

1 Introduction
Marine sponges provide an extraordinary supply of bioactive metabolites, among them, bryostatins. The bryostatins are a novel family of macrolides first isolated by Pettit et al. from the bryozoan invertebrates Bugula neritina Linnaeus and Amathia convulata. Eighteen bryostatins have been isolated so far from these two organisms which grow in the Gulfs of Mexico, California, and Sagami. All these bryostatins possess a complex 20-membered ring macrolactone that is highly functionalized with three tetrahydropyran rings interconnected by a (E)-disubstituted alkene and a methylene bridge. It is worth noting that bryostatins show remarkable in vitro and in vivo antitumoral activities against a range of mouse tumours such as P388 lymphocytic leukemia, ovarian sarcoma, and B16 melanoma. The biological trials demonstrated that bryostatin 1 (Fig. 1) has considerable potential for the treatment of ovarian cancer and relapsed low-grade non-Hodgkin's lymphoma. A remarkable review has been published recently by Hale et al. describing the isolation, structure elucidation, biosynthesis, mechanisms of antitumor action and synthesis of bryostatins [1] (Fig. 1). The synthesis of bryostatins, as well as simplified bryostatins analogues, is a synthetic challenge, due to the complexity of the molecules and in general more than 30 steps are necessary to obtain the desired compounds. However, chemists and Nature, can produce simplified compounds and, recently, a related compound to the bryostatins has been isolated: exiguolide (Fig. 2).

The bryostatin family of antitumor macrolides.
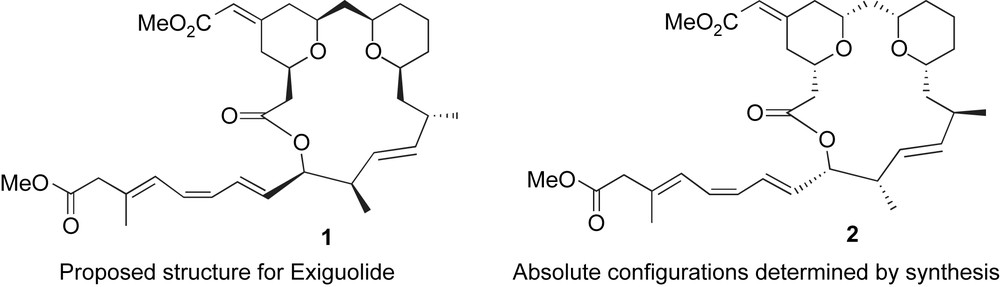
2 Isolation and structural determination of exiguolide
Exiguolide was isolated from the marine sponge Geodia exigua Thiele (order Astrophorida, family Geodidae) [2], and in 2006, the structure of exiguolide was reported on the basis of careful NMR and (+)-FABMS studies. By using DEPT, HMQC spectral data, nOe correlation, and NOESY experiments as well as a J-based configuration analysis method, the relative stereochemistry between the different substituents was determined but the absolute configuration could not be established. Exiguolide is a 20-membered ring lactone which incorporates only two cis-2,6-disubstituted tetrahydropyran rings, instead of three as in the case of the bryostatins. One of the tetrahydropyran rings is substituted by an exocyclic enoate appendage which is also present in the bryostatins. The first structure that was reported in 2006 corresponds to structure 1, and two years later, the absolute configuration was established by synthesis by Lee et al. [3] (compound 2, Fig. 2).
3 Biological properties
Exiguolide inhibits the echinoderm fertilization from marine sponges [4–7]. When sea urchin (Hemicentrotus pulcherrimus) gametes were treated with exiguolide, at a concentration of 21 μM or higher, fertilization was prevented as the egg could not form the fertilization envelope after insemination. However, exiguolide at a concentration of 100 μM did not effect the development of fertilized eggs up to the gastrula stage. Due to these results, it seems that exiguolide is a promising new antitumor compound which should be much easier to synthesize than bryostatins as its degree of structural complexity is less important [3].
4 Synthesis
No synthesis of exiguolide has been reported up to this point, however, Lee et al. [3] have reported the synthesis of the enantiomer of exiguolide which confirmed the structure of exiguolide established by Ikegami et al. [2] and allowed the absolute configuration of the stereogenic centers to be determined.
In order to synthesize exiguolide, Lee et al. have envisioned, as the key steps: a ring-closing metathesis (RCM) to introduce the C16–C17 double bond in the macrolide, a Prins reaction to construct one of the tetrahydropyran rings and a radical cyclization to build the second tetrahydropyran ring. In order to introduce the trienic side chain, a Sonogashira coupling was envisaged (Scheme 1).

Retrosynthetic analysis.
The synthesis of the fragment containing the two tetrahydropyran rings started with the known aldehyde 3 [8] which was converted to compound 4 (Fragment C10–C16) by using a Brown allylation [9] to control the stereogenic center at C13, then a hydroboration–oxidation and protection of the resulting primary alcohol (TBSCl, imid., DMAP, CH2Cl2). Compound 4 was then transformed to iodide 5 in three steps (1,4-addition to methyl propiolate, deprotection, and iodination) in order to perform the radical cyclization to obtain one tetrahydropyran ring. The radical cyclization [10] of 5 in the presence of 1-ethyl piperidinium hypophosphite and triethyborane [11] proceeded efficiently to produce 6. In order to obtain the second tetrahydropyran ring through a Prins reaction [12], compound 6 was transformed to 7 in three steps involving a DIBAL-H reduction, a Brown allylation to produce an homoallylic alcohol, and a 1,4-addition of this latter alcohol onto ethyl propiolate (NMM, CH2Cl2). The Prins cyclization [12] proceeded (TFA, CH2Cl2) and, after hydrolysis (K2CO3, MeOH) and oxidation (DMP, CH2Cl2), ketoester 8 was formed [13]. Ketoester 8 was then converted to carboxylic acid 9 by dimethyl ketalization–hydrogenolysis, Dess–Martin oxidation, rhodium-catalyzed methylenation [14], and hydrolysis-deketalization. This carboxylic acid corresponded to the C1–C16 fragment of exiguolide (Scheme 2).

Synthesis of the C1–C6 fragment of exiguolide.
The synthesis of the C17–C20 fragment was achieved from the known aldehyde 10 [15]. After a Brown crotylboration [16], the homoallylic alcohol 11 was produced in low enantiomeric excess which was improved by using a Sharpless kinetic resolution (Scheme 3, Equation 1) [17]. Esterification of keto-acid 9 with alcohol 11 (Scheme 4) was achieved and ester 12 was produced (70% yield). The crucial intramolecular ring-closing metathesis was realized by using the Hoveyda–Grubbs catalyst, and macrolide 13 was formed in 44% yield. The exocyclic enoate with the correct configuration was then introduced by treatment of 13 with the chiral phosphonate 14 [18,19]. The synthesis of exiguolide was completed by using a Sonogashira [20] coupling reaction [Pd(PPh3)4, CuI, TEA, THF] between the macrocycle 15 and acetylenic 17, [prepared from vinyl iodide 16 (Scheme 3, Equation 2], and partial hydrogenation of the obtained product (Lindlar catalyst). The resulting functionalized macrocyclic compound corresponded to the enantiomer of exiguolide, as the specific rotation of the final synthetic product was +119 (c = 0.0011 g. cm−3, CHCl3) instead of −92.5 (c = 0.00069 g. cm−3, CHCl3) for the natural sample [1]. The [α]D of the synthesized compound confirmed that the latter was the enantiomer of the natural product.

Synthesis of 11 and 17.

Endgame.
By using the same strategy but replacing (+)-(Ipc)2allylborane and (+)-(Ipc)2crotylborane respectively with (−)-(Ipc)2allylborane and (−)-(Ipc)2crotylborane, natural exiguolide will be obtained. The challenge now for the synthesis of (+)-exiguolide is to develop a short and efficient route to obtain analogues that are more active than the natural product itself and more active than the bryostatins.