

1 Introduction
1.1 Structures
The plant-derived maytansinoids [1] and their microbial counterparts, the ansamitocins [2], are extraordinarily potent antitumor agents in vitro and in tumor-bearing animals, blocking the assembly of tubulin in functional microtubules [3]. The maytansinoids as well as ansamitocins are members of the ansamycin group of natural products [4] and are characterised by 19-member ansamacrolide structures attached to a chlorinated benzene ring chromophore. Kupchan and coworkers isolated the unique parent compound maytansine (1) in 1972 [1]. Its structure and stereochemistry were determined based on an X-ray crystallographic analysis of the 9-(3-bromopropyl) derivative. Selected examples of the natural maytansinoids (2–13) of different origins are shown in Figs. 1 and 2. A majority of about 50 known naturally occurring members of this class were discovered and reported in the decade following the report on maytansine. Since the mid-1980s only a few more new members of this group have been reported [5–8]. The major structural alterations are located at the 3-hydroxy group where a variety of other acids or amino acids can be attached. Other points of modifications are the amide nitrogen, the methyl group at C-14 and the benzylic position at C15. Additionally, a 10-epi derivative (5) has been reported. The latter two modifications refer to oxygenations while the amide nitrogen can be methylated or be part of a second cycle which forms a ring with the ester side-chain. Also N-demethylated derivatives have been isolated from plants as well as from microorganisms. Only very recently, the first N-demethyl-N-β-d-glucopyranosyl ansamitocins P-1 and P-2 (14) [9a,b] as well as ansacarbamitocins (produced by a strain of the Pseudonocardiaceae family) (15) [9c] have been reported (Figs. 3 and 4).

Selected examples of maytansinoids from plant origin.

Selected examples of maytansinoids missing the ester group at C3.

Selected examples of maytansinoids from microbial sources (Actinosynnema pretiosum).
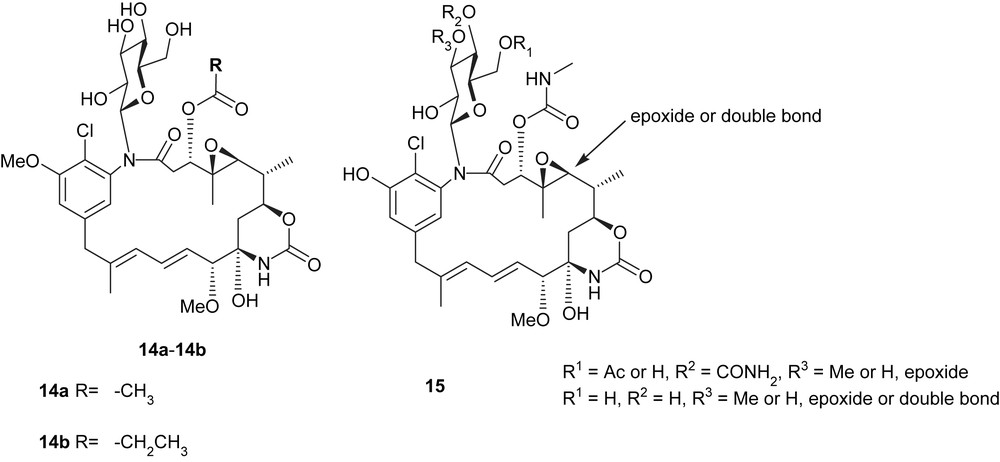
Selected examples of maytansinoids from microbial sources.
1.2 Biological activities
Maytansine (1) shows a very strong cytotoxic activity against the human KB cells in vitro (ED50 = 10−4–10−5 μg/mL), the Lewis lung carcinoma and the B-16 melanocarcinoma solid murine tumor systems [1,10]. Furthermore, antileukemic activity against P388 lymphocytic leukemia, over a 50–100 fold-dosage range at a remarkable low dose per kg body weight, was encountered [11]. Likewise, the bacterial maytansinoids, the ansamitocins, exhibit an activity spectrum and wide effective dose range similar to maytansine. They show antitumor activity against P388 leukemia at daily doses as low as 0.8 μg/kg, with a maximum at 25 μg. Ansamitocin P-3 (11d) and ansamitocin P-4 (11e) are also effective against B-16 melanoma, sarcoma 180, Ehrlich carcinoma, and P815 mastocytoma but less active against leukemia L1210 [2]. The antitumor activity of the maytansinoids has been covered extensively in earlier review articles [3b–d,12].
Maytansine [13–15] and the ansamitocins [16] bind to tubulin which results in the inhibition of microtubule assembly [13,14]. One typical property of ansamitocins is their ability to prevent the polymerisation of tubulin, instead leading to depolymerisation of the polymeric tubulin. In contrast to colchicine, the maytansinoids bind to β-tubulin monomers at a site overlapping the vinca alkaloid binding site.
1.3 Structure–activity relationships (SARs)
The pharmacologically active part has not yet been fully elucidated. The first speculative thoughts came from Kupchan [10]. He proposed that the ester residue is oriented in a manner which would sterically hinder the approach of reactants to the hydrophilic face. The ester function in the antileukemic maytansinoids may play a key role in the formation of highly selective molecular complexes with growth-regulatory biological macromolecules. Such molecular complex formation may be crucial for the subsequent selective alkylation of specific nucleophiles by the carbinolamide and/or epoxide function.
Investigations of the structure–activity relationships revealed that indeed the ester side-chain plays a key role. Maytansinol esters such as maytansine (1) and ansamitocin P-3 (11d) or P-4 (11e) are highly cytotoxic agents. In contrast, compounds carrying a double bond (maysine (8a), trewiasine (8b) and maysenine (9)) or hydroxy function (maytansinol (7)) at C3 lack cytotoxic activity including antileukemic activity (1/10 000 compared to maytansinol esters) [10,17]. Additionally, the change from the (S)- to the (R)-configuration at C3 leads to the loss of inhibitory activities [18]. Maytansinoid analogues bearing a variety of C3 acyloxy side-chains were prepared by semisynthesis, revealing that a change in the C3 ester group modulates the biological activity [19]. Functional groups at the hydrophobic (“lower”) side of the molecule have been spotted to be important for contributing to the biological activity. Thus, it was suggested that the C9 carbinolamide serves as a function that is able to alkylate. Indeed, when the carbinolamide is alkylated at the C9 alcohol, a marked decrease in antitumor activity and cytotoxicity against KB cells was encountered [17]. 10-Epitrewiasine (5), the C10 epimer of common maytansinoids, also shows a strong biological activity similar to 10-trewiasine (3b) [20]. A hydroxy group at C15, as in colubrinol (3a), is compatible with potent antitumor activity [21,22]. Surprisingly, the epoxide function is not essential for the antitumor activity. Neither the amide N-methyl group [23] nor the 20-O-methyl group [24] seem to be necessary for biological activity.
1.4 Clinical trials
Phase II clinical trials with maytansine alone and in combination with other agents were carried out from about 1977 to 1984 with more than 800 patients [25]. Only one patient with islet cell carcinoma showed a complete response, while about 20 patients showed partial responses [26]. Although maytansine (1) itself proved ineffective in phase II clinical trials, the maytansinoids currently attract high clinical interest as “warheads” in tumor-targeted immunoconjugates [27,28].
2 Semisynthetic studies on maytansinoids
Further diversity of maytansinoid compounds has been generated by chemical derivatisation of the natural ansamitocins obtained from plants or by fermentation of microorganisms. Modifications have been introduced by chemical means in the context of semisynthesis, or by making use of the biosynthetic potential of microbes as part of a bioconversion approach. Fig. 5 summarises the important semisynthetic modifications carried out with ansamitocins.

Important semisynthetic modifications of ansamitocins (microbial transformations marked in italic).
During chemical degradation experiments directed towards the structure elucidation of ansamitocins, the deacylated core-structure of ansamitocin P-0 (7) (synonym:-maytansinol) was prepared by reductive cleavage of the ester using LiAlH4 and the compound could then be re-acetylated with Ac2O, resulting in semisynthetic ansamitocin P-1 (11a) (synonym:-maytanacine) [29]. A related sequential deacylation and re-acylation protocol represents the essence of today's semisynthetic approaches in industrial synthesis of ansamitocin derivatives [19,30]. Modern ansamitocin chemistry relies on LiAlH(OMe)3 as the reducing agent (THF, −30 to −40 °C, 81% yield) and DCC/ZnCl2-mediated re-esterification for the incorporation of the desired acyl side-chain (≈30% yield) [27]. The new ester side-chain can also be modified so that the attachment of tumor-directed antibodies via disulfide linker formation becomes feasible [27]. Ansamitocin-based “warheads” such as DM1 (16) and DM4 (17) are currently undergoing clinical trials [28]. Other chemical modifications including dehalogenation using LiAlH4 [31] and removal of the epoxide using the reagent system TiCl3/LiAlH4 have been reported [32].
In addition to chemical modifications, conversion with Gram-positive bacteria such as various members of the genus Streptomyces and a strain of Bacillus megaterium was employed for the removal of functional groups that are bound to hetero atoms [33]. Depending on the strain, typical biotransformations are 20-O-demethylation, deacylation and N-demethylation. Alternatively, ansamitocins were also hydroxylated at C15 by this approach. However, it was reported that this oxidation occurs in a non-stereoselective fashion, leading to 15-hydroxyansamitocin P-3 (10a) as well as 15-epi-hydroxyansamitocin P-3 (10b) [33c], a derivative whose formation has also been reported to occur during fermentations of Actinosynnema pretiosum [34,35]. It has to be noted that several of the compounds available via biotransformation or chemical degradation are in principle directly available by fermentation of A. pretiosum mutants blocked in distinct post-polyketide synthase tailoring steps. For instance, a mutant strain with an inactivated N-methyltransferase has been shown to accumulate N-demethylansamitocin P-3, while another one lacking the respective 20-O-methyltransferase activity produced mainly 20-N,O-didemethylansamitocin P-3 and in lower amounts the expected compound lacking the phenolic methoxy group [36]. Finally, modifications generated by means of biotransformation also include acylations of the hydroxyl groups at C15 and C20 [37].
Interestingly, apart from the presence and stereochemistry of the 3-acyl moiety, neither the presence or absence of the N-methyl group, the 20-O-methyl group, the epoxide or a 15-hydroxy group exerts a large influence on the activity of ansamitocins [3a].
3 Total synthesis of the maytansinoids
The complex structures of the maytansinoids coupled with their potent cytotoxicity and potential for cancer treatment stimulated several total synthesis programs, particularly in the 1980s. By 1984, total syntheses of naturally occurring maytansinoids had been reported by three different groups. To date they have accumulated five: Meyers [38] (1980), Corey [39] (1980), Isobe [40] (1982), Gao [41] (1988) and Huu [42] (1996). Additionally, the syntheses of fragments were published by Götschi [43], Ganem [44], Vandewalle [45], Barton [46], Ho [47], Fried [48] and Parsons [49]. Furthermore, the Confalone group reported studies on a new methodology for the ring closure using a nitrile oxide–olefine [3 + 2] cycloaddition [50]. The major synthetic challenges are by all means the position of ring closure, the installation of the carbamoyl group and the diene unit which is in deconjugation with the arene ring. Although macrolactamisation evidently appears to be the first choice for ring closure, difficulties have been encountered to smoothly carry out this transformation in the syntheses of ansamycin antibiotics [51]. This can be ascribed to conformational reasons because many ansamycin antibiotics bear E-configured olefinic double bonds in the ansa chain. Additionally, it needs to be noted that anilines are less reactive in amide bond forming processes than aliphatic primary amines.
3.1 Retrosynthetic analyses of the total syntheses and fragments
Naturally, the macrolactam can be dissected into two fragments: the ansa chain and the aromatic moiety. While the groups of Corey [39], Isobe [40], Gao [41] and Huu [42] relied on a macrolactamisation, Meyers and coworkers [38] achieved the cyclisation by an intramolecular Claisen reaction thus forming the bond between C2 and C3 (Fig. 6). Common to all five syntheses is the use of organometallic transformations for setting up the diene moiety.

Strategic bonds in successful total synthesis approaches.
More detailed retrosynthetic analyses of the successful syntheses are summarised in Schemes 1 and 2. While Meyers, Isobe and Huu targeted the biologically not active maytansinol (7), the groups of Corey and Gao achieved to prepare the cytotoxic natural product maytansine (1).
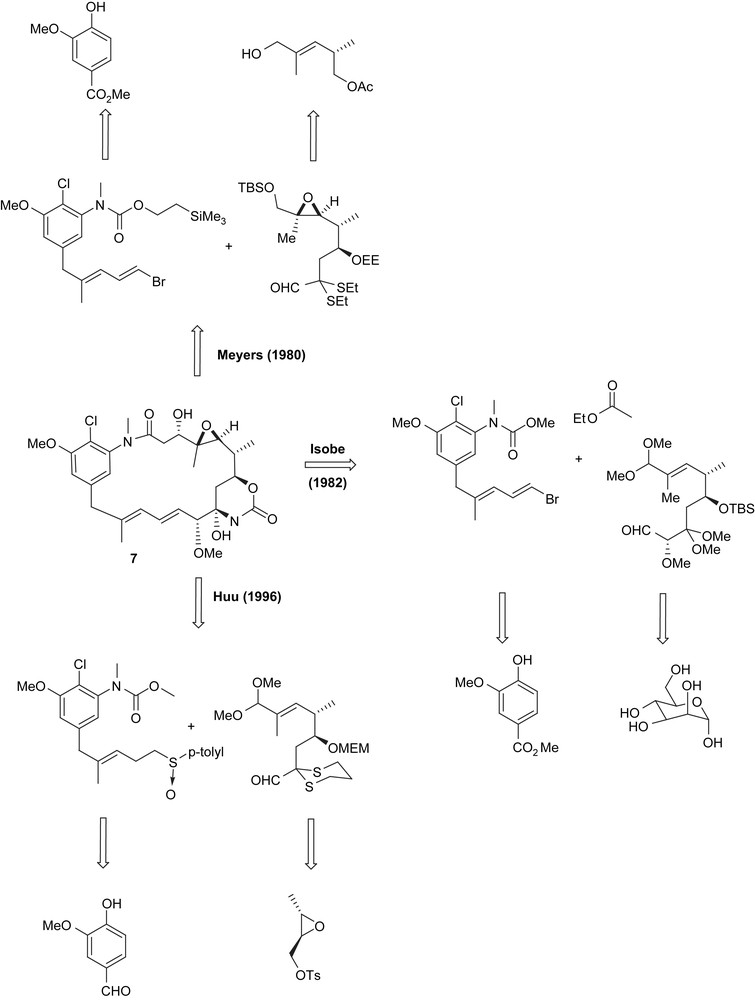
Retrosynthetic approach of maytansinol (7) by Meyers [38], Isobe [40] and Huu [42].

Retrosynthetic approach of maytansine (1) by Corey [39] and Gao [41].
Studies on the synthesis of fragments appeared very shortly after the first discovery of maytansine (1). Thus, the aromatic moiety was prepared by Götschi [43], Meyers [38], Ganem [44], Corey [39] and Ho [47]. Ganem [44], Ho [47], Fried [48] and Parsons [49] dealt with the construction of the carbamoyl system in the eastern fragment and Vandewalle [45] and Barton [46] directed their efforts mainly on the installation of the correct stereochemistry at C6 and C7 (Fig. 7).

Reported syntheses directed towards selected fragments [43–49].
Confalone et al. investigated a new method of ring closure via the carbamoyl group which they expected to be a key reaction in the total synthesis of maytansine (1) (Scheme 3) [50]. The intramolecular [3 + 2] cycloaddition reaction of a nitro alkane with an alkene in 18 yielded isoxazoline 19 which was hydrogenated in the presence of Raney-Ni, and further transformed into an amide to form the desired carbamide 20.
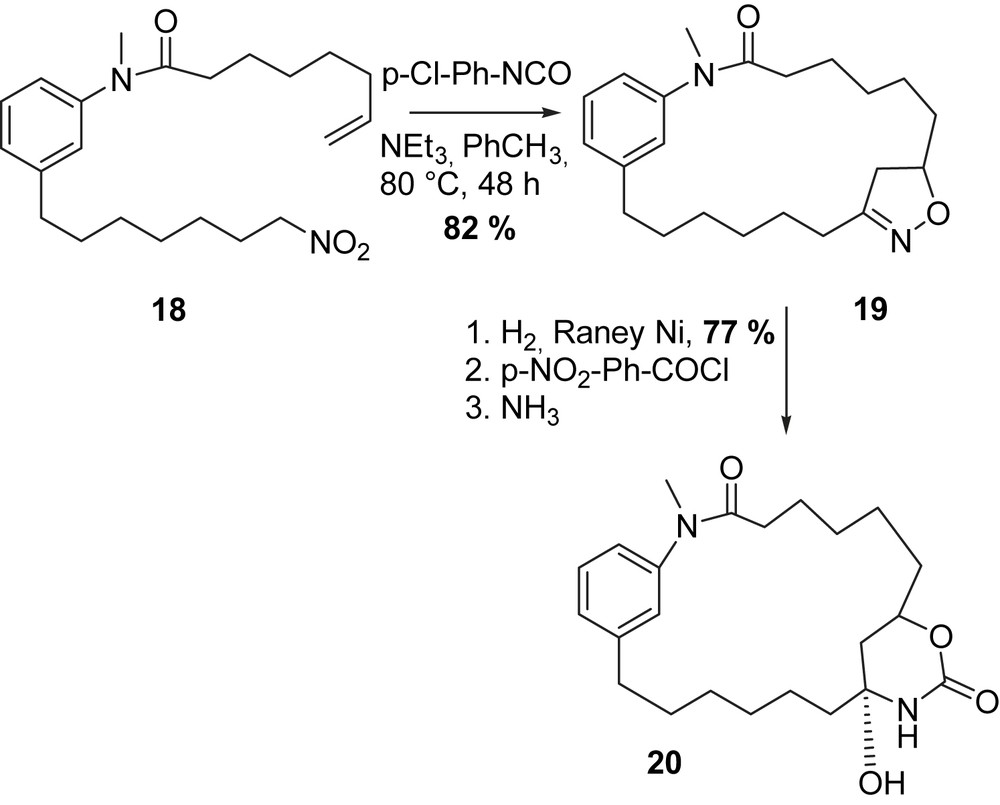
Confalone's nitrile oxide–olefine [3 + 2] cycloaddition route to maytansinoids [50].
The following detailed description will only cover the four successful total syntheses of the Meyers, Corey, Isobe and Huu groups. The Gao synthesis is very close in identity with that reported by Corey and therefore will not be discussed in detail. The individual syntheses are not described one by one but are rather divided into subsections that discuss fragment syntheses, fusion of fragments and the end games. From our point of view this approach allows us and certainly also the readers to better compare and evaluate the four strategies. As a period of almost two decades is covered the direct comparison is able to reveal (historical) the developments and changes in the strategy and methodology until the mid-1990s. In chapter 4 the total synthesis of proansamitocin published last year is described. Again, new reactions can be found unknown during the time of the first wave of synthetic interest in the maytansinoids. This fact is clearly indicating the ongoing explosive growth of new methodologies that are still being added to the synthetic chemist's arsenal.
3.2 Synthesis of the aromatic moiety including coupling to the diene unit
Related to Götschi's first studies, Meyers, Isobe and Gao devised synthetic accesses towards the tetra-substituted benzene moiety by starting from ethyl or methyl vanillate, respectively. Huu's approach is very closely related in the fact that they started from vanillin. Finally, Corey's synthesis is based on a completely different and unique sequence. Once the aromatic ring was in hand, the diene unit was introduced in miscellaneous ways. Meyers and Corey linked this functionalised fragment via a cuprous intermediate, Isobe by organolithium chemistry, while Gao and Huu have chosen a Wittig approach.
3.2.1 The Meyers synthesis
Meyers synthesis started with methyl vanillate 21 (Scheme 4) which was nitrated and chlorinated as described before [52]. Subsequent bromination, reduction of the nitro group and transformation of the intermediate aniline into the corresponding silylethylurethane and finally N-methylation yielded fully functionalised aromatic key intermediate 22. Metal–halogen exchange and cuprate formation allowed them to couple with mesylate 24 (obtained from β-bromoacroleine 23 by a set of standard reactions) to yield vinyl bromide 25.
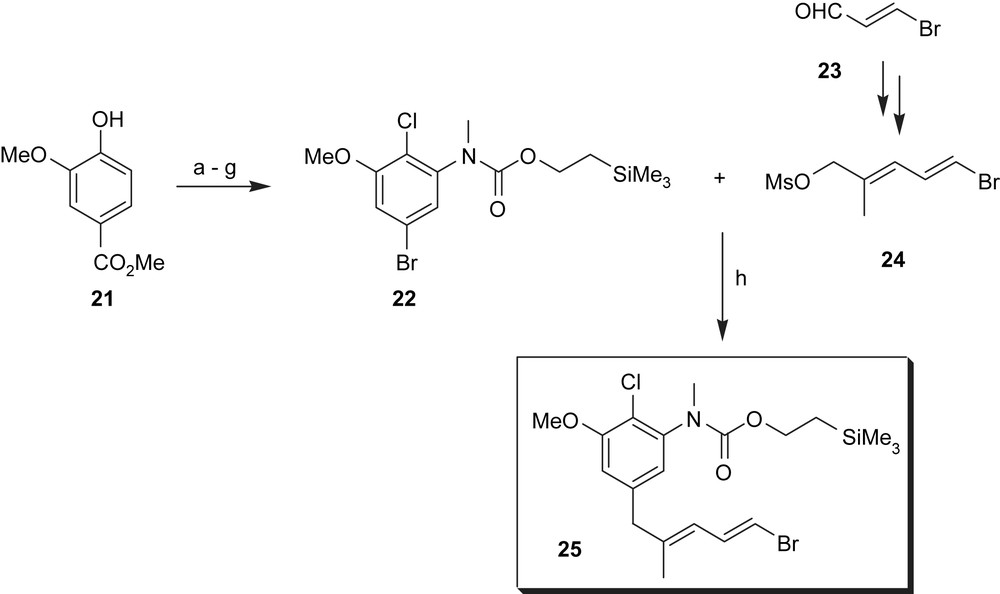
(a) HOAc–HNO3, 0 °C, 86%; (b) SOCl2–DMF, Δ, 61%; (c) 10% aq. KOH, 93%; (d) HgO–Br2, CCl4, Δ, 55%; (e) SnCl2–HOAc, 75%; (f) PhOCOCl, pyridine; (g) β-(trimethylsilyl)ethanol, 0–25 °C, THF, tBuOK; then tBuOK–MeI, 80% over three steps; (h) n-BuLi, −78 °C, C3H7CCCu·[(Me2N)3P]2, Et2O, −78 °C, 40–45%.
3.2.2 The Corey synthesis
The starting point of Corey's synthesis relies on gallic acid 26 which was dearomatised by Birch reduction (Scheme 5). Treatment of enone 27 with N-methylbenzylamine introduced the amino functionality. Chlorination, rearomatisation and N-methylation and reductive removal of the N-protection group furnished amino ester 28. A set of standard transformations gave benzyl iodide 29 which was cross-coupled with the mixed Gilman cuprate 30 to selectively form E-configured trisubstituted alkene 31. Aldehyde 32 (from 31 after deprotection and allylic oxidation) was transformed into dienal 34 using enolate equivalent of acetaldehyde 33.
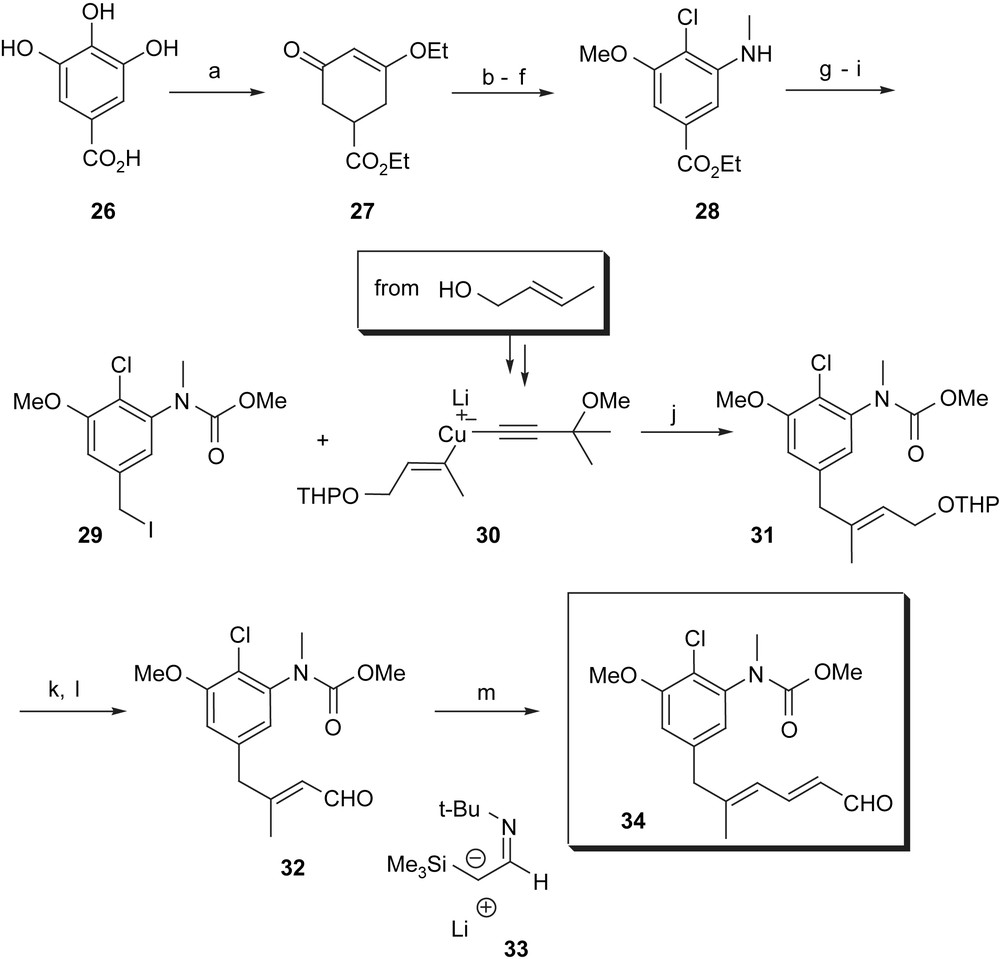
(a) Birch reduction, then EtOH–AcOH, 90%; (b) MeNHBn, 85 °C, 18 h, 85%; (c) tBuOCl, CHCl3, −50 °C, 80%; (d) LDA, THF, −78 °C, BnSeBr, 80%; (e) MeI, K2CO3, acetone, 95%; (f) Pd–C, EtOH, quant.; (g) LAH, THF, 0–5 °C, 30 min, then 23 °C, 45 h; (h) ClCO2Me, K2CO3, Δ, 12 h, then 4% NaOH in MeOH, 25 °C, 2 h, 90%; (i) MeSO2Cl, NEt3, THF, −78 °C to −25 °C, 30 min, then NaI, DME, −25 °C, 15 min, 23 °C, 2.5 h, 94%; (j) THF, −78 °C, 30 min, −25 °C, 4 h, 90%; (k) PTSA, MeOH, 23 °C, 12 h, 93%; (l) MnO2, CH2Cl2, 0 °C, 97%; (m) Et2O, 33, −78 °C, 1 h, then AcOH–NaOAc, 23 °C, 3 h, 80%.
3.2.3 The Isobe synthesis
Isobe's route for the construction of the western fragment completely resembles the synthesis disclosed by Götschi et al. [43] (Scheme 6). Benzyl iodide 29 was converted into phosphorous ylide 36 using 4-lithio-(phenylsulfonyl)pent-1-ene for elongation. The synthesis was finalised after oxidative cleavage, reduction to yield intermediate alcohol, bromination and final nucleophilic substitution with triphenyl phosphine and deprotonation.
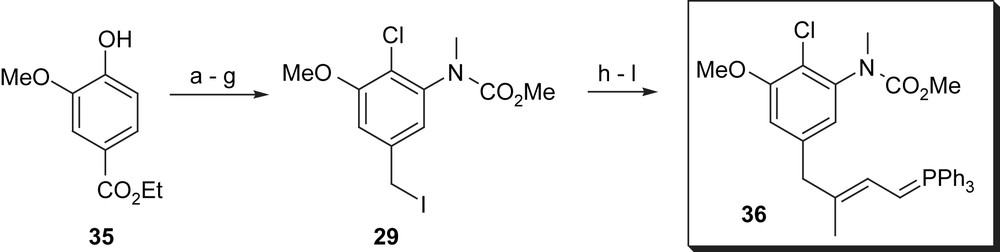
(a) HOAc–HNO3, 0 °C, 86%; (b) SOCl2–DMF, Δ, 61%; (c) SnCl2–HOAc, 75%; (d) MeI, K2CO3, acetone, 95%; (e) LiAlH4, THF, 0–5 °C, 30 min, then 23 °C, 45 h; (f) ClCO2Me, K2CO3, Δ, 12 h, then 4% NaOH in MeOH, 25 °C, 2 h, 90%; (g) MeSO2Cl, NEt3, THF, −78 °C to −25 °C, 30 min, then NaI, DME, −25 °C, 15 min, 23 °C, 2.5 h, 94%; (h) 4-lithio-4-(phenylsulfonyl)pent-1-ene, THF, −78 °C, 15 min; (i) O3, then Et3N (E:Z = 5:1); (j) NaBH4, EtOH; (k) PBr3, LiBr, collidine; (l) PPh3, CH3NO2, then tBuLi, THF–DMF, 45% for five steps.
3.2.4 The Huu synthesis
Finally, vanillin 37 served as a starting point in Huu's synthesis of the western maytansinol fragment (Scheme 7). It was nitrated, chlorinated and the aldehyde was converted into the corresponding protected aldehyde. After reduction, the aniline derivate was carboxylated to yield acetal 38 which was further transformed into benzaldehyde 39 (N-methylation, aldehyde deprotection). Introduction of a functional chain was realised by using phosphine oxide 40 in a Horner–Wittig olefination. Acetal hydrolysis yielded intermediate aldehyde which forced the olefinic double bond into conjugation. The diastereomeric E/Z-ratio was determined to be 3:1. After reduction the isomeric allyl alcohols were separated and the E-isomer was transformed into homoallylic sulfone 42 after bromination and substitution with lithiated methylphenylsulfoxide.

(a) HNO3, Et2O, 10 °C then rt, 4 h, 88%; (b) LiCl, TsCl, HMPA, 80 °C, 18 h, 72%; (c) HOCH2CH2OH, TsOH cat., PhH, Δ, 16 h, 100%; (d) H2NNH2·H2O, EtOH, Raney nickel, rt, 40 min, 99%; (e) ClCO2CH3, pyridine, rt, 2 h, 99%; (f) NaH, THF, 0 °C then MeI, rt, 3 h, 99%; (g) 1 N HCl, acetone, rt, 1.5 h, 100%; (h) LDA, THF, 10 min, 0 °C to −78 °C, 40 in THF, −78 °C to rt, extraction, NaH, DMF, 50–60 °C, 1 h, 89%; (i) 1 N HCl, acetone, 24 h, rt; (j) NaBH4, EtOH, 0 °C; (k) LiBr, Et2O/THF, PBr3, collidine, 0 °C then rt, 50 min, 95%; (l) LDA, THF, methylphenylsulfoxide, −78 °C, then HMPA, −78 °C to −20 °C, 90 min, 68%.
3.3 Synthesis of the ansa chain
The retrosynthetic cuts of all total syntheses are very similar. Thus, besides the construction of the aromatic ring with the attached diene unit, the preparation of the ansa chain bearing several stereogenic centers had to be envisaged by all groups which again opens the door for easy comparison. The Meyers, Gao and Huu syntheses all are based on a small (C4) starter building block and introduce the stereocenters by enantioselective methods or small chiral molecules [38,41,42]. All three groups rely on organolithium species for the key C–C bonding forming process. Corey's and Isobe's strategies pursue a “Chiron approach” [52] utilising carbohydrates as chiral starting material. The Isobe synthesis may seem to be lengthy but three stereogenic centers are derived from the carbohydrate while Corey's and Gao's syntheses only install two stereocenters.
3.3.1 The Meyers synthesis
The Meyers group started from protected methallyl alcohol 43 which was subjected to an oxidative hydroboration (Scheme 8). Pfitzner–Moffat oxidation yielded intermediate aldehyde which was elongated and condensed using lithiated cyclohexylimine based on propanal which afforded the corresponding α,β-unsaturated aldehyde 44. Reduction to the allylalcohol and protection via silylation were followed by epoxidation with m-chloroperbenzoic acid.

(a) B2H6, NaOH–H2O2 then hydrolysis; (b) DCC, DMSO, TFA, pyridine, 79% for two steps; (c) C6H11NCHC2H5, LDA, −78 °C, THF; (d) oxalic acid–H2O–THF, 25 °C, 18 h, 70% for 2 steps; (e) 5% HCl–THF (1:1), 100%; (f) CH3COCl, pyridine, CH2Cl2, 0 °C, 95%; (g) NaBH4, EtOH, 25 °C; (h) TBSCl, DMAP, imidazole, CH2Cl2; (i) m-CPBA, CH2Cl2, 0 °C, 53:47; (j) MeMgCl (2 eq), THF, 0 °C; (k) Collins oxidation; (l) lithiodithioacetate, −78 °C, THF, 6 h then HOAc, −78 °C, 3:1; (m) ethyl vinyl ether, p-TsOH·H2O, 25 °C, 1 h; (n) EtMgI, −45 °C, THF, 2 h; (o) 2-(N-methyl-N-formyl)-aminopyridine, 7% for nine steps.
Various epoxidation procedures were attempted, but all gave unsatisfactory mixtures of diastereomers. Finally, m-chloroperbenzoic acid furnished a 53:47 mixture in favour of the desired isomers which could be separated after conversion into the corresponding alcohols. The desired diastereomer 45, which was obtained in 25% yield from aldehyde 44, was treated with Collins reagent followed by ethyl lithiodithiodiacetate which resulted in the formation of diastereomeric β-hydroxy dithioesters (dr = 3:1). After separation the major isomer was protected as an ethoxyethyl acetal. Nucleophilic S-attack with ethyl-Grignard and formylation of the metallated dithioacetal yielded α-formyl dithioacetal 47.
3.3.2 The Corey synthesis
Commercially available tri-O-acetyl-d-glucal 48 was first transformed into the trityl protected derivative 49 via six standard steps in Corey's synthesis (Scheme 9). Treatment of 49 with propane-1,3-dithiol resulted in the cleavage of the pyranose ring and the trityl ether to give trihydroxyalkyl-1,3-dithiane in a very good yield. The intermediate dithiane was selectively protected as a cyclopentyl acetal and subsequently as methoxyethoxymethyl ether to afford dithiane 50.
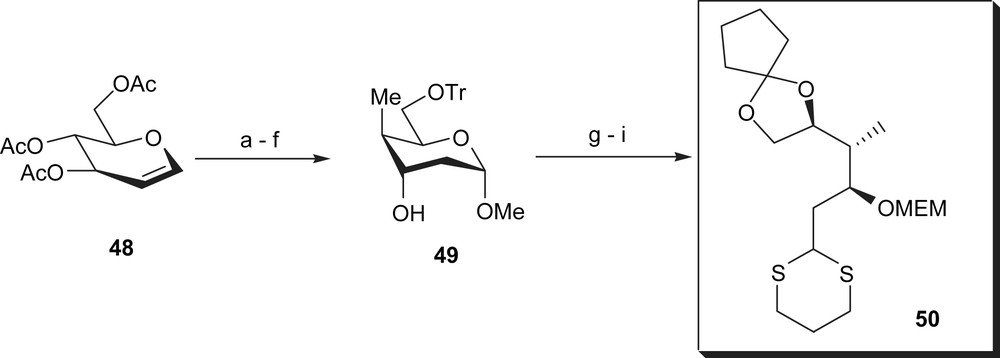
(a) Cat. NaOMe in MeOH, 23 °C, 1.5 h; (b) HgOAc, 23 °C, 2.5 h, 75% for two steps; (c) NaCl, MeOH, 23 °C, 15 min then 0 °C, NaBH4, i-PrOH, 30 min then HCl, 99%; (d) TrCl, pyridine, 23 °C, 16 h; (e) NaH, HMPT, 5 °C then 23 °C, 30 min, then THF, −25 °C, triisopropylbenzenesulfonylimidazole, −25 °C to −5 °C, 3 h, 90%; (f) MeLi, CuI, Et2O–toluene, −78 °C, 48 h and −45 °C, 48 h, 95%; (g) propane-1,3-dithiol, CHCl3–HCl (12 N), 0 °C, 15 min, 96%; (h) 1-ethoxycyclopentene, BF3–Et2O, THF, −30 °C, 30 min, 86%; (i) MEMCl, DIPEA, CH2Cl2, 20 °C, 20 h, 99%.
3.3.3 The Isobe synthesis
Isobe and coworkers also relied on a chiral carbohydrate precursor, namely d-mannose 51 (Scheme 10). It needs to be noted that the unfunctionalised CH2 group at C8 (maytansine numbering) derives from C4 (carbohydrate numbering) and not from C2 as in Corey's synthesis. Based on an established sequence, epoxide 52 was first prepared and reductive ring opening was then effected with lithio triethylborohydride. The resulting alcohol was O-benzylated, the trityl group was removed and the resulting alcohol at C6 was oxidised to the corresponding aldehyde 53. This set the stage for chain elongation by condensing it with [bis(trimethylsilyl)(phenylsulfenyl)methyl]lithium followed by oxidation with m-CPBA. The resulting vinylsilylsulfone was stereoselectively methylated in a Michael-type fashion and desilylated with potassium fluoride. After removal of the benzyl ether and reprotection as THP ether, sulfone 54 was obtained as a single isomer. It took another six steps (α-alkylation of sulfonyl group, THP removal, oxidative acetalisation, transglycosidation, substitution of the chloro group by phenylthiol and oxidation to the corresponding bissulfone) to achieve the synthesis of homoallyl sulfone 55. Sodium borohydride-mediated reduction yielded the corresponding open chained diol which was monoacetylated and silylated. Finally, ozonolysis (which led to sulfinic acid elimination) and reductive workup afforded the α,β-unsaturated aldehyde which was protected as acetal, deacylated and oxidised to the desired aldehyde 56.
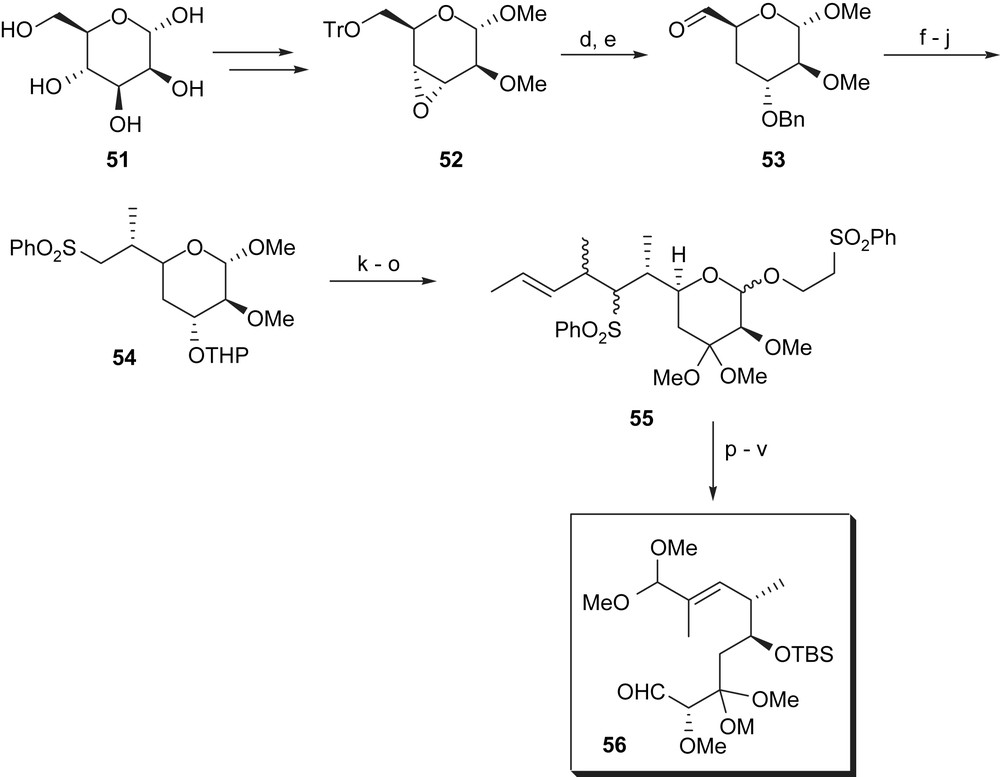
(a) i. (CH3)2C(OMe)2–PPTS, DMF, 5 °C, 36 h; ii. TsCl, pyridine–CH2Cl2; iii. NaH–MeI, THF, 60% for three steps; (b) Dowex 50 W, MeOH, 100%; (c) i. TrCl, pyridine; ii. tBuOK, THF, 0 °C, 30 min, 86% for two steps; (d) i. LiBHEt3, −20 °C, 5 d, then rt; ii. BnBr, NaH, THF, DMSO, rt, 1 d; iii HCl, CHCl3, 15 h, rt, 67% for three steps; (e) CrO3/2-pyridine, CH2Cl2, 69%; (f) PhS(Me3Si2)CLi, THF–hexane, −45 °C, 5 min then rt, 30 min; (g) m-CPBA, 72% over two steps; (h) i. MeLi, THF, −78 °C, 30 min; ii. KF, MeOH–CH2Cl2, Δ, 100% for two steps; (i) H2, Pd-black, EtOH–AcOH, Δ, 2 d, 98%; (j) DHP, PPTS, CH2Cl2; (k) n-BuLi/4-bromopent-2-ene, THF–HMPA; (l) i. HOCH2CH2Cl, HC(OMe)3, CSA; ii. CrO3/2-pyridine, CH2Cl2; (m) HOCH2CH2Cl, HC(OMe)3, CSA, MeOH; (n) PhSNa; (o) m-CPBA; (p) NaBH4, EtOH, Δ then THF; (q) AcCl, pyridine; (r) tert-BuMe2SiCl, imidazole, DMF; (s) O3, CH2Cl2, then Et3N, 38% for 11 steps; (t) PPTS, CH(OMe)3, MeOH; (u) NaOMe, MeOH; (v) CrO3/2-pyridine 91% for three steps.
3.3.4 The Huu synthesis
The starting material 57 of Huu's approach was obtained by Sharpless epoxidation of (E)-crotyl alcohol followed by tosylation (Scheme 11). Regioselective ring opening with a propynyl alane species yielded alkyne 58 which was subjected to regioselective hydrostannylation according to Zhang's procedure [53] and base-mediated epoxide formation. The resulting vinyl stannane 59 was treated with lithiated dithiane which led to epoxide ring opening. After O-protection with the MEM-group, the stannyl group was transmetallated into the organocopper species and malonaldehyde monodiethyl acetal was added. Silyl protection afforded dithiane 60 which was followed by a formylation to yield aldehyde 61. This step was achieved after dithiane deprotonation and reaction with DMF.

(a) H3CCCAlEt2, 50%; (b) Bu3SnH, PdPh2Cl2, rt, THF, 70%, (c) MeONa, MeOH, rt, 1 h, 100%; (d) LiHCS(CH2)3S, rt, THF, 16 h, 81%; (e) MEMCl, DIPEA, DMAP, CH2Cl2, rt, 72 h, 85%; (f) (CH3)2CuCNLi2, 0 °C, 30 min then malonaldehyde monodiethyl acetal, 30 min, −70 °C to −40 °C, 77%; (g) TBSCl, imidazole, DMF, rt, 12 h, 80%; (h) n-BuLi, TMEDA, −30 °C, THF, −20 °C, 3 h then −78 °C, DMF then 0 °C, 30 min, 97%.
3.4 Coupling of the aromatic moiety with the ansa chain and formation of seco-maytansinoids
The Meyers [38], Corey [39], Gao [41] and Huu [42] groups have carried out the merging of the two major fragments by organolithium chemistry. Isobe's [40] approach relies on the Wittig olefination for linking the major building blocks.
3.4.1 The Meyers synthesis [38]
The Meyers group coupled the two major fragments 25 and 47 by first carrying out a lithium–bromide exchange, followed by the addition of 47 to yield carbinol and subsequently the methyl ether (Scheme 12). After removal of both silyl protecting groups, the primary alcohol 62 was oxidised, using azodicarbonyldipiperidine and tert-butoxymagnesiumbromide. The resulting aldehyde was N-acylated and then oxidised to the carboxylic acid (seco acid). Finally, methylation yielded the corresponding methyl ester 63.
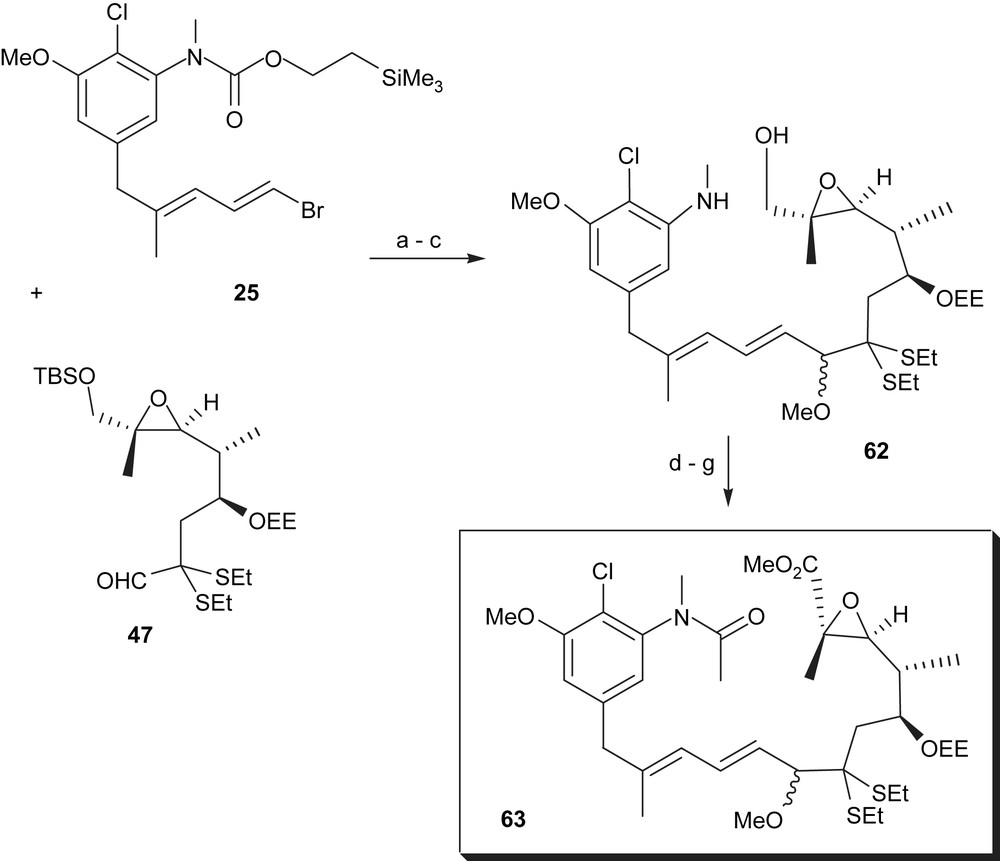
(a) tBuLi, −120 °C to −90 °C, THF–Et2O–pentane, then 47, −120 °C to −60 °C, 30 min; (b) NaH, THF, CH3I, 0–25 °C, 2 h, 70% for two steps; (c) TBAF, THF, 4 h, 100%; (d) 1,1-(azodicarbonyl)dipiperidine, tert-BuOMgBr, 71%; (e) H3CCOCl, pyridine, 0 °C, 3 h, 97%; (f) AgNO3, NaOH, THF–H2O, 25 °C, 2 h; (g) H2CN2, Et2O, 0 °C, 65%.
3.4.2 The Corey synthesis
The Corey synthesis continued with the lithiation of the dithiane moiety 50 and nucleophilic addition to the aldehyde group in 34 (Scheme 13). As a result, a 1:1 mixture of the epimers was formed. The wrong epimer was transformed into the desired one by an oxidation–reduction sequence. The resulting coupling product was methylated at C10 to yield 64, N-deprotected and liberated from the cyclopentanone protection. Oxidative 1,2-diol cleavage with lead tetraacetate afforded aldehyde 65, which was transformed into the hydroxy phenylester 66.
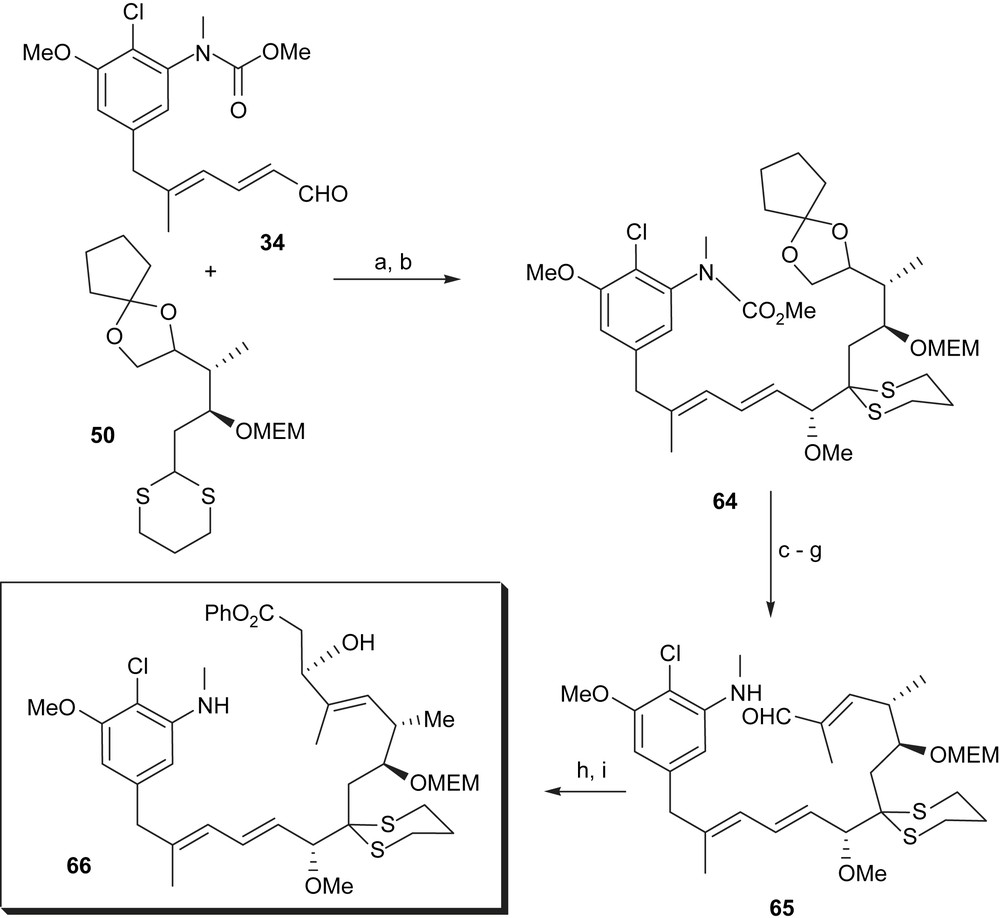
(a) n-BuLi, TMEDA, THF, −25 °C, 3 h then 34, THF, −78 °C, 0.5 min, 90% (1:1; 44% of the right enantiomer); (b) NaH, MeI, THF, 20 °C, 15 min, 91%; (c) LiOMe, DMF, 0 °C, 40 h, 90%; (d) HClO4, MeCN–H2O, 0 °C, 40 min; (e) Pb(OAc)4, MeCN, KOAc, −25 °C, 5 min, 95%; (f) α-lithio-α-TMS-propionaldehyde-N-tert-butylimine, −110 °C, 5 min then −78 °C, 10 min then SiO2 aq; (g) pyridinium chloride, CH2Cl2, 20 °C, 2 h, 82%; (h) (+)-p-tolyl phenoxycarbonylmethyl sulfoxide, tert-BuMgCl, −78 °C, 25 min, then 65 in THF, −78 °C, 15 h; (i) Al–Hg, THF–H2O, 23 °C, 2 h, >80% for two steps.
3.4.3 The Isobe synthesis
The Wittig reaction was the key reaction of the Isobe group to fuse the two major fragments 36 and 56 (Scheme 14). After deprotection of the amino group and liberation of the aldehyde functionality at C1, the synthesis was continued with aldehyde reduction, epoxidation of the allylic alcohol and reoxidation of the primary hydroxyl group to furnish epoxy aldehyde 67 as a single diastereoisomer. Aldol reaction using lithium ethyl acetate as enolate component yielded the corresponding ethyl ester as the enriched desired isomer. Finally, silylation at C3 afforded the advanced intermediate 68.

(a) THF, −63 °C to rt, 15 h, 78%; (b) THF–H2O–AcOH (4:4:1), −10 °C, 2 d; (c) NaBH4, MeOH, 0 °C, 15 min; (d) KOH–EtOH (1:3), Δ, 22 h, 84%; (e) tert-BuOOH, Ti(O-i-Pr)4, CH2Cl2, −20 °C, 1.5 h, 70%; (f) SO3–pyridine; (g) lithium ethyl acetate, −78 °C, 30 min; (h) TBSCl, imidazole, DMF, 35 °C, 12 h.
3.4.4 The Huu synthesis
The Huu group utilised another olefination reaction, for fusion of the two major fragments S-(−)-p-tolyl sulfoxide 42 and aldehyde 61. The alcohol 69 was generated as a 4:1 mixture in favour of the desired epimer (Scheme 15). These isomers could be separated by chromatography at this stage.

(a) LDA, DME–pentane, −78 °C, 1 h; (b) toluene, 80 °C, 73% (4:1) for two steps.
3.5 Ring closure and end games
Four of the five end games, namely those reported by Corey, Isobe, Gao and Huu are close in identity as they go via the seco acid and achieve ring closure by classical macrolactamisation. Therefore, the last steps are summarised for one of the four syntheses only. We have chosen to describe Corey's end game in detail because it represents the first successful end game of a maytansine total synthesis and most other groups have followed this beaten track. Only the approach developed by the Meyers group differs substantially from the other four and is therefore also included in this article.
3.5.1 The Meyers end game
The final cyclisation relies on an intramolecular Claisen reaction and was accomplished in 58% yield. After sequential deprotection at C9 and C7, the alcohol at C7 was activated as carbonate, which was immediately transformed into the cyclic carbinolamide by treatment with ammonia. The final step of the Meyers synthesis comprised ketone reduction at C3 using sodium borohydride, which yielded an epimeric mixture of four compounds. The major isomer (−)-maytansinol (7) was isolated in 45% yield (Scheme 16).
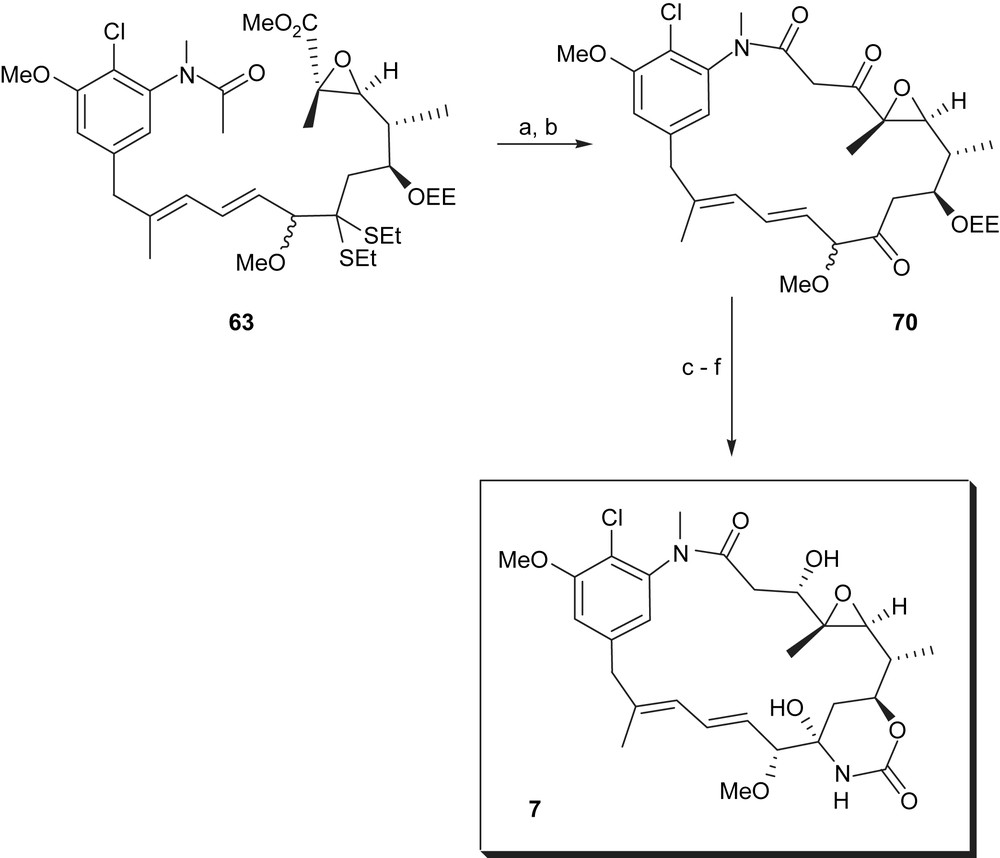
(a) LiHMDS, −78 °C, THF, 4 h, 58%; (b) HgCl2, CaCO3, MeCN–H2O (4:1), 25 °C, 1.5 h, 98%; (c) 1 N HCl, THF, 0 °C, 2 h; (d) PhCOCl, pyridine, Et2O–THF (1:1), 0 °C, 1 h; (e) NH3, THF, −78 °C, 15 h, 50–55%; (f) NaBH4, THF–MeOH (1:1), −40 °C, 30 min, 45%.
3.5.2 The Corey end game
The cyclisation of seco acid 71 was smoothly effected by using the mixed anhydride with mesitylenesulfonic acid for carboxyl activation and yielded macrolactam 72 in 71% yield. Removal of the methoxymethyl ether and subsequent carbamidation of the C7 alcohol yielded dithiane 73 (Scheme 17). Dithiane hydrolysis led to the desired cyclic urethane in good yield. After desilylation at C3 the liberated hydroxy group at C9 was selectively methylated and the allylic alcohol epoxidised to furnish (−)-9-methyl-maytansinol, which could be hydrolysed to (−)-maytansinol (7). The following ester formation to maytansine (1) was accomplished as was first described by Takeda [54].

(a) TBAOH, PhH, then MsCl, DIPEA, PhH, 40 °C, 28 h, 71%; (b) i-PrSH, CH2Cl2, −78 °C, BF3·Et2O, 5 min, then TBAOHaq–PhH–i-PrSH (8:8:1), 27 °C then AgNO3, 2,6-lutidine, THF–H2O (4:1), 25 °C, 1.75 h; (c) p-NO2PhCOCl, pyridine, 27 °C, 20 min, then 15 N NH4OH–H2O–t-BuOH (1:2.3:5), 27 °C, 2 h; (d) HgCl2, CaCO3, MeCN–H2O (5:1), 25 °C, 12 h, then DIPEA, 60% for three steps; (e) MeCN–HF–H2O, 0 °C, 45 min, 83%; (f) p-TsOH, MeOH, 25 °C, 30 min, >90%; (g) tert-BuOOH, VOacac2, 2,6-lutidine, toluene–PhH (10:7), 25 °C, 3.5 h then Me2S, 25 °C, 30 min; (h) NaBH4, EtOH–H2O (pH 7), −5 °C, 87% for two steps; (i) hydrolysis, >90%.
The other three groups have utilised the Corey route for ring closure. The remaining steps are only trivial transformations for deprotection, carbamoylation and cyclisation.
4 Mutasynthetic studies on ansamitocins
It has to be noted that in principle any kind of derivative can be synthesised by total synthesis. However, none of the total synthesis approaches described above resulted in novel analogues suitable for SAR studies. With a few exceptions1 [55] this is a typical problem of total syntheses aiming at complex natural products. As a consequence, total syntheses are commonly considered to be inefficient for the generation of natural product derivative libraries. On the other hand, semisynthesis approaches are usually limited by the different functional groups present in natural products. Their inherent reactivity profiles clearly limit the possibilities for subsequent modifications and result in the fact that only certain types of derivatives are available by semisynthesis.
In recent years, advances in metabolic engineering have given rise to a new approach offering access to novel types of derivatives. The basic idea of thisconcept called mutational biosynthesis (synonym:-mutasynthesis) is the introduction of modifications before the natural product is assembled during biosynthesis. This is made possible by the construction of mutants of the microbial producer strains blocked in the biosynthesis of a key building block required for the synthesis of the target natural product. Analogues of the respective building block can then be fed to cultures of the mutants and can lead to unnatural derivatives not available via semisynthesis. For a recent review the interested reader is directed to Ref. [56].
Due to the uniqueness of the starter unit 3-amino-5-hydroxybenzoic acid 74 used for the biosynthesis of ansamycin antibiotics, the internal biosynthesis of this essential building block could be disrupted without affecting other pathways, leading to the mutant strain A. pretiosum HGF073 unable to produce ansamitocin without external starter unit addition [57]. A series of 3-aminobenzoic acid analogues 76–78 were fed to this strain and as a result 19-halogenated 20-demethoxyansamitocins bearing fluorine, chlorine or bromine substituents 80–82 could be generated (Scheme 18) [58]. When a fermentation of the mutant strain was supplemented with methoxy-aminobenzoic acid 75 19-dechloroansamitocin P-3 79 was formed.

Successful mutasyntheses of ansamitocin derivatives.
All new derivatives exhibited strong antiproliferative activity against several tumor cell lines (IC50 values in pg mL−1 range). In a second study the ansamitocin core-macrolactam proansamitocin 83 was prepared by chemical total synthesis employing a diene–ene ring-closing metathesis as the key macrocyclisation step (Scheme 19) [59a].

Total synthesis of proansamitocin 83 and transformation to ansamitocins. (a) NH3, NH4Cl, 180 °C, 40 h; MeOH, AcCl, 75% over two steps; (b) Boc2O, THF, H2O, NaHCO3; TBDPS–Cl, CH2Cl2, imidazole, 4-DMAP; Dibal, THF, −78 °C to −30 °C; CBr4, PPh3, CH2Cl2, 85% over four steps; (c) 90, Pd(pddf)Cl2 (2 mol%), THF, Δ, 5 h, 96%; (d) TBAF, THF, −10 °C, 88%; (e) AlMe3 (9 eq), Cp2ZrCl2 (3 eq), CH2Cl2, rt, 72 h; I2, THF, −30 °C, 58% over two steps; (f) Teoc–Cl, py, CH2Cl2, 89%; (g) 91, Pd(PPh3)4, DMF, 55 °C, 79%; (h) ZnCl2, MeNO2, ultrasound, 48%; (i) TBDPS–Cl, DMAP, imidazole, CH2Cl2, 89%; (j) TBDPS–Cl, imidazole, 4-DMAP, DMF; Dibal, CH2Cl2, −78 °C to −30 °C; Dess Martin-periodinane, CH2Cl2; 92, CH2Cl2, 89% over four steps; (k) Dibal, CH2Cl2, −78 °C to −30 °C; Dess Martin-periodinane, CH2Cl2; 93, TiCl4, NEt3, 86% over three steps, dr > 20:1; (l) TBSOTf, 2,6-lutidine, CH2Cl2, 95%; (m) LiBH4, H2O, Et2O, 92%; (n) PMBOC(NH)CCl3, TrBF4, CH2Cl2; NaOH (10%), MeOH, 66% over two steps; (o) Dess Martin-periodinane, CH2Cl2, 84%; (p) 93, N-ethylpiperidine, Sn(OTf)2, CH2Cl2, 99%, dr > 99:1; (q) HN(OMe)Me·HCl, AlMe3, CH2Cl2; TBSOTf, 2,6-lutidine, CH2Cl2; Dibal, toluene, 88% over three steps; (r) 94, BF3·OEt2, CH2Cl2, 87%, dr > 95:5; (s) DDQ, CH2Cl2, H2O, 97%; (t) DMSO, (COCl)2, NEt3, −78 °C to −45 °C, 83%; (u) NaOCl2, NaH2PO4, tBuOH, H2O, 99%; (v) BOP–Cl, NEt3, 41%; (w) Grubbs I, CH2Cl2, rt, 23%; (x) HF·py, THF, 94%.
The macrocycle is the first free intermediate formed during ansamitocin biosynthesis and is liberated from the polyketide synthase by a macrolactam-forming amide synthase. Consequently, it could be readily transformed by the mutant strain A. pretiosum HGF073 into ansamitocin P-3 11d, with 19-dechloroansamitocin P-3 79 being the minor by-product [59a].
5 Conclusions
Despite the fact that the maytansinoids are an “old” and well-established group of ansamycin antibiotics with very strong antiproliferative activity, they attract renewed interest in academic research as well as in laboratories of pharmaceutical industry. In their “early days” research was mainly devoted to total and semisyntheses, accompanied by their biological evaluation as drugs for the treatment of solid tumors. Having made it to the clinical phase, studies with maytansinoids had to be stopped due to toxic side-effects. Despite all efforts, this first wave of interest did not provide compound libraries of sufficient size and with appropriate structural diversity to fully appraise the maytansinoid class's potential.
More than 10 years have passed since the last major synthetic work on maytansine by Huu [42] has been published. Especially driven by pharmaceutical companies investing in the development of drugs based on the fusion of maytansine with tumor-associated antibodies, the renewed interest in this group of ansamycin antibiotics has inspired new research projects aiming at the generation of maytansinoid libraries. A highly attractive strategy for this approach is the utilisation of mutant organisms blocked in key biosynthesis steps for the generation of derivatives by mutational biosynthesis.
Undoubtedly, the chapter on maytansinoids has been reopened again.
1 The highly active epothilone derivative ZK-EPO was part of a compound library of about 300 active analogues which were exclusively obtained by total synthesis.