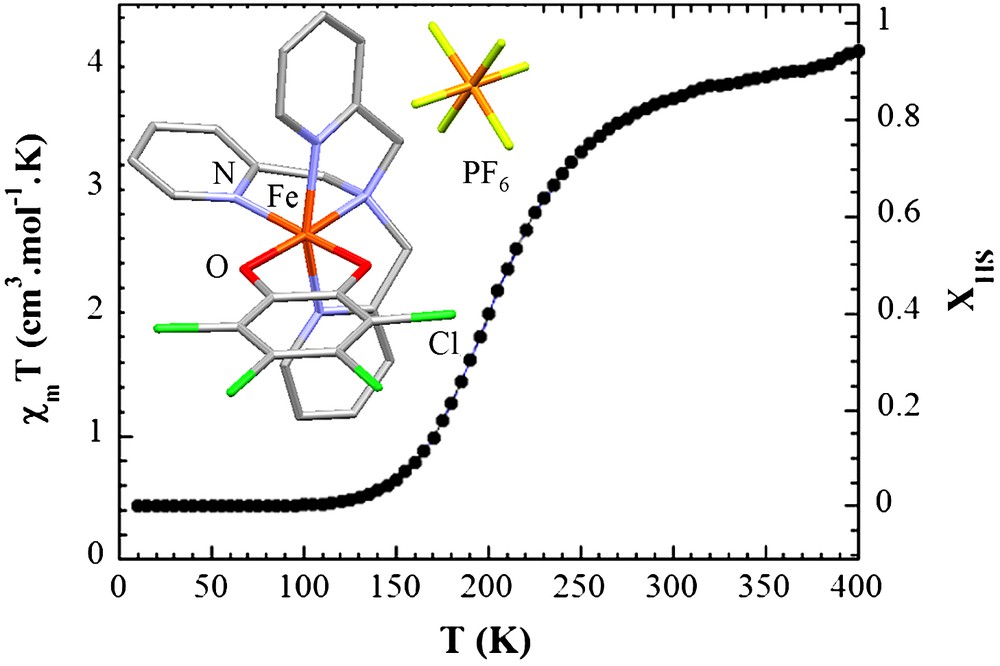
Temperature dependence of the magnetic susceptibility (χMT, left axis) associated with the variation of the fraction of HS molecules XHS (right axis). Inset: structure of the molecular complex.
It is well known that in this family of molecular materials, the change of spin (electronic) state is associated with significant variations of other physical properties, optical in particular. Indeed, many of these systems undergoing the LIESST effect are also thermo- and photo chromic [2]. Fig. 2 shows photographs of a 15 μm thick single crystals taken in transmission mode on a standard optical microscope Laborlux 12 pol from Leica equipped with CCD camera Sony Digital Hyper HAD SSC-DC38P. All of the optical experiments presented here were performed with light polarization parallel to the long crystal axis (crystallographic axis a in Ref. [15]), which yields maximum transmission. It is clear from these photographs that the transmission in the visible range is lower in the HS state as the crystal darkens. This is in agreement with Vis–NIR absorption data reported on a powder [16]. This change of optical properties is also well illustrated by the temperature dependence of the transmission shown in Fig. 2 for 480 nm (wavelength for which the transmission is maximum) and 600 nm, i.e. a region where different catechol-to-Fe(III) charge-transfer transitions take place. For clarity the transmission at both wavelengths was normalized to 1 at 100 K. The occurrence of significant variations correlates well with the average molecular switching from LS to HS. The transmission across VIS wavelengths is higher for LS [16] and it decreases with raising temperature, whereas the transmission change upon temperature increases with wavelength. The apparent colours are light blue and dark-purple blue for the LS and HS states, respectively. Therefore, optical properties can be used as indirect probes of the spin-state, especially on short and ultra-short time-scales, where measuring the magnetic signal is technically not feasible. It was demonstrated this way that near-infrared excitation of this orthorhombic solid in the LS state with ns laser pulses generates a transient HS state, with a lifetime in the ms range at temperatures close to T1/2 [16]. Because ns laser pulses forbid access to ultra-short time resolutions and consequently elementary transformation processes, we set up an ultrafast optical pump–probe experiment to track the transient response of the material down to 100 fs with excitation at 800 nm.
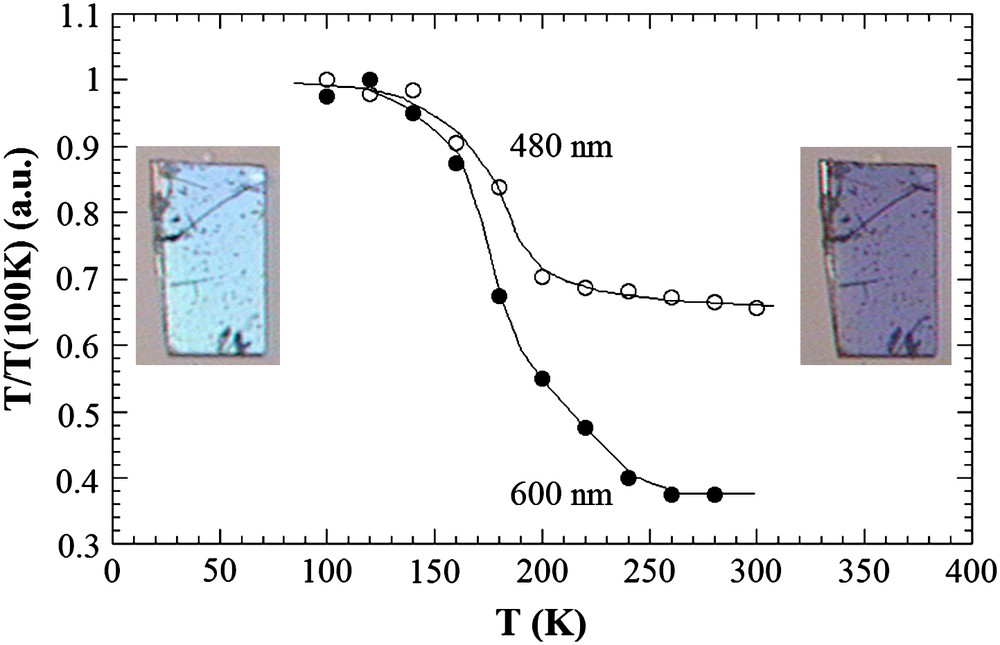
Temperature dependence of the optical transmission (normalized to the values at 100 K) around the spin-crossover, plotted at 480 nm (○) and 600 nm (•). Insets show photographs of the single crystals of the orthorhombic polymorph observed with a microscope in transmission geometry (vertical polarization) in the LS state (left at 80 K) and HS state (right 350 K).
3 Hundred femtosecond pump–probe experiments
The basic principle of pump–probe experiments is to trigger a transformation by an ultra-short laser pulse (pump) and interrogate the dynamics by using a second delayed pulse (probe). The out-of-equilibrium evolution can be reconstructed by taking several “snapshots” at different delays. The experiment is run stroboscopically to build up statistics.
The laser (Fig. 3) generates 50 fs pulses at 800 nm, amplified to 2.5 mJ at 1 kHz. These are separated into twin pulses seeding two optical parametric amplifiers (OPA), tunable in the range between 400 and 2000 nm. We control the delay between pump and probe by varying the optical pathlength with a delay line, the accuracy of which is better than 1 μm (it takes 1 ps to a photon to travel 300 μm). Therefore the time resolution (70 fs for zero intersection angle between pump and probe) is limited by the spatio-temporal convolution of the two pulses and not by the accuracy of the mechanical delay stage. The measurements of the variations in probe pulse intensity, either through transmission or through reflectivity, are performed using a lock-in amplifier, which significantly enhances the detection sensitivity through band-width narrowing in frequency domain.
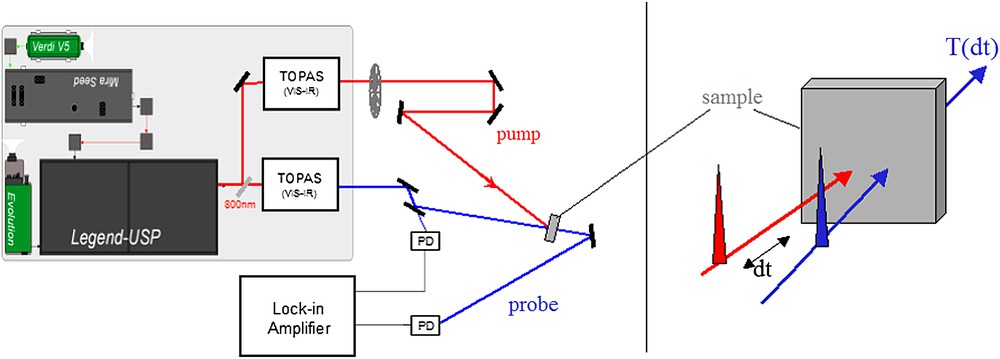
Optical pump–probe experimental set-up. The femtosecond pulses generated by the Mira laser are amplified with the Legend regenerative amplifier. The beam splits into two: pump (red pass) and probe (blue pass). The TOPAS allow selecting the wavelength of the pump and probe beams, which arrive with a relative delay dt on the sample. The sample is probed in transmission geometry. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4 Results and discussion
In order to investigate the ultrafast stage of the photoinduced process evidenced with microsecond time resolution [16], we performed femtosecond pump–probe experiments. A 15 μm thick single crystal of [(TPA)Fe(TCC)]PF6 similar to the one presented in Fig. 2 was excited by a femtosecond flash at 800 nm of 40 μJ/mm2 excitation density. The corresponding ratio between the number of photons in the laser pulse to the number of molecules in the excited volume is roughly 1 photon/300 molecules, as the penetration depth reaches 8 μm at 800 nm. We probed the transition at two wavelengths identified as sensitive to the thermal switching: 480 and 600 nm (pump and probe polarizations were parallel to the long crystal axis a). Experiments were performed at 160 K, where the best compromise is struck between the relaxation rate allowing stroboscopic experiment at 0.5 kHz and a high enough fraction of photo-triggered molecules from the LS state (Fig. 1). The time dependence of transmission following 100 fs laser irradiation is shown in Fig. 4a and b for probing wavelengths at 480 and 600 nm, respectively. On the picosecond time-scale, a decrease of transmission is observed between negative (before) and positive (after laser irradiation) delays. The decrease of transmission is higher at 600 than at 480 nm, as observed for the thermal conversion (Fig. 2), and it provides a clear fingerprint of the ultrafast photogeneration of the HS molecular states.
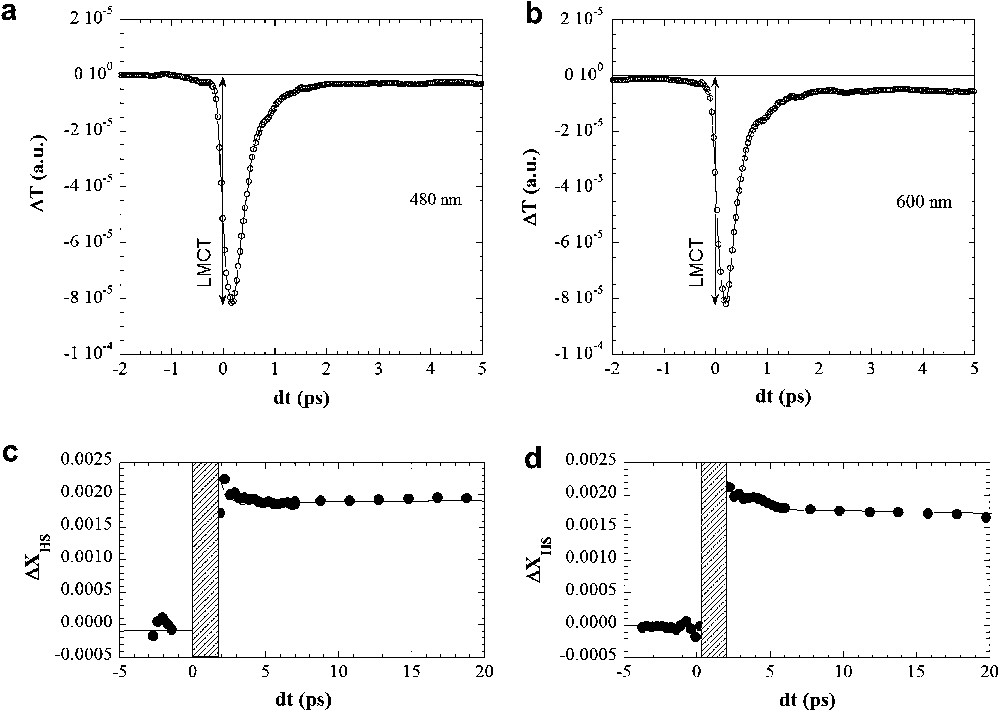
Time dependent variation of the transmission ΔT at 480 nm (a) and 600 nm (b). The decrease of transmission, more pronounced at 600 nm, is the direct signature of the generation of HS molecules within 1 ps. An intermediate state is observed on the sub-ps time-scale. The time dependence of the photo-converted HS fraction ΔXHS in (c) and (d) is deduced from the comparison of transmission at 480 nm (a) and 600 nm (b) to the temperature dependence of the transmission (Fig. 2).
It is well known that a direct transition from the LS to the HS state is a highly forbidden direct process, as it involves ΔS = 2 change of spin [10,13]. The change of spin-state is only possible through one or several intermediate state(s), here a Ligand-to-Metal Charge-Transfer state [16] (LMCT state). The occurrence of this intermediate state is associated with a transient absorption peak, which appears within the experimental resolution, as observed at 480 and 600 nm (Fig. 4a and b). The subsequent relaxation process down to the low-lying HS state is a hotly debated matter, and we observe here an exponential population of the HS state from the LMCT state with τ = 300 fs time constant. The conclusions we draw for the early stages of transition are in very good agreement with the reports on molecules in solution [10–12]. These results are also consistent with the ones obtained in the monoclinic polymorph of [(TPA)Fe(TCC)]PF6 [19].
In order to obtain a quantitative description of the molecular transformation rate, we scaled the time dependence of the variation of the transmission to the variation of transmission observed during the thermal conversion (Fig. 2) between the LS and HS states. The fractional change of HS (ΔXHS) thus obtained is plotted in Fig. 4a and b. However, scaling ΔXHS(t) against ΔT is only possible in the time domain where only HS and LS states coexist, as we have no information about the transmission spectra of LMCT which is not a thermally equilibrated state. For this reason ΔXHS is not given in the time domain from 0 to 2 ps, where LMCT dominates the spectral variations. The results presented in Fig. 4 indicate an increase of HS fraction by ≈0.18 ± 0.01% on the picosecond time-scale. It shows with a good confidence level that such a very small concentration of photo-excited molecules can be detected with stroboscopic techniques. Given the excitation density used, the transformation efficiency is close to one HS molecule per incident photon. In other words, there is no indication of cooperative effect in the early stage of the transformation. We could not significantly increase the excitation density to enhance the transformation rate as the sample threshold damage occurs just above 100 μJ/mm2. Therefore, single shot experiments will be required to observe the dynamics at higher excitation densities, as already performed to study relaxation [20]. A recent report [21] promises entirely new possibilities to run single shot optical experiments with 100 fs time resolution.
5 Conclusion
We have demonstrated the ultrafast spin-state switching of a spin-crossover solid, by using optical pump–probe techniques. The optical response resulting from the spin-state change takes place on the 100 fs to 1 ps time-scale. Coupling such investigations with time-resolved X-ray experiments, which now offer 100 fs to 100 ps time resolutions [22–26,27,28], will provide unprecedented insights into the mechanisms driving this photo-switching, as well as the nature of the intermediate LMCT state. Comparison of both polymorphs may help in better understanding of the general mechanism driving spin-state switching, and extension of this work to Fe(II) solids is underway.
Acknowledgments
This work was supported by the Ministry of Research of France (ACI JC E. Collet, ANR ‘Fast-switch’ NT05-3_45333), Région Bretagne (CREATE 4146) and Rennes Métropole.