

1 Introduction
Over the last decade, the design of linear molecular architectures exhibiting electronic properties such as electronic or photonic conduction has been a constantly growing field of interest, aiming for new concepts and applications in molecular electronics. Illustrated by carefully selected examples, this non-exhaustive overview aims to emphasize the synthetic strengths, weaknesses, and complementary features of both the covalent and self-assembly approaches that lead to internally structured molecular wires, fibers, and cables, with respect to their electronic properties. Even though the use of single molecular wires to connect applied devices seems rather unrealistic, many uses for linear anisotropic molecular structures can be foreseen. However, the association of molecular wires, cables and fibers in devices with classical electronic components in transistorized devices requires the understanding and the mastering of the electronic properties and more generally the physical properties of linear, anisotropic, single molecules.
The scope of this update is deliberately limited to organic species incorporating iterative units, and will focus primarily on their preparation methods. Molecular wires, fibers, cables, and ropes will be distinguished in a way that recalls the stepwise structuration of collagene (single wire) into triple strands (fiber), hair (cable), and finally into ligaments (rope). Carbon nanotubes are also deliberately ignored because of their well-known properties, and commercial availability have already led to the development of research towards their integration in devices, grafting, and manipulation at the micro- and nanoscale.
In a wire that comprises an iterative monomer unit, no matter what the assembling method, the targeted properties are extrapolated on the basis of the physical properties of the monomer. The linear architecture is also defined by the chemical properties (bond angles, conjugation, steric constraints) of the monomers and their ability to connect with one another in a defined geometry, which, if linear, is far from the thermodynamically favored discrete reaction product. The controlled stepwise connection of monomer units into single molecular wires of low polydispersity will be examined first and followed by the polymer approach. The interest will then shift to non-covalent synthetic methods, after a brief recall of the weak intermolecular interactions that are essential to the structural organization of the linear species. In all cases, the linear structures examined will be distinguished depending on whether they remain single wires or they associate in bundles or fibers spontaneously or in the presence of a template. Last but not the least, it should be noted that the properties will be examined independently of the nature of the molecule/electrode junction.
2 Single molecular wires
2.1 Covalent wires
Like in any covalent approach, linearity is usually achieved by the introduction of links at the opposite positions on an aromatic (Fig. 1, top) [1] or polyaromatic ring (Fig. 1, bottom) [2]. Relatively short species, like those presented, allow an obvious geometrical control on the molecular shape that is suitable for single molecule studies by break-junction techniques. This method and its multiple applications were recently reviewed by a pioneer in the field [3].
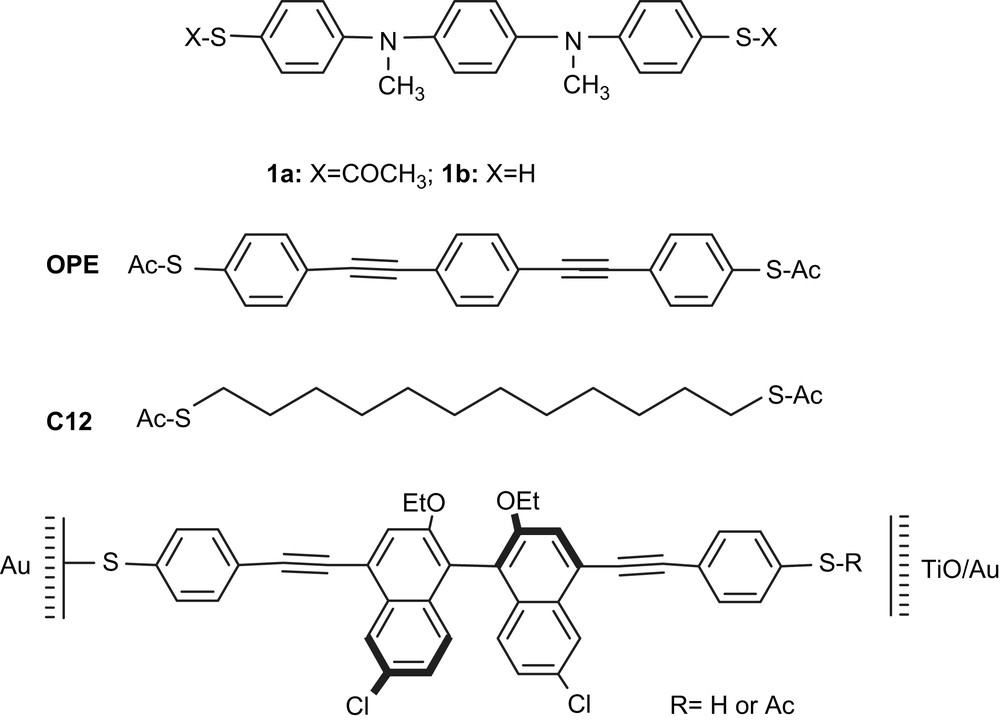
Examples of rigid linear molecules introduced into nanoelectrodes or break-junctions. Top: (1a) oligo pyridine dithioacetate; (1b) oligo aniline-dithiol, OPE: oligo phenylethynyl dithioacetate, (C12 (used as a control)). Bottom: first optically active molecular wire.
It should be noted that an alternate route to break-junction studies was developed recently by Nuckolls, using plasma abrasion to generate gaps in single walled carbon nanotubes (SWCNT) in a controlled manner (Fig. 2) [4]. Carboxylic groups generated at the periphery of the gap allow the insertion of various bridges tested for the conducting properties.
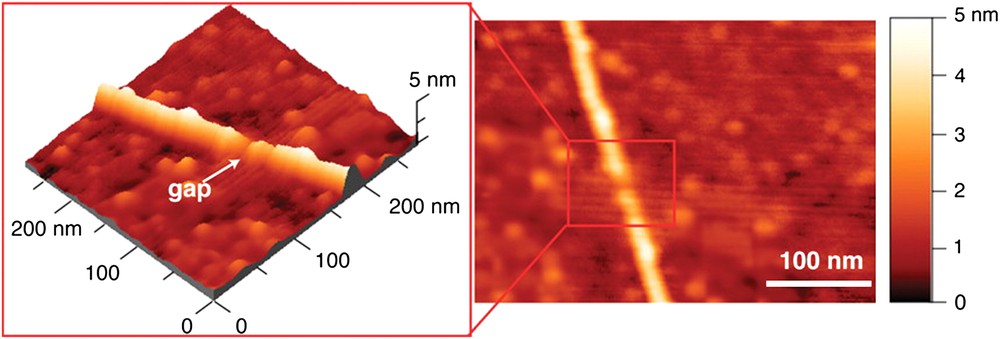
Alternate approach to break-junctions: plasma abrasion of controlled gaps in SWCNTs. Reprinted with permission from Ref. [4], copyrights (2006) AAS Publishers. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The gaps can be bridged by conjugated oligomers, and the strategy can be extended to bridges bearing functional groups, thus providing access to devices for single molecule detectors [5]. Recently, reconstruction of junctions by insertion of two gold nanoparticles bound by molecular wires into the nanogap between two gold electrodes was performed. Conceptually, it can be considered very close to the break-junction approach [6]. In general, these approaches seem very well adapted for studying and tuning electronic properties, for example as a function of conformational changes [7–10]. The length of the wires used is necessarily short, with recent reports of measurements on 7-nm-long conjugated wires (Fig. 3) [11]; however, the shortness allows the exploration of concepts that will be detailed later on.
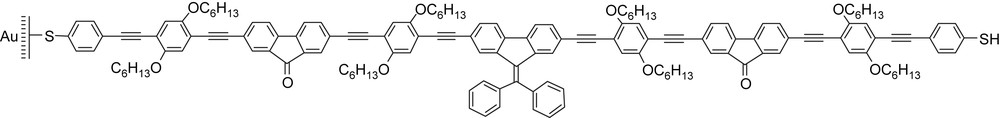
A 7-nm-long molecular wire studied by Lambert et al. [11].
Intuitively, the synthesis of linear species such as that described above (Fig. 3) can be foreseen as an intrinsic limitation to the potential applications of single molecules. With a few exceptions, such species will be limited to the exploration of concepts, and to the rationalization of structure–property relationships in single molecular wires. These findings will then be the basis for the design of self-assembled species that theoretically bear fewer synthetic drawbacks. Indeed, beyond the size or length of a few nanometers, just like peptide helices, most single molecular wires will usually deviate from linearity because such distortion is energetically costless and may be easily induced by interaction with a solvent or a surface. For this reason, additional structuring factors will frequently be required to obtain long linear structures. The addition of a rigidifying external insulation layer to the molecular wires (Fig. 4) has been envisioned. This topic was recently reviewed exhaustively by Anderson [12]. Conceptually, this insulating templated approach is very close to the utilization of nanoporous solids and membranes to template the growth of nanofibers that will be described later.
Insulation directed synthesis of single molecular wires. Reprinted with permission from Ref. [12], copyrights (2007) Wiley-VCH Publishers.
In the field of molecular wires, porphyrin derivatives are extremely attractive due to both their extended delocalized π-electron systems and their structural features as flat, well-defined and functionalizable building blocks. Due to their various electro- and photo-chemical properties, numerous linear arrangements of porphyrin macrocycles have been investigated since the pioneering work of Lindsey (Fig. 5) [13].
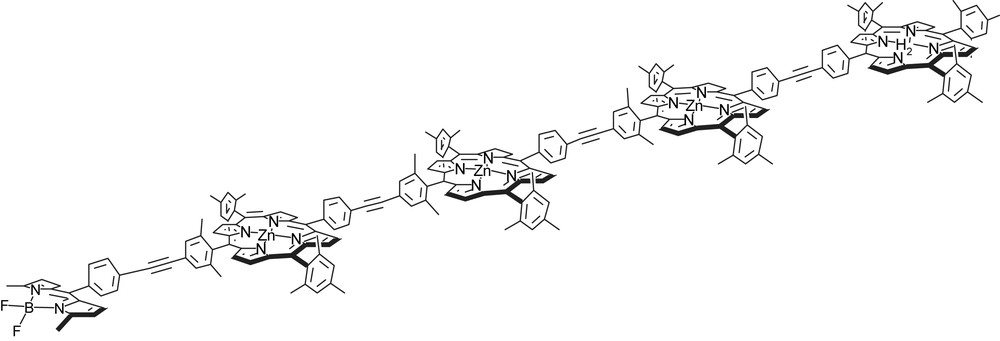
First photonic wire built on porphyrins. Energy is transported over 90 Å with an efficiency of 76%.
Several review articles exhaustively collect covalent synthetic approaches to porphyrin-based molecular wires, but practically applicable strategies are restricted to the iterative oxidative coupling of monomeric species (Fig. 6) [15]. Beside the iterative aspects, this method opens access to rather rigid oligomers such as that represented in Fig. 6a. However, there is still a clear difference between the idealized geometries (Fig. 6a and d) inherent to their design and the observed geometries, as far as the linearity (Fig. 6b and c) or the planarity (Fig. 6e) of the scaffolds are concerned. Indeed, linearity of the single molecular multi-porphyrin wire seems to be restricted to rather short species of less than 50 nm.
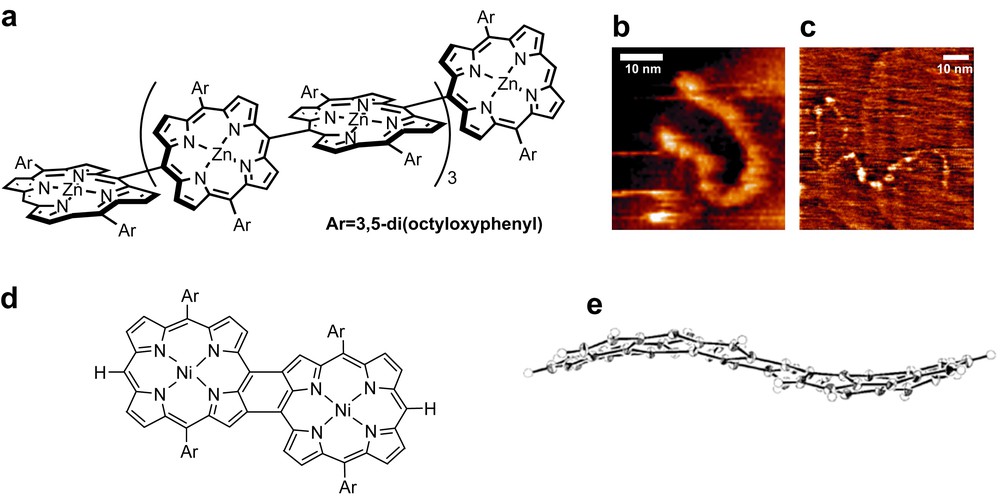
Linear porphyrin oligomers obtained by iterative oxidative couplings of dimer into tetramer, octamer. (a) Octamer n = 8; (b) STM image of n = 48; (c) STM image of n = 128; (d) ideal representation of a rigid dimer; (e) ruffled structure of the rigid dimer. Reprinted with permission from Ref. [14a], copyrights (2005) American Chemical Society, and Ref. [14b], copyrights (2005) Wiley VCH.
At this stage, it is quite easy to conceive the limitations of pure covalent approaches to molecular wires, and the growing interest for strategies that provide linear species more rapidly and efficiently. As mentioned earlier, the templating insulation may be an efficient tool to achieve this goal by adding an “external” structuring factor. Before examining approaches based on a guided linearity, it is necessary to emphasize approaches based on self-assembling, which already offers many advantages demonstrated by the rapid production of more or less linear structures.
2.2 Self-assembled molecular wires
The previous examples are based on covalent species. One of the many interests of such species is their high degree of conjugation and electronic delocalization that can be achieved by the use of ethylenic or acetylenic connectors between the monomers. Historically, these derivatives have drawn early attention for information transport and storage [15]. As we have seen, a major drawback lies in the difficulties inherent to the synthesis and the purification. These difficulties increase gradually with the size and the sophistication of the targetted wire or molecular electronic device. Although approaches based on self-assembly renders the synthetic requirements a lot simpler, many limiting parameters have to be considered: the self-assembly process needs to be controlled regarding the dimensions of the generated objects, and a significant degree of electronic communication has to be reached despite the non-covalent, and thus non-conjugated, bonding between each monomeric unit.
Various approaches were recently described and are discussed hereafter. As for the other approaches, no attempt is made to cover the field exhaustively, but the examples discussed were selected mostly because they are representative of concepts, but also based on their interesting properties. The selected examples hereafter were built by the use of multiple weak interactions; therefore, a brief overview of the weak interactions panel is necessary.
2.2.1 Weak interactions
Prior to the discussion of typical examples, it should be realized that contrary to a single covalent bond, a single weak interaction is less directional. For this reason, a successful self-assembly process will usually require a combination of interactions, resulting in a geometrical control of the assembled structure, but also allowing the correction of mismatches during the thermodynamically driven assembling process. The general approach will then be based on the use of a main weak interaction (Fig. 7), which will be modulated or tuned by auxiliary weaker interactions, introducing a subtle tuning of the geometric control during the assembly process.
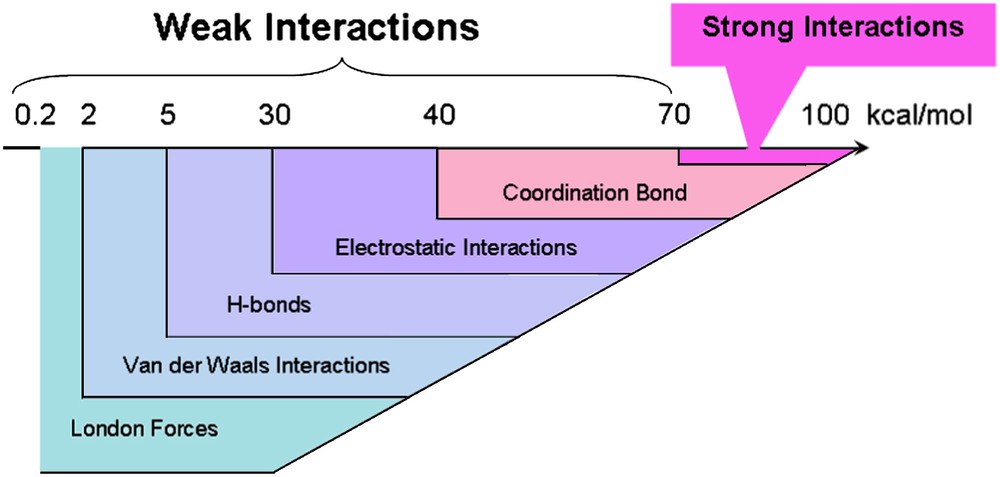
Energy range of weak intermolecular interactions. Values are indicative values and can be strongly dependent on the solvent. (1 kcal/mol = 2.6 × 1022 eV/mol).
Many possibilities are available to adjust the strength of these interactions, and the nature of the solvent will play a crucial role. Electrostatic character may also be introduced to tune either van der Waals interactions and π–π-stacking, such as in electron-rich/electron-poor π-stacked aromatic rings, or the hydrogen bonds between complementary charged species.
2.2.2 Directed aromatic stacking
The complementarity of aromatic stacking with hydrogen bonding is illustrated in Fig. 8. Examples of stacking interactions controlled by H-bond auxiliary interactions were reported for peptide type interactions [16], purine–pyrimidine (DNA type) interactions [17,18], and intermolecular steroid interactions [19].
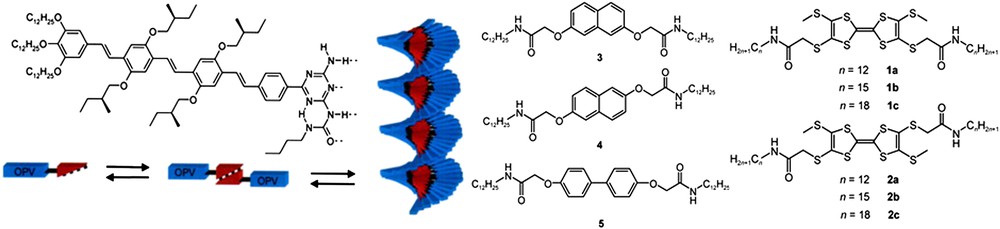
Individual wires built on H-bonding and aromatic stacking (main interactions) of a polar core (in red). A fine tuning is based on the lipophilic interactions in the protective alkyl coating. Fibers of a similar type can be obtained using compounds 1–5 without prior assembly through H-bonding. Reprinted with permission from Ref. [30], copyrights (2006) AIP Publishers. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
For solubility reasons, peripheral, long, linear alkyl chains are usually incorporated in the structures. In the prospect of auxiliary weak interactions, such peripheral substitution and the lipophilic association of the alkyl chains adds a lipophilic coating to the wire formed by strong π–π-stacking interactions. This coating is conceptually connected with the use of the insulating templates mentioned previously, and most probably has a more significant contribution to the structuration of the scaffolds than to their cohesion. An additional role of the lipophilic coating is the protection of the H-bonded central core against hydrophilic perturbations due to polar solvents.
A general tendency for such self-assembled single stranded molecular wires is to associate in higher hierarchical structures to form fibers with a significantly larger section. Whereas this could be considered as an unwanted property, the properties of these fibers are of high interest, and such objects will be examined in a specific section of this document. However, when single stranded molecular wires are the desired target, these inter-wire interactions must be avoided. Thus, an interesting approach based on the use of perfluoro-alkyl chains around a hexabenzocoronene (HBC) core was developed by Aebischer et al. [20] These HBC derivatives can then form non-associated molecular wires with a uniform 10 nm section.
Virtually, these assembly strategies have no limitations regarding their applications, but are still preferentially applied to π-systems presenting some photo- or electro-chemical reactivities, and targeting photo- or electro-conduction. Thus, examples using perylene diimide [21,22] polyethynyl aryl [23,24], tetra thiofulvalene [25,26] and porphyrin [27] derivatives were recently reported.
2.2.3 Coordination bonds
The use of coordination bonds as weak interactions for self-assembled coordination polymers has been applied mostly to porphyrin-based building-blocks. A major reason is the presence of axial coordination sites on most central metal cores used in porphyrins. Whereas the nitrogen–metal bonds maintaining the metal in the macrocycle are strong, the axial ligation, especially in the case of a zinc metal center and an exogenic nitrogen ligand, is rather labile. The major player in this field is Kobuke, who described the formation of linear coordination polymers built on rigid zinc porphyrin dimers bearing imidazole substituents (Fig. 9) [28].

Coordination polymer built using axial base binding on pentacoordinated zinc porphyrins. Linearity is controlled by the head-to-tail arrangement of the porphyrin dimers.
The polymer formation provides the main structuring interaction, which can then be tuned by side chain interactions. When the side chains contain appropriate functional groups, it is then possible to lock the structure. This aspect will be detailed later on, as it can be generalized to various self-assembled species. Nuckolls, in an extension of his previously described SWCNT version of the break-junction (vide supra), utilizes metal coordination to establish the junction between two metallic nanoelectrodes nanopatterned according to a subtle process [29].
2.2.4 Metal–metal bonds
Although more energetic than a coordination bond, the metal–metal bond is remarkably suitable for the formation of molecular wires possessing a strong inorganic character. Although this is not within the scope of this review, it is worth pointing out the important role that can be played by the organic wrap around the metallic nanowire, as is the case for the platinum nanowires represented in Fig. 10 [30].

Wires built on Pt–Pt bonds tuned by the presence of a hydrophobic core. In solution, the wires are single molecular objects, but aggregate as fibers in the solid state (right). Reprinted with permission from Ref. [17], copyrights (2006) Wiley-VCH Publishers.
In this case, due to the presence of the organic side chains, it is possible to use the influence of the surrounding medium to modulate the luminescence of the wires. Even though the X-ray diffraction solid-state structure of the nanowire itself suggests the existence of single stranded molecular species, it should be noted that the properties reported and discussed for the platinum wires concern fibers comprising several wires. In a similar approach, the formation of DNA-type structures around a hydroquinone copper(II) [31] or mercury(II) [32] has been described by Shionoya. The formation of the central coordination complex elegantly replaces the formation of base pairs.
2.2.5 Locking scaffolds a posteriori
The definitive stabilization of structures after a directed, self-assembly process is an elegant combination of approaches based on weak interactions or covalent bond. The very simple principle is based on the use of monomer species that bear reactive functional groups such as ethynyl [33] or olefin [34] for polymerization or metathesis, as illustrated in Fig. 11.
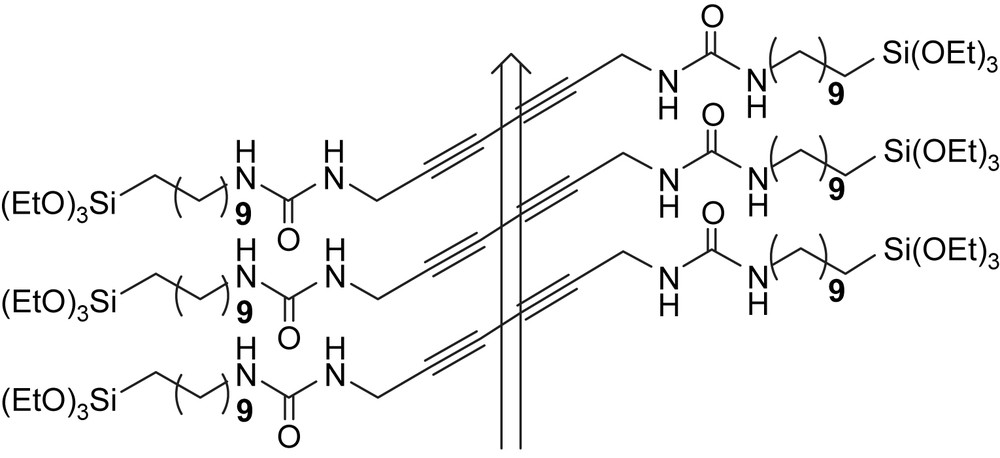
A posteriori locking of scaffolds. Polymerization proceeds along the arrow and geometry is controlled by peripheral H-bonds.
2.3 Spontaneous hierarchical structuration: from wires to fibers
All of the scaffolds examined so far comprise an outside component or coating that is more or less voluminous or structured, rendering control of linearity more or less successful. Hence, for either covalent or self-assembled wires, this coating will play a major role in the way such wires may interact together, or self-organize at higher hierarchical levels. When the outer component (e.g., alkyl side chain) is weakly structured, the induction of linearity is poor, and higher orders of organization are scarcely observed, as shown in Fig. 6 (similar images have been reported for species in Fig. 8). On the contrary, a dense coating will significantly enhance linearity, and will also promote the self-association of wires into bundles or fibers. Sometimes, the effects related to the coating will be detected only when the objects are deposited on a substrate and subjected to epitaxial orientation [16], or in liquid crystalline phases [21] or simply in the solid state [35]. When such additional structuration takes place in solution, the formation of bundles and fibers is extremely interesting as it allows the direct handling of the objects due to their relatively large size (Fig. 12) [36].
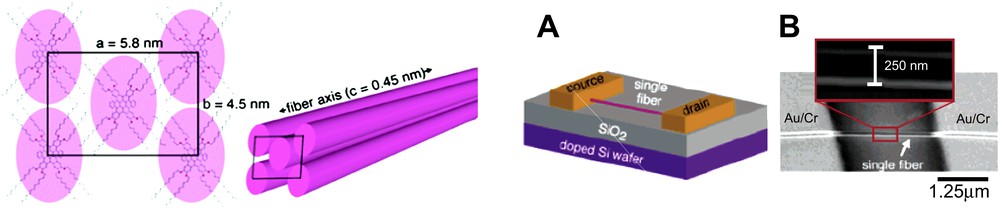
(Left) Fibers formed from HBC wires by external lipophillic interactions. (Right) Device incorporation after wet-stamp handling of a single fiber. Reprinted with permission from Ref. [36], copyrights (2006) American Chemical Society.
In self-assembled species, primary interactions for strong associations of monomers and weaker interactions for the fine tuning of the architectures may also be combined with reactive peripheral groups suitable for a posteriori locking of wire-like species assembled under thermodynamic control. Such an approach was applied to structures obtained in solution (e.g., Fig. 13, left) [37,38], or deposited in the solid state when a surface influence was necessary to control the geometry of the objects [39], as illustrated in Fig. 13 (right).
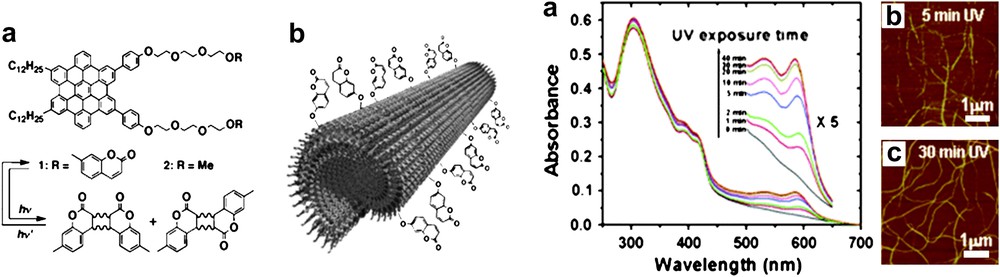
(Left) Tubular fibers obtained by torque assembly of monomers bearing coumarins on the outside, and a posteriori locking of the structure by coumarin photodimerization (Ref. [38]); a similar locking can be performed with in peripheral olefinic groups [38]. (Right) Fibers obtained by photopolymerization of ethynyl groups after spin-coating deposition [39]. Reprinted with permission from Refs. [38a,b,39], copyrights (2005) and (2006) American Chemical Society.
2.4 Templated methods: assisted linear organization
Using approaches that are conceptually close to those leading to the growth of aligned carbon nanotubes in mats [40], several research teams have explored the possibility of using an “external” assistance to promote linear association of wires that are initially coiled or in random orientations. This concept can be illustrated by two selected examples. The first examples report the incorporation of bidentate binding moieties within the strands of non-rigid polymers. These coiled structures are then organized by molecular clamps which interact with the bidentates under thermodynamic control (Fig. 14) [41,42].
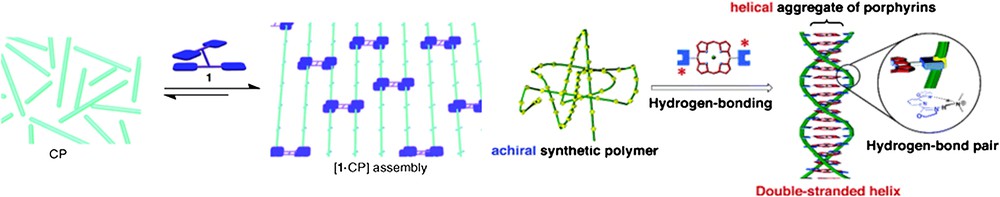
(Left) Formation of mats with polymers containing bidentates using molecular clothespins [41]. The final arrangement can be locked using olefin metathesis [41b]. (Right) Double-helix structure formed from two flexible polymers around a porphyrin template [42]. Reprinted with permission from Refs. [41a,42], copyrights (2006) Wiley-VCH, and (2007) American Chemical Society.
The second approach, although mainly used for polymeric species that are out of the scope of this update, consists in the growth of polymers within mesoporous templates [43,44]. After dissolution of the template, the organic materials retain the shape imposed by the matrix, and can be eventually manipulated as single objects (Fig. 15) [45].

(Left) Nanorods (∅ = 200–400 nm) of polyphenylene nanotubes obtained by Diels–Alder reaction of tetraphenylcyclopentadienones within a mesoporous template [44b]. (Center and right) Poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(bithiophene)] (F8T2) nanorods and single nanorod isolated and used as fluorosensor [45]. Reprinted with permission from Refs. [44b,45], copyrights (2005) and (2006) Wiley-VCH.
This approach was used by the group of Müllen for the organization of HBC derivatives possessing highly extended, delocalized π-systems. Using peripheral, long alkyl substituents, these derivatives display strong self-organization behavior over long distances. This was observed in capillary glass tubes (∅ = 2 mm) as well as in aluminium oxide nanopores. In the latter case, the nanoporous alumina template can be dissolved in soft conditions, and the linear organization can be preserved in the scaffolds that further organize into bundles (Fig. 16) [46].
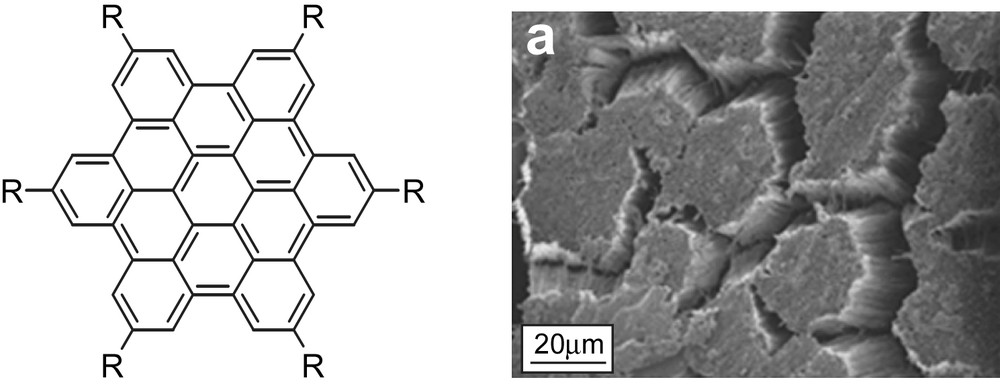
(Left) Structure of the HBC core. (Right) Microphotographs (MEB) of nanorods organized in bundles after soft dissolution of the aluminium oxide template [46]. Reprinted with permission from Ref. [46], copyrights (2006) American Chemical Society.
Historically, nanoporous membranes have been widely used since the pioneering work of Martin [47] to perform electrosynthesis in confined media. These concepts are still explored for the in situ preparations of polypyrroles or polythiophene nanowires. The HBC example illustrates the application of this concept to fields other than electrosynthesis.
3 Electronic conduction in organic nanowires
All of these approaches leading to a large variety of nano objects, with various shapes, morphologies, and dimensions certainly illustrate the successful development of synthetic concepts. However, the modest performances of their electronic properties or chemical stability represent serious limitations in terms of application of organic nanostructures. This is especially true when comparing the performance of organic nanostructures with inorganic analogs. Even though band structures similar to those found in inorganic solids can be found in most covalent organic nanowires due to significant electronic overlaps, a strong degree of communication in self-assembled structures, in which the overlap is limited, is hardly envisioned. In the following evaluation of performances, the properties of linear alkane thiols in self-assembled monolayers (SAMs) have been deliberately excluded, as a recent update on these properties is available in the literature [48]. Our collection of significant performances will be limited to the type of objects previously examined in this update.
Regarding the data concerning the conductivity in polyaromatic covalent linear wires, aside from the results reported for the structures presented earlier [4,5,29,36], an interesting study performed at Columbia University needs to be mentioned. As illustrated in Fig. 17, the dihedral angle between directly linked aromatic rings has a direct influence on their conducting properties. Measurements were compared with theoretical calculations: the conductance through oligomeric π-conjugated species varies as a function of the square cosinus of the dihedral angle between two adjacent monomers in the scaffold.
The molecules studied are oligomers of two aromatic rings, and the dihedral angle between the rings is determined by the substitution on each ring. By fine tuning electrostatic and steric interactions, dihedral angle values ranging from 0 to 88° were reached for seven biphenyl derivatives. Conductance measurements were performed in a break-junction type set up using a STM tip and a gold surface [49]. The reference value in these conditions is probably the linear single molecular wire represented in Fig. 3 for which currents of 0.35 nA were measured under application of a 0.3-V potential. The corresponding resistance value of 0.86 × 103 MΩ is consistent with values of 0.12–0.67 × 103 MΩ reported by Osuka for the linear covalent oligoporphyrins represented in Fig. 6 [14c]. It should be noted that, due to their photoactive character, the conductivity of oligoporphyrins can be significantly enhanced under irradiation. However, in general, the limitations in covalent conducting wires are due to their heterogeneity, and more specifically to their conformational heterogeneity and its effect on the electronic communication along the architecture of the wires. When self-assembled species are considered, the limitations are usually generated by incomplete control of the electronic overlap between monomers. As a consequence, the electronic structure is very different from the band structure, and the electronic interactions are ruled by the Marcus and Hush theories. The performances of self-assembled structures in the case of single crystalline materials will thus remain lower or equal to that of amorphous silicon as far as charge carrier mobility is concerned. Recently, conductivities in the range of 9.7 × 10−4 S cm−1 were measured in the crystalline state for self-assembled thiazyl radicals, and can be compared to values of 6.4 × 10−4 S cm−1 measured in the amorphous state for the same species in order to illustrate the strong dependence of the properties on the level of organization [50].
Thus, one of the main challenges in self-assembled materials is the design of anisotropic non-covalent assembled arrangements that maximize the electronic overlap between the monomers and also allow easy manipulation of the objects. As a recent successful example, the approach developed by Nuckolls on distorted HBC derivatives which produces linear (∅ = 5 mm) columnar and directional stacks of HBC cores in dodecane deserves to be highlighted. Several thousands of these wires self-organize in bundles, and then into orthorhombic assemblies of fibers (∅ = 200 mm) that are relatively easy to characterize by TEM and X-rays and that can be manipulated with a PDMS stamp [36]. The properties were explored in OFET devices and h+ mobilities in the 10−4 to 10−2 cm2/Vs range were measured for approximately 100 devices. It should be noted that this represents the first utilization of a PDMS stamp to manipulate objects that are neither CNTs nor inorganic wires. In a similar approach, Aida has developed the manipulation of macro-fibers based on an amphiphilic HBC derivative (Fig. 17), and measured changes in the resistivity from 200 MΩ cm along the main axis to 20 Ω cm, after doping by exposure to I2 vapors [38b].
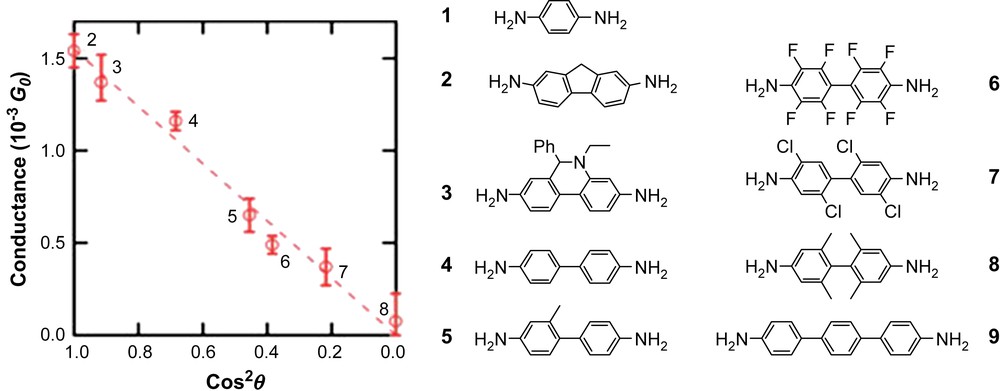
Conductance changes in a molecular junction as a function of the torsion angle (θ) between two aromatic planes in a molecule. Reprinted with permission from Ref. [49], copyrights (2006) Nature Publishing Group.
In addition to incomplete electronic overlap between π-electron densities in aggregates, the presence of long alkyl chains can also explain weak performances in these materials. Even though the presence of chains is necessary for architectural reasons, these peripheral groups introduce large insulating domains in the structures, and thus energetic barriers against conduction. This is the main conclusion of research groups who have prepared devices based on OPV self-assemblies presented in Fig. 8 [17].
Thus, a promising strategy consists in the preparation of fibers without alkyl chains. In this case, classical crystal design approaches can be used. A recent report describes the remarkable properties of nanocrystalline fibrils of hexathiapentacene, that becomes an organic semi-conductor once placed in the channel of an OFET [51]. Indeed, h+ mobility of 0.27 cm2/Vs was measured for a single nanofibril (Fig. 18), which represents a significant breakthrough for organic electronics. The templated growth of linear objects using nanoporous membranes is also a good alternative if unidirectional aggregation is not spontaneous.
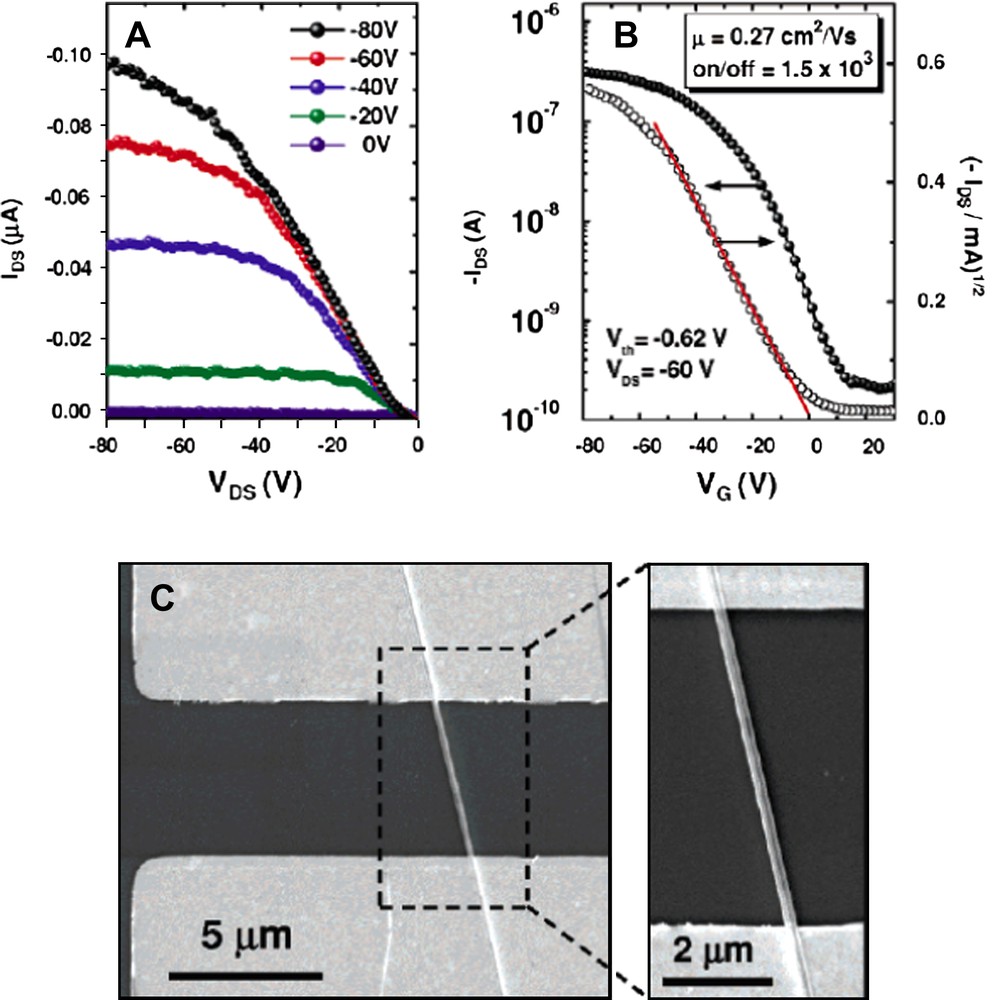
I/V curves of a single fiber of hexathiapentacene. Reprinted with permission from Ref. [51], copyrights (2007) American Chemical Society.
Finally, it should be noted that most conducting and semi-conducting nanofibers are usually of the p-type. This fact renders extremely exciting and interesting recent work on fibrils of perylene diimide materials, for which n mobilities in the range of 0.42 cm2/Vs were obtained.
Hexaazatrinaphthylene (HAN) derivatives represent another family of electron accepting aromatics. These compounds were recently used to prepare n-type semi-conducting fibers [39], in studies that demonstrate that microfibrils are suitable for microcontact printing on Si/SiO2 surfaces. Last but not least, to circumvent the low availability of n-type semi-conducting materials, the n-doping of organic nanofibers appears quite promising [52].
4 Conclusion
The main advantage of a covalent linear molecular wire is the architectural control on both its morphology and its electronic properties. In a fundamental approach, the studies on covalent linear species are useful in order to establish structure–property relationships, and they are facilitated by the characterization of structures that can be observed in both solution and the solid state. The preexistence of the objects in solution also allows easier manipulation and incorporation in break-junction setups for physical measurements. However, the main limitations concern the suitability of large-scale production of covalent single molecular wires, and their practical use or incorporation in functional devices. It should be mentioned that conducting fibers obtained by electropolymerization in nanoporous solids or other templates are suitable for a wide range of applications, but this established method is conceptually rather far from a “molecular” approach [53] and thus has not been included in the scope of this review.
Based on the information obtained by studying covalent materials, self-assembly and polymerization- based approaches can be designed to obtain readily available functional materials. Both approaches provide wires of rather high polydispersity which usually complicates their solution structure determination by methods other than surface deposition. In that case, the linearity of the object observed on the surface can be more or less non-existent in solution, due to the absence of surface/object interactions that renders the assembling and the architecture entropy controlled. Thus, the characterization of self-assembled objects by imaging on a surface involves additional interactions that are developed between the object and a highly organized surface (mica, HOPG, SiO2).
Aside from single device production for physical studies (OFET, break-junction), this stage of development is still close to utopia for organic nanowires in the current state of the art, while some pre-applicative research is already under progress concerning inorganic nanowires. It thus seems legitimate and necessary to assess the potential practical advantages of organic nanowires. As far as the physical properties are concerned, the covalent and self-assembled approaches clearly compare in favor of controlled covalent linear structures in terms of performances. This comparison points out that, in self-assembled species, structural defects are usually responsible for low performances. There is obviously a great deal of ongoing research that targets better efficiencies of the self-assembly processes. It could be anticipated that the electronic or photonic communication between existing building blocks of molecular electronics will be optimized soon. The development of self-assembled materials will be then conditioned by the controlled growth of single supramolecular strands or wires. In other words, it will be necessary to design species for which the growth can be controlled and tuned by surface materials commonly used in electronic devices, either as dielectric materials or as contact electrodes. These species will then provide access to devices that will be grown between preset boundaries and contacts patterned on a given surface. Non-covalent organic wires and fibers are among the ideal candidates for the nanojunction production, which is very difficult to carry out in soft conditions with carbon nanotubes and inorganic nanowires. Towards this goal, perylene di-imides, HBC, HAT, and other flat aromatic species like porphyrins and phthalocyanines [54] are highly attractive candidates due to their peculiar geometries, their established functionalization patterns and strategies, and the mobility of their widely delocalized π-electron density.
Acknowledgements
The realization of this minireview has been made possible with the help and encouragements of the “Observatoire des micro- et nanotechnologies” (OMNT: www.omnt.fr). We are indebted to Stéphane Fontannel and Dominique Vuillaume for initiating this literature coverage, and to Gérard Bidan for stimulating discussion and helpful comments.