

1 Introduction
The interaction of organic multidentate ligands with transition-metal ions can lead to a variety of molecular architectures, including macrocycles, polyhedra and linear and helical polymers [1–3]. As might be expected, ligand structure plays a critical role in the type of structure formed. Flexible ligands favor the formation of chelates or macrocyles, while stiff ligands often result in polymeric complexes. Bifunctional P,O ligands, which combine a soft phosphine moiety and a hard oxygen function, such as ester, amide or ketone, remain the subject of numerous studies, owing to the various properties and/or applications of their transition-metal complexes [4–7].
In metal complexes with these P,O molecules both P-monocoordination and P,O chelation are commonly found. The P,O chelate ligands give stable metal complexes and due to the different properties of the P and O ligand atoms they can strongly influence the stereoelectronic properties at the metal center [6]. Furthermore, in late transition-metal complexes, the greater lability of the O-ligand atom favours fluxional processes resulting from reversible O-atom dissociation and recoordination [4–7].
The O-atom dissociation in these hemilabile ligands creates a vacant coordination site, and conversely weak O-association can give rise to stabilisation of an otherwise coordinatively unsatured species.
For this reason complexes with P,O ligands have been studied for their catalytic properties [8–16], especially in reactions involving the activation of small molecules [17–22].
An increased interest in the chemistry of α-diimine ligands and their complexes [23–26] was stimulated by the recent discovery by Brookhart and co-workers of Ni(II) and Pd(II) α-olefin polymerisation catalysts containing bulky α-diimine ligands [27,28]. α-Diimine ligands exhibit a rich coordination chemistry [29]. In fact, they can formally act as 2-, 4-, 6-, or 8-electron donors. Metal coordination of the heterodiene is possible in different conformations. The most frequent case is the chelation of two imine nitogens by one metal.
Recently, we have been interested in the synthesis of divalent complexes supported by the α-diimine ligands [30–32]. Other works in our laboratory reported the synthesis of cationic methallyl complexes containing α-diimine ligands [33–36].
Son and co-workers [37] describe the synthesis of one such bis(pyridylether) ligand and its coordination preferences with Ag(I), Cu(II) and Co(II).
Pyridine-alkoxide ligands have been shown to be useful in polymerisation reactions involving early transition-metals [38,39]. Osborn and co-workers reported preliminary experiments indicating possible applications of molybdenum(VI) and vanadium(IV) complexes, containing the chiral pyridine-alkoxy ligand [40], in asymmetric oxidation reactions.
Aminoalcohols are important synthetic targets because of their versatile utility as building blocks in organic synthesis [41]. More precisely, chiral 1,2-aminoalcohols obtained by chiral aminoacid reduction have been used as chiral auxiliaries and as source of sterogenic center.
A series of new modular Ru/aminoalcohol systems have been used as enantioselective catalysts in the asymmetric transfer hydrogenation reaction in both water and 2-propanol [42].
The synthesis of a new enantiopure, conformationally constrained 1,4-aminoalcohol was reported by Guarna and co-workers [43].
Chiral P,N ligands have been found widespread application in a variety of asymmetric catalysis in past decade [44]. As an important class of P,N ligands, chiral phosphine-imine compounds have been attracting considerable attention recently since they are easily prepared and readily modified in electronic and steric properties.
Zheng and co-workers [45] have reported the synthesis of ferrocenylphosphine-aldimine ligands which are easily prepared in high yields from ferrocenylphosphine amines with m-substituted benzaldehydes in refluxing ethanol in the presence of anhydrous MgSO4.
Some specific procedures reported for the synthesis of aldimine ligands include the reaction of pyrrole-2-aldehyde and 2-aminophenol in hot ethanol [46], reaction in-situ of 2-pyridinecarboxaldehyde and the appropriate primary aromatic amine in refluxing ethanol [47] and reaction of aldehyde with the primary amine in dichloromethane in the presence of anhydrous magnesium sulphate [48].
Recently, Fossey and co-workers have reported the first chiral bis-aldimine NCN–pincer complexes [49]. 2-Bromoisophthalaldehyde or commercially available isophthalaldehyde has been converted to the corresponding aldimines by reaction with (S)-α-methylbenzylamine in ethanol heated at reflux.
Considerable interest has been shown in metal complexes of thiocarbazate derivatives especially those having NNS donor sequences [50]. These compounds warrant further study because they provide an interesting series of ligands whose properties can be greatly modified by introducing different organic substituents, thereby giving variation in donor properties. Furthermore, limited studies have been done on ligands having ONS donor sequences [51,52]. Therefore, Crouse and co-workers [53] have reported the synthesis of isomeric bidentate ligands having nitrogen–sulphur donor sequence by condensing S-benzyldithiocarbazate with 5-methyl-2-furylaldehyde in refluxing ethanol.
Molecular sieves have been used as an effective agent for the condensation of aromatic and heterocyclic aldehydes with various amines [54,55]. The aldimine ligands N-(hetarylmethylidene) and N-(arylmethylidene)anilines have been obtained by condensation of furan, thiophene and benzene aldehydes with aniline and its derivatives in dry benzene in the presence of freshly backed molecular sieves [56]. The reaction was conducted at room temperature or with heating on a water bath at 80 °C in an atmosphere of argon. After a specific time interval, depending on the substrates, they are almost completely converted into the aldimine ligands.
We report herein a simple synthetic route as compared with previous ones of furfuryl imine ligands starting from aldehyde and primary aromatic amines in methanol at 0 °C.
2 Results and discussion
A series of furfuryl imine compounds (1–5) were synthesized in one step from 2-furfuraldehyde and the appropriate aromatic amine in methanol with high yields (Scheme 1). All furfuryl imine 1–5 used in this work were obtained in the solid form as yellow powders except for cyclohexylamine, we obtain a yellow oil product. They are slightly sensible to the air and moisture. These compounds were characterized by IR, 1H and 13C NMR spectroscopies. Selected examples were also characterized by mass spectroscopy.
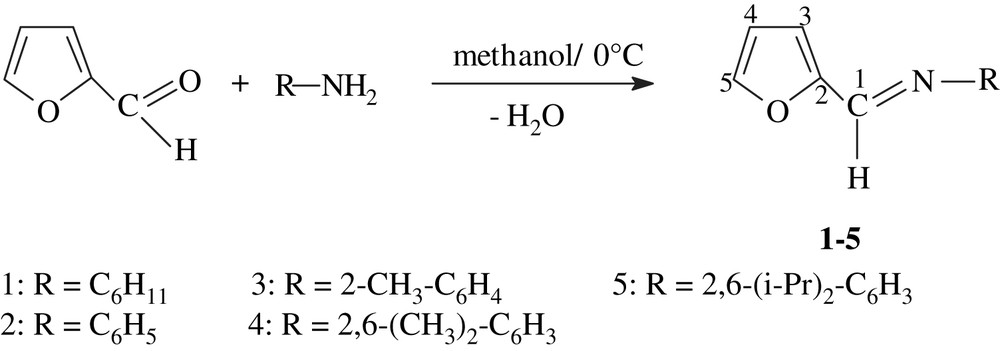
Synthesis of furfuryl imines.
The formation of furfuryl imine ligands 1–5 could be concluded from IR spectroscopy, where only CN stretching vibrations were observed in the range 1619–1646 cm−1 and no CO stretching vibrations of the starting aldehyde at 1779 cm−1. The absorption peak near 1580 cm−1 appears especially when benzene is conjugated with other groups (CN) by π–π conjugation.
The NMR data of ligands are consistent with furfuryl imine structure shown in Scheme 1.
The 1H NMR spectrum of the furfuryl imine ligands presents a singlet around 8.02 ppm, which corresponds to iminic proton. The furyl protons H3 and H4 of the furfuryl imine ligands 1–5 appear as a doublet and doublet of doublets around 7.51 and 6.44 ppm, respectively.
The 13C NMR spectra of furfuryl imine ligands 1–5 present a signal characteristic of the iminic carbon around 150 ppm.
The mass spectra show the molecular peak ([M+]) at m/z = 177, 171 and 185 for compounds 1–3, respectively. The principal fragmentations are also instructive, since they show the presence of furyl (C4H3O) group and C5H4NO moiety which indicate the formation of CN bond. Cyclohexyl, phenyl and 2-methylphenyl fragments have been also detected.
The compound 4 crystallizes in P21/a space group with a monoclinic system, it has one imine group with bond distance N–C1 1.292(2) Å which is bound to a furyl group such that C1–C2 = 1.490(3) Å (Table 1 and Fig. 1). The imine plane is essentially planar as indicated by the torsion angle of −2.3° for N–C1–C2–O. The o,o′-dimethylphenyl groups make an angle of approximately 90° to the plane of the CN bonds, due to the presence of the o-methyl substituents, as appears from the torsion angle of 78.3° and −104.3° for C1–N–C7–C8 and C1–N–C7–C6, respectively.
Selected bond lengths [Å] and angles [°] for 4
| Bond lengths (Å) | Angles (°) | ||
| N–C1 | 1.292(2) | N–C1–C2 | 119.78(15) |
| C2–C1 | 1.490(3) | C1–N–C7 | 113.64(15) |
| C2–C3 | 1.358(2) | C3–C2–C1 | 130.03(18) |
| C3–C4 | 1.378(3) | C2–C3–C4 | 105.80(19) |
| C5–C4 | 1.327(3) | C6–C7–N | 120.71(16) |
| N–C7 | 1.440(2) | C8–C7–N | 117.80(20) |
| C7–C6 | 1.419(3) | C11–C6–C12 | 119.50(20) |
| C7–C8 | 1.432(2) | C9–C8–C13 | 124.08(16) |
| C6–C12 | 1.511(3) | C11–C6–C7 | 121.07(18) |
| C8–C13 | 1.477(3) | C9–C8–C7 | 114.50(20) |

Perspective ORTEP diagram of furfuryl imine 4. Thermal ellipsoids are at 50% probability.
The benzenic ring is planar with a maximum deviation of 0.0158(18) Å for the C9 atom. The bond angles in the benzenic group deviate significantly from the idealized value of 120°. This is the effect of the three substituents, the angular deformation can be described as a sum of the effects of the different substituents [57].
3 Conclusion
In summary, we have developed a straightforward synthesis of new furfuryl imine ligands in high yields. Their coordination to palladium is currently under study. It gives new complexes supported by N,O ligands.
4 Experimental
4.1 General
All manipulations were carried out under an air atmosphere. Cyclohexylamine, aniline, 2-methylaniline, 2,6-dimethyllaniline, and 2,6-diisopropylaniline were distilled from potassium hydroxide prior to use. Methanol was purchased from SDS and used without any further purification. NMR spectra were recorded on a Bruker AMX 300 spectrometer. H and C chemical shifts are given in parts per million and referenced to the residual solvent resonance relative to TMS; infrared spectra were recorded on a Bio-Rad FTS-6000 spectrometer. Mass spectra were recorded on a Hewelet–Packard P5890A instrument. The atom labelling scheme for the furfuryl imine ligand 1–5 is shown below:

4.2 General procedure for the preparation of furfuryl imine ligands
To a stirred solution of amine (1 equiv.) in methanol at 0 °C, was added the pure 2-furfuraldehyde (1 equiv.) drop wise. The resulting suspension was stirred for 24 h. The pure yellow ligand 2–5 was obtained after crystallization in isopropanol. Ligand 1 is purified by filtration through a 5-cm silica gel column, using n-hexane as eluent. All furfuryl imine ligands obtained are slightly sensitive to air.
4.2.1 Synthesis of N-(2-furylmethylene)(cyclohexylamine) (1)
Following the general procedure, from cyclohexylamine (6.5 mL, 5.6 mmol), 2-furfuraldehyde (4.7 mL, 5.6 mmol) and methanol (13 mL) was obtained 6.23 g of 1 as a yellow oil. Yield 62%. IR (KBr): νCN = 1639 cm−1.
MS (EI): 177 (M+; calculated for C11H15NO 177), 148 (C10H14N+), 134 (C10H14+), 122 (C9H14+), 107 (C6H5NO+), 94 (C5H4NO+), 81 (C4H3NO+), 67 (C4H3O+), 55 (C3H3O+).
1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.49–1.50 (m, 10H, Har); 3.1–3.02 (m, 1H, H–C–N); 6.39 (dd, 1H, H4, JH4H3 = 1.8 Hz; JH4H5 = 3.3 Hz); 6.66 (d, 1H, H5, JH5H4 = 3.3 Hz); 7.44 (d, 1H, H3, JH3H4 = 1.8 Hz); 8.06 (s, 1H, H1).
13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 24.46–33.97 (Car); 69.74 (C–N); 111.12 (C4); 113.41 (C3); 144.07 (C5); 146.69 (C2); 151.35 (C1).
4.2.2 Synthesis of N-(2-furylmethylene)(phenylamine) (2)
Following the general procedure, from phenylamine (5 mL, 5 mmol), 2-furfuraldehyde (4.53 mL, 5 mmol) and methanol (10 mL) was obtained 5.62 g of 2 as a yellow solid. Yield 60%. IR (KBr): νCN = 1620 cm−1.
MS (EI): 171 (M+; calculated for C11H9NO 171), 142 (C10H8N+), 104 (C7H6N+), 77 (C6H5+), 63 (C5H3+), 51 (C4H3+).
1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 6.41 (dd, 1H, H4, JH4H3 = 1.7 Hz; JH4H5 = 3.5 Hz); 6.81–7.23 (m, 6H, Har + H5); 7.48 (d, 1H, H3, JH3H4 = 1.7 Hz); 8.15 (s, 1H, H1).
13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 112.00 (C4); 116.15 (C3); 120.85–129.00 (Car); 145.46 (C5); 147.55 (C2); 151.19 (C–N); 151.91 (C1).
4.2.3 Synthesis of N-(2-furylmethylene)(2-methylphenylamine) (3)
Following the general procedure, from 2-methylphenylamine (6.44 mL, 6 mmol), 2-furfuraldehyde (5 mL, 6 mmol) and methanol (12 mL) was obtained 9.28 g of 3 as a yellow solid. Yield 83%. IR (KBr): νCN = 1619 cm−1.
MS (EI): 185 (M+; calculated for C12H11NO 185), 175 (C11H8NO+), 156 (C11H10N+), 130 (C9H8N+), 117 (C8H7N+), 914 (C7H7+), 65 (C5H5+).
1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 2.36 (s, 3H, CH3); 6.52 (dd, 1H, H4, JH4H3 = 1.7 Hz; JH4H5 = 3.4 Hz); 6.86–7.21 (m, 5H, 4Har + H5); 7.58 (d, 1H, H3, JH3H4 = 1.7 Hz); 8.14 (s, 1H, H1).
13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 17.80 (CH3); 111.96 (C4); 115.52 (C3); 117.59–131.83 (Car); 145.35 (C5); 147.50 (C2); 150.91 (C–N); 152.26 (C1).
4.2.4 Synthesis of N-(2-furylmethylene)(2,6-dimethylphenylamine) (4)
Following the general procedure, from 2,6-dimethylphenylamine (5 mL, 4 mmol), 2-furfuraldehyde (3.34 mL, 4 mmol) and methanol (10 mL) was obtained 6.43 g of 4 as a yellow solid. Yield 80%. IR (KBr): νCN = 1641 cm−1.
1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 2.04 (s, 6H, CH3); 6.44 (dd, 1H, H4, JH4H3 = 1.8 Hz; JH4H5 = 3.6 Hz); 6.84–6.99 (m, 4H, 3Har + H5); 7.52 (d, 1H, H3, JH3H4 = 1.8 Hz); 7.89 (s, 1H, H1).
13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 18.71 (o,o′-CH3); 112.42 (C4); 116.04 (C3); 124.24–128.44 (Car); 145.00 (C5); 145.96 (C2); 151.30 (C–N); 152.04 (C1).
4.2.5 Synthesis of N-(2-furylmethylene)(2,6-diisopropylphenylamine) (5)
Following the general procedure, from 2,6-diisopropylphenylamine (6 mL, 6 mmol), 2-furfuraldehyde (5 mL, 6 mmol) and methanol (12 mL) was obtained 10.93 g of 5 as a yellow solid. Yield 71%. IR (KBr): νCN = 1646 cm−1.
1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.06–1.11 (m, 12H, CH3–i–Pr); 2.62 (sept, 2H, CH–i–Pr); 6.47 (dd, 1H, H4, JH4H3 = 1.8 Hz; JH4H5 = 3.4 Hz); 7.01–7.06 (m, 4H, 3Har + H5); 7.54 (d, 1H, H3, JH3H4 = 1.8 Hz); 7.89 (s, 1H, H1).
13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 24.02 (CH3–i–Pr); 28.24 (CH–i–Pr); 112.41 (C4); 115.83 (C3); 123.92–138.25 (Car); 145.97 (C5); 147.76 (C2); 150.78 (C–N); 152.07 (C1).
The supplementary material has been sent to the Cambridge Crystallographic Data Center, 12 Union Road, CB2 1 EZ, UK (CCDC No. 687539) and can be obtained by contacting the CCDC.