

1 Introduction
In 2007, Wright et al. reported the isolation of a new marine natural product, neopeltolide, from a deep-water specimen of a sponge of the family Neopeltidae which was collected off the northwest coast of Jamaica [1]. The sponge was not identified, but it most closely matches the taxonomic description for the genus Daedalopelta [2], and described as a close relative member of Callipelta which is a rich source of biologically active marine compounds [3].
2 Biological activity
Biological assays of neopeltolide revealed potent in vitro cytotoxicity toward various cancer cell lines such as A-549 human lung adenocarcinoma (IC50 = 1.2 nM), NCI-ADR-RES human ovarian sarcoma (IC50 = 5.1 nM), and P 388 murine leukemia (IC50 = 0.56 nM). Neopeltolide is also a strong inhibitor of PANC-1 pancreatic and colorectal cell lines, both of which have p53 mutations. It is worthy of note that only 50% cell death was observed over an extended dose range, suggesting a cytostatic rather than a cytotoxic effect [1]. Preliminary investigations into the mechanism of action of neopeltolide suggest that it does not act via interaction with tubulin or actin. In addition, neopeltolide is a potent inhibitor of the growth of the fungal pathogen Candida albicans with a MIC value of 0.62 μg mL−1 in liquid cultures [1b].
In a recent report, Kozmin et al. [4] reported that neopeltolide may inhibit mitochondrial ATP synthesis. They have evaluated the activity of the four mitochondrial electron transfer chain complexes in yeast and mammalian cells and found cytochrome bc1 complex as the principal cellular target. These results provide the molecular basis for the potent antiproliferative activity of this compound.
3 Structural determination
The structure of neopeltolide was established on the basis of NMR, HRMS [1], and by total synthesis. By NMR and HRMS studies, neopeltolide was identified as a 14-membered macrolactone possessing an anti-1,3-oxygenated moiety, a trisubstituted tetrahydropyran, and an oxazole-bearing side chain identical to the side chain of leucascandrolide A [5,6], a compound with a related structure and similar bioactivity to neopeltolide. By using NOESY, and a series of double-pulsed field gradient spin echo (DPFGSE) NOE experiments, and by carefully analyzing the coupling constants by NMR, the relative stereochemistry between the different substituents was determined and the absolute configuration was established, at first, to be (11R,13R). However, a reassignment of the absolute configuration at C11 and C13 was achieved by total synthesis which has established that the C11 and C13 stereocenters actually possessed the (S) configuration (Scheme 1) [7].
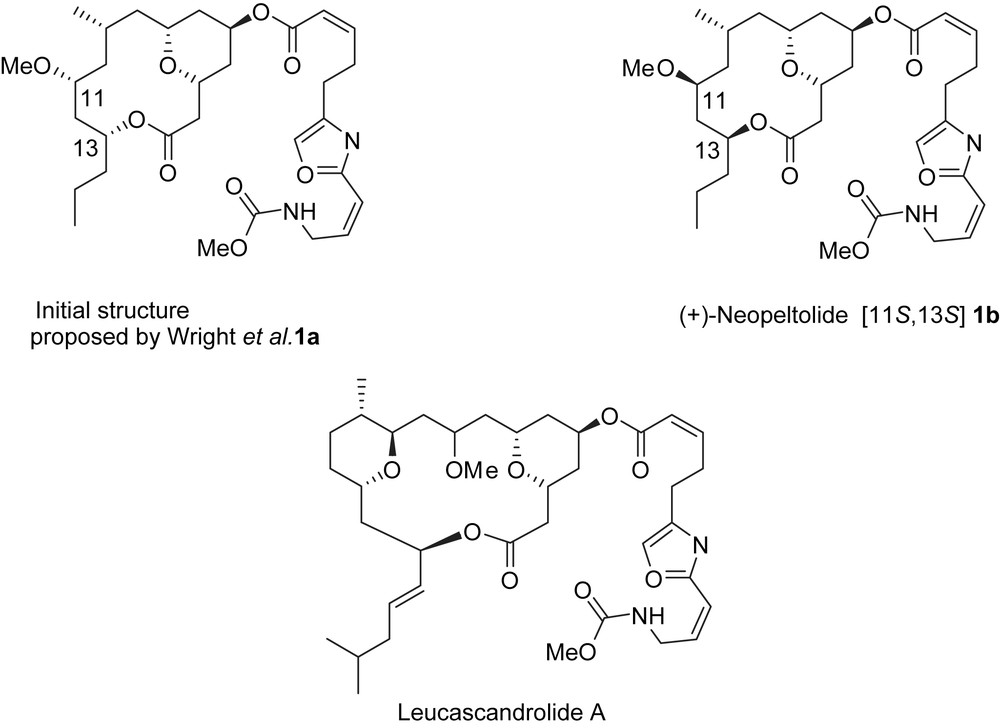
4 Synthesis
To date, six syntheses have been published. The first total synthesis of (+)-neopeltolide was achieved by Panek et al. in 2007 [7]. Their initial efforts focused on the synthesis of the proposed neopeltolide structure, and they obtained a compound that showed several 1H and 13C NMR spectroscopic discrepancies with the data from natural neopeltolide. In order to identify the correct stereochemical configuration of neopeltolide, Panek et al. undertook the synthesis of a set of diastereomers. The spectroscopic comparison of the prepared diastereoisomers allowed them to attribute the (S) configuration to the stereogenic centers at C11 and C13 instead of the (R) configuration initially proposed by spectroscopic analyses [1].
4.1 Panek et al. total synthesis [7]
Panek et al. synthesized (+)-neopeltolide by coupling the macrocyclic lactone A and the oxazole side chain B using a Still–Gennari olefination. The key steps involved the construction of the macrocyclic lactone by using a Yamaguchi esterification, a [4 + 2]-annulation strategy with an allylsilane to install the cis-disubstituted tetrahydropyran, a ring opening of an enantio-enriched epoxide to control the stereogenic center at C13, and a modified Evans–Tishchenko reduction to control the C11 sterogenic center (Scheme 2).
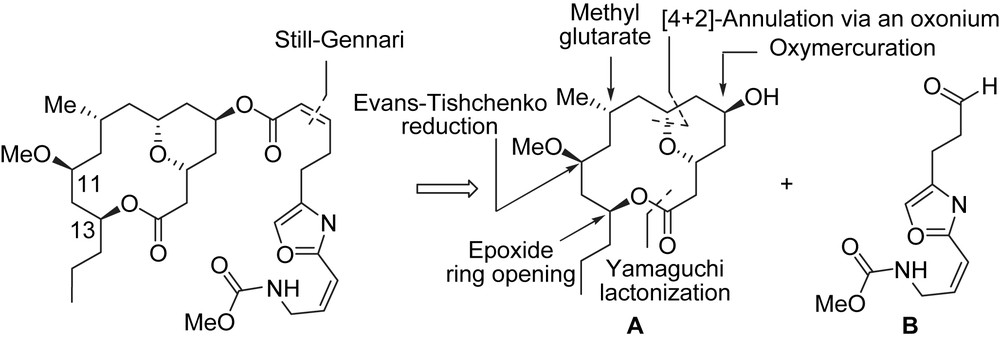
The synthesis of (+)-neopeltolide started with the preparation of the C7–C11 fragment from the commercially available methyl (R)-(+)-3-methyl glutarate 2 (Scheme 3). The chemoselective reduction of the carboxylic acid functionality in 2 (BH3·Me2S, THF) [8] to the corresponding primary alcohol, followed by TBDPS protection (TBDPSCl, imidazole) provided silyl ether 3, which was then transformed to dithiane 4 in two steps (reduction of the ester into an aldehyde and thioacetalization). Coupling of dithiane 4 with epoxide 5 [9] led to the C7–C16 fragment of (+)-neopeltolide, e.g. compound 6. Removal of the dithioketal (CaCO3/MeI) and stereodirected reduction of the resulting β-hydroxy ketone with a modified Evans–Tishchenko reduction [10] resulted in the anti-relationship between the ester group at C13 and the hydroxy group at C11 (dr = 14/1). The etherification of the hydroxy group at C11 (Me3O·BF4, proton sponge, molecular sieves 4 Å) [11] afforded 7. After a deprotection/oxidation sequence (HF, H2O then Swern oxidation) aldehyde 8 was produced and then involved in a [4 + 2]-annulation with allylsilane 9 [6h] (TfOH, CH2Cl2, PhH) thus leading to cis-dihydropyran 10 in good yield (75%) and with good diastereoselectivity (dr = 10/1) [6h]. The transformation of 10 to the macrocyclic lactone 11 was achieved in five steps and required the transformation of the mesylate group into a carboxylic acid. Mesylate 10 was treated with NaCN (DMF), followed by a first treatment with DIBAL-H in Et2O to selectively reduce the ester group at C13, and then by a second treatment with DIBAL-H in CH2Cl2 to reduce the cyano group to the corresponding aldehyde, which was oxidized using a Pinnick oxidation [12] (NaClO2) to the desired carboxylic acid. The obtained seco-acid was then transformed to macrolactone 11 through a Yamaguchi macrocyclization (44% yield). In order to introduce the hydroxyl group at C5, a regioselective oxymercuration [6h] of the intracyclic double bond yielded the axial C5 alcohol A as a single isomer.
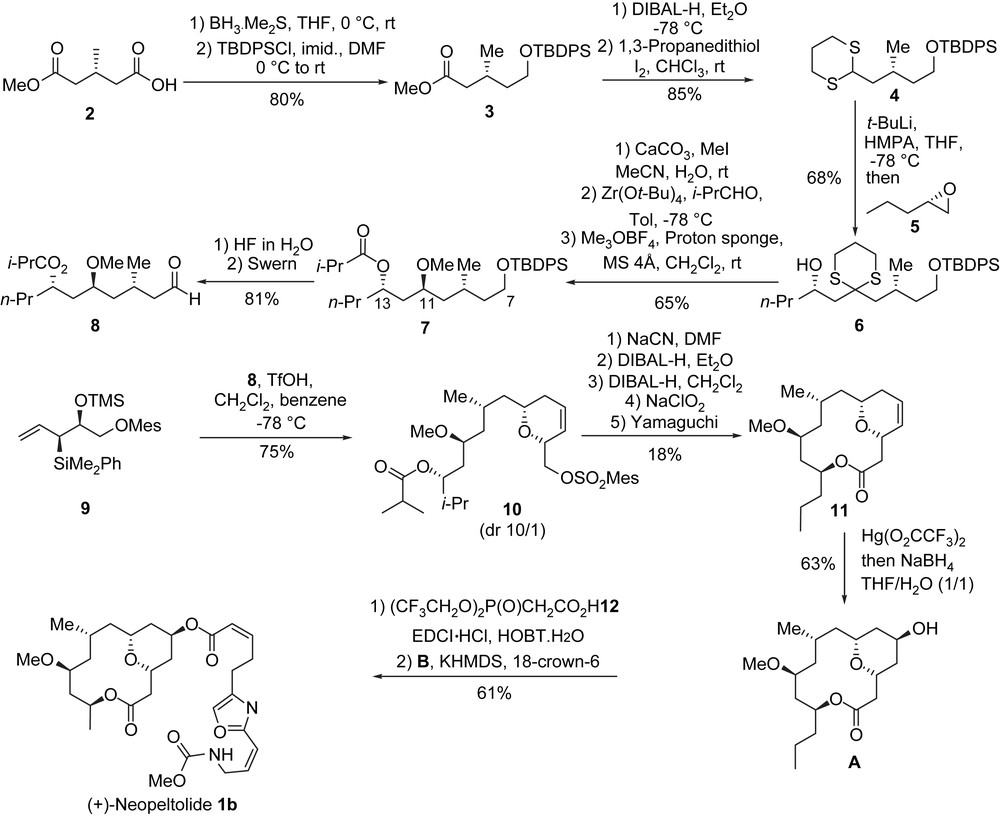
To complete the synthesis of (+)-neopeltolide, a Still–Gennari olefination [6a] was used to establish the cis-enoate of the side chain. At first, acylation of A with bis-(2,2,2-trifluoroethyl) phosphonoacetic acid (12) [13] gave the corresponding phosphonoacetate, which was then treated with KHMDS (THF, 18-crown-6), and the generated anion was quenched with aldehyde B providing a mixture of (+)-neopeltolide 1b and the corresponding (E)-olefin in a 7/1 ratio (62% yield).
The first enantioselective total synthesis required 19 steps for the longest linear sequence. (+)-Neopeltolide was obtained with an overall yield of 1.3%, starting from the commercially available methyl (R)-(+)-3-methyl glutarate 2 (Scheme 3).
4.2 Scheidt et al. total synthesis [14]
At the beginning of 2008, the syntheses of 1a and 1b were reported by Scheidt et al. [14]. Only the total synthesis of the natural product 1b will be described herein as the strategy and the key steps are identical for the two diastereomers.
The synthesis of (+)-neopeltolide was accomplished by involving an intramolecular Lewis acid-catalyzed cyclization [15] between a dioxinone and an in situ generated oxocarbenium ion (transition state A″) to establish the tetrahydropyran moiety and the macrocyclic ring in a single step. The oxazole side chain was introduced by using a Mitsunobu reaction between carboxylic acid B′ and the hydroxy group present on the cis-substituted tetrahydropyran at the C5 position. The synthesis of A′ is highly convergent and was achieved from the two compounds A1 and A2 (Scheme 4).
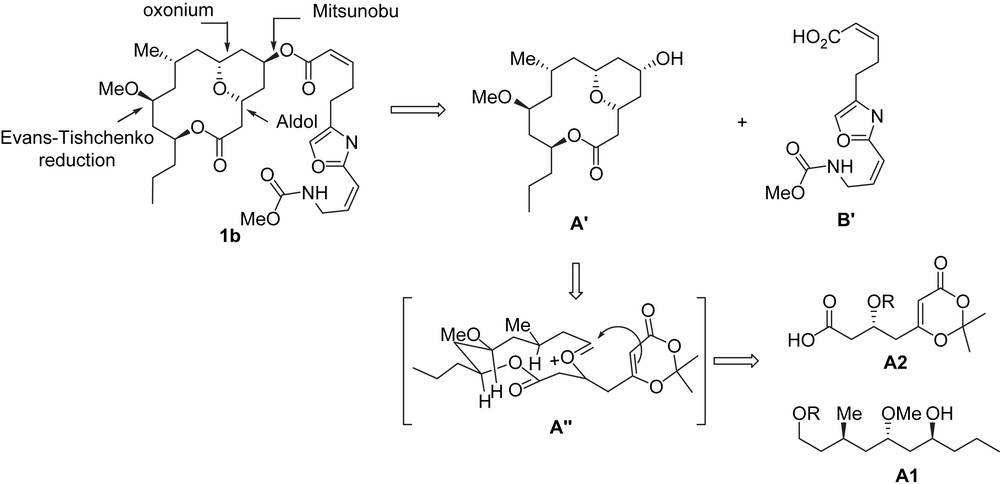
The synthesis of (+)-neopeltolide started with the construction of dioxinone 16 which was synthesized from the simple aldehyde 13 and dienoxysilane 14 in four steps. Ti(IV)/(R)-BINOL-catalyzed aldol reaction between dienoxysilane 14 [16] and aldehyde 13 afforded secondary alcohol 15 [17] (63% yield, 88% ee). Alcohol 15 was transformed to the corresponding carboxylic dioxinone 16 through a protection/deprotection/oxidation sequence. The synthesis of the second fragment which corresponds to fragment A1 (compound 21) began with the two-step conversion of β-hydroxyester 17 to Weinreb amide 18 [18]. Its transformation to the extended ketone 20 was achieved by adding the alkyllithium derived from 19. The removal of the PMB group in 20 by DDQ was followed by an Evans–Tishchenko reduction, thus creating the requisite anti-sterochemistry between the hydroxy groups at C11 and C13. The obtained hydroxyl group was then transformed to a methyl ether (MeOTf, DTBMP), and finally the benzoate group was cleaved to yield alcohol 21 (Scheme 5).
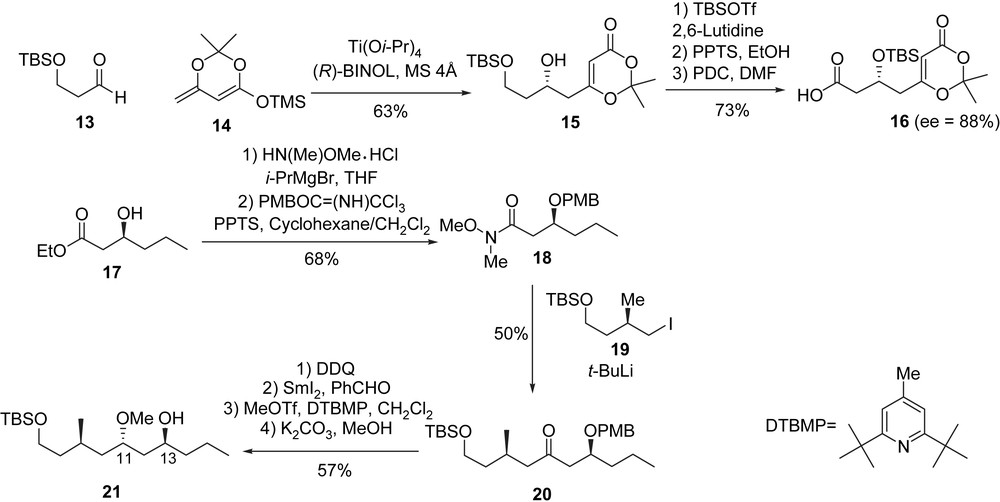
The coupling of 16 with 21 was accomplished using a Yamaguchi esterification [19], which was followed by a deprotection and an oxidation (TEMPO) [20] steps leading to dioxinone aldehyde 22, which is a precursor of the macrocyclic lactone. The key step in this synthesis is an intramolecular Prins reaction which was achieved via an oxonium species of type A″ generated from 22 using Sc(OTf)3 (CaSO4, MeCN). This single step afforded a tricyclic dioxinone intermediate in a modest yield but with a high degree of complexity. Treatment of the dioxinone with wet DMSO formed the bicyclic ketone 23. After diastereoselective reduction of ketone 23 to the corresponding equatorial alcohol with NaBH4, a Mitsunobu reaction with acid B′1 produced (+)-neopeltolide (1b) (Scheme 6).
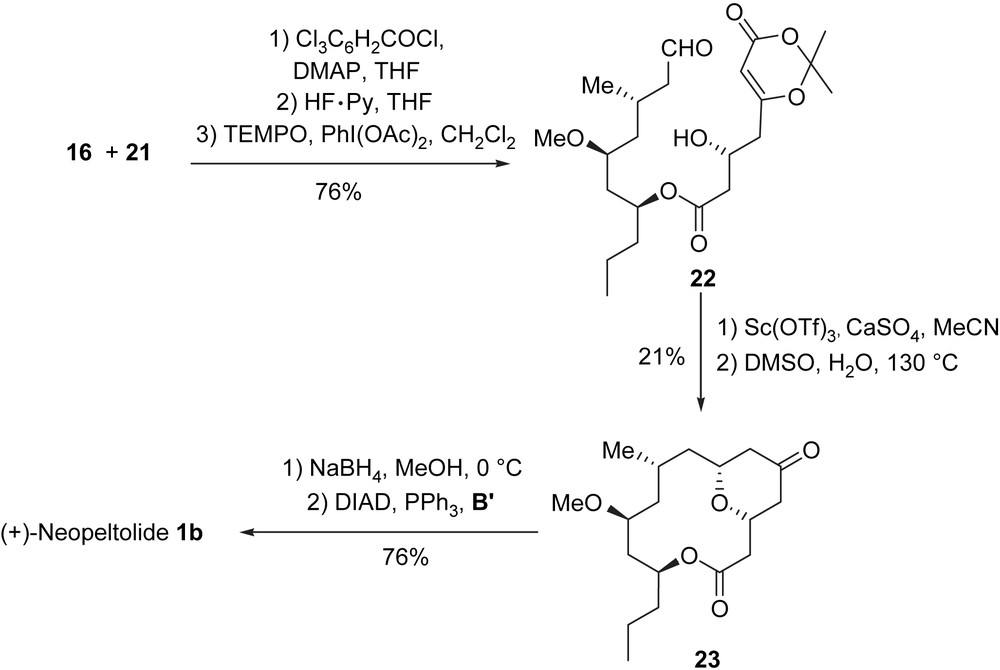
This synthesis of (+)-neopeltolide required 15 steps in the longest linear sequence from the chiral hydroxyester 17 and produced (+)-neopeltolide in 2.3% overall yield.
Following these two first syntheses, four other syntheses were reported in the literature in 2008, and for three of them, a Prins cyclization was used as the key step to establish the pyran ring.
4.3 Maier et al. formal synthesis [21]
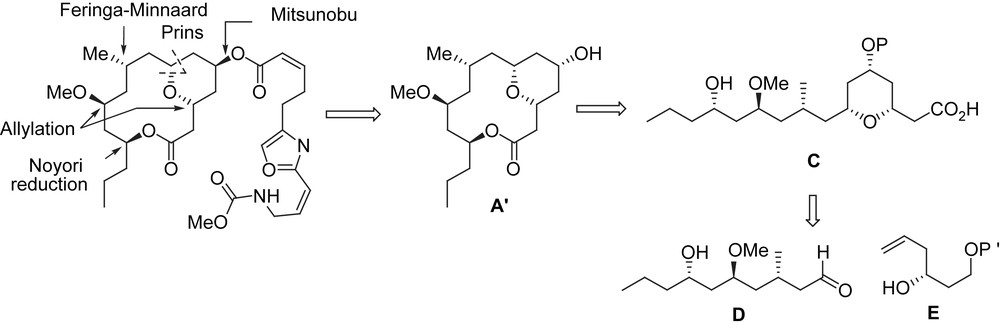
Similar to the synthesis described by Scheidt et al., the synthesis of (+)-neopeltolide by Maier et al. utilizes a Mitsunobu reaction to attach the oxazole side chain to the macrocyclic lactone. Their synthesis relied on the obtention of the macrocyclic lactone by using a Yamaguchi macrolactonization. The stereogenic centers were controlled by applying a Leighton allylation (C11), a Noyori reduction (C13), a Feringa–Minnaard asymmetric methyl cuprate addition on an unsaturated thioester (C9), and a Prins reaction [22–26] as the key steps. The seco-acid C was synthesized from D and E (Scheme 7).
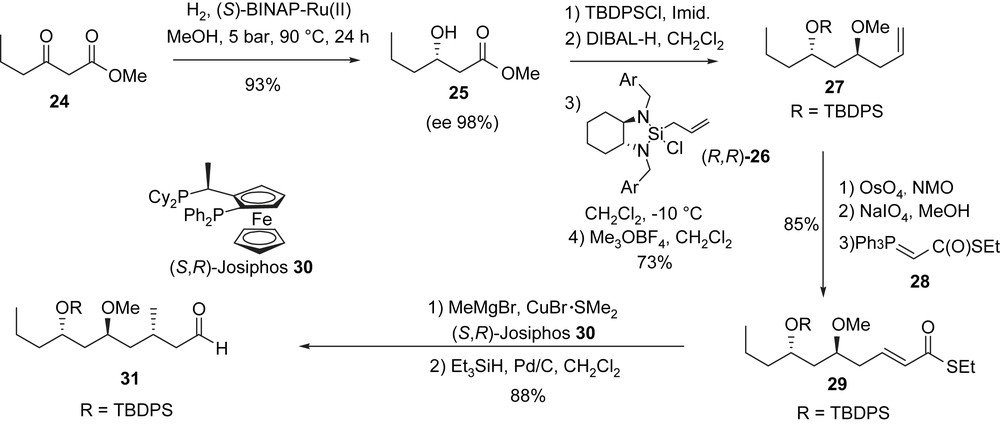
The synthesis started with β-ketoester 24 which was subjected to an enantioselective Noyori hydrogenation [27,28] using (S)-BINAP/Ru(II) to produce hydroxyl-ester 25 (98% ee). After protection (TBDPSCl, imidazole, DMF) and reduction of the ester (DIBAL-H), the obtained aldehyde was treated with Leighton reagent (R,R)-26 [29] to produce an 1,3-anti-diol pattern, and the free hydroxyl group at C11 was etherified using Meerwein's salt [30] in the presence of a proton sponge to afford 27. Oxidative cleavage of the double bond (OsO4, NMO, then NaIO4), followed by an extension of the obtained aldehyde using the Wittig reagent 28 [31] led to the α,β-unsaturated thioester 29. A conjugated addition of methylmagnesium bromide to 29 in the presence of CuBr·Me2S and the chiral diphosphine (S,R)-Josiphos 30 [32] produced compound 31 after Fukuyama reduction [33] of the thioester (Et3SiH, Pd/C) (Scheme 8).
The homoallylic alcohol 33 required for the Prins reaction was available by allylation of aldehyde 32 with the Leighton reagent (S,S)-26 [29]. The crucial Prins reaction was then performed between aldehyde 31 and homoallylic alcohol 33. By treatment with TFA (10 equiv), the desired trifluoroacetate 34 was isolated in 72% yield (major/minor 8/1) via the formation of an oxonium species of type F. After a saponification/protection/deprotection sequence, alcohol 35 was isolated. Its transformation to the seco-acid 36 was achieved in two steps, after an oxidation (Dess–Martin periodinane and sodium chlorite oxidation) [12,34] of the primary alcohol and a cleavage of the silyl ether (TBAF). Seco-acid 36 was obtained and transformed to the macrocyclic lactone A′ by using a Yamaguchi protocol followed by the final cleavage of the MOM ether.
The macrocyclic core of (+)-neopeltolide was obtained in 17 steps from the β-ketoester 24 with an overall yield of 23% (Scheme 9).
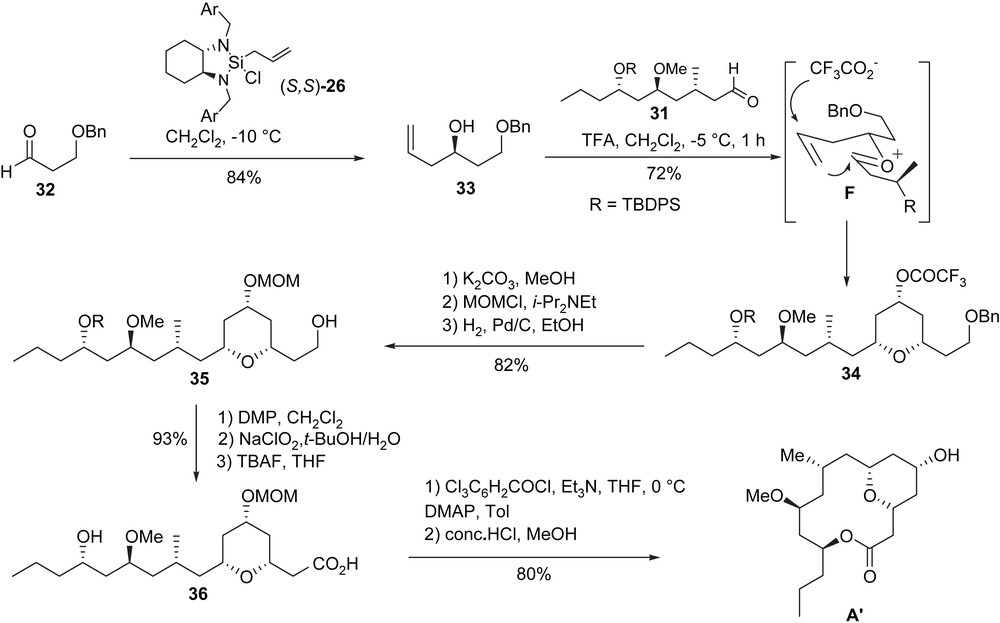
4.4 Lee et al. total synthesis [35]
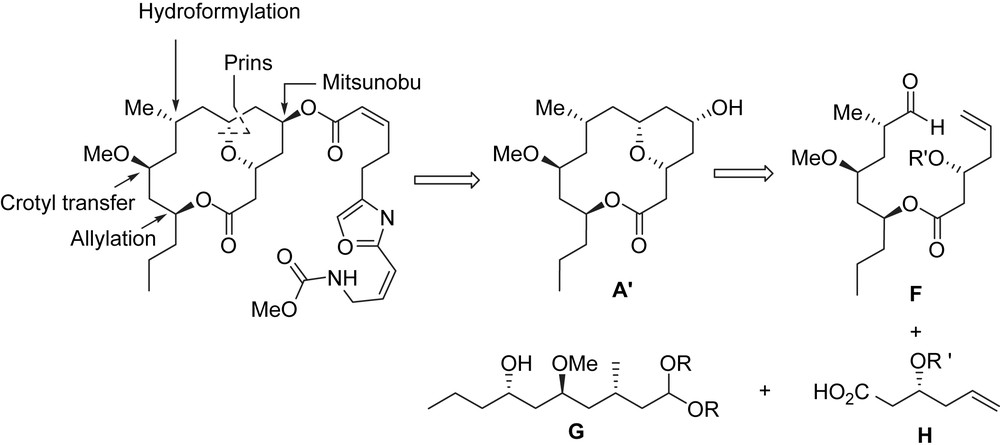
In a similar fashion to Maier et al., Lee et al. used a Mitsunobu reaction in between macrocyclic lactone A′ and carboxylic acid B′ to introduce the oxazole side chain, and an intramolecular Prins reaction was also used to form the functionalized tetrahydropyran moiety. An asymmetric crotyl transfer reaction and a titanium-mediated methallylation were considered to control the stereogenic centers at C11 and C13, and a substrate-directed hydroformylation was applied to control the sterogenic center at C9. A convergent synthesis using a Prins reaction was conceived from F which would be obtained after a Yamaguchi esterification between G and H (Scheme 10).
After a CSA-mediated Loh crotylation [36] of aldehyde 37, followed by the protection of the hydroxyl group at C13 using BnBr, the ozonolysis of the double bond afforded aldehyde 39 (49%, three steps). A diastereoselective methallylation in the presence of TiCl4 led to a homoallylic alcohol which was acylated with 2-diphenylphosphinobenzoic acid 40 to produce ester 41. After the diastereoselective substrate-directed hydrofomylation of 41, aldehyde 42 was formed in a 5/1 ratio and then transformed in three steps (acetalization, saponification, then O-methylation) to dimethyl acetal 43. This compound corresponds to the C7–C16 fragment (compound of type G) of (+)-neopeltolide. The second fragment, carboxylic acid 45, was prepared from the known olefin 44 [37] after removal of the silyl protecting group and oxidation of the resulting primary alcohol (Scheme 11).
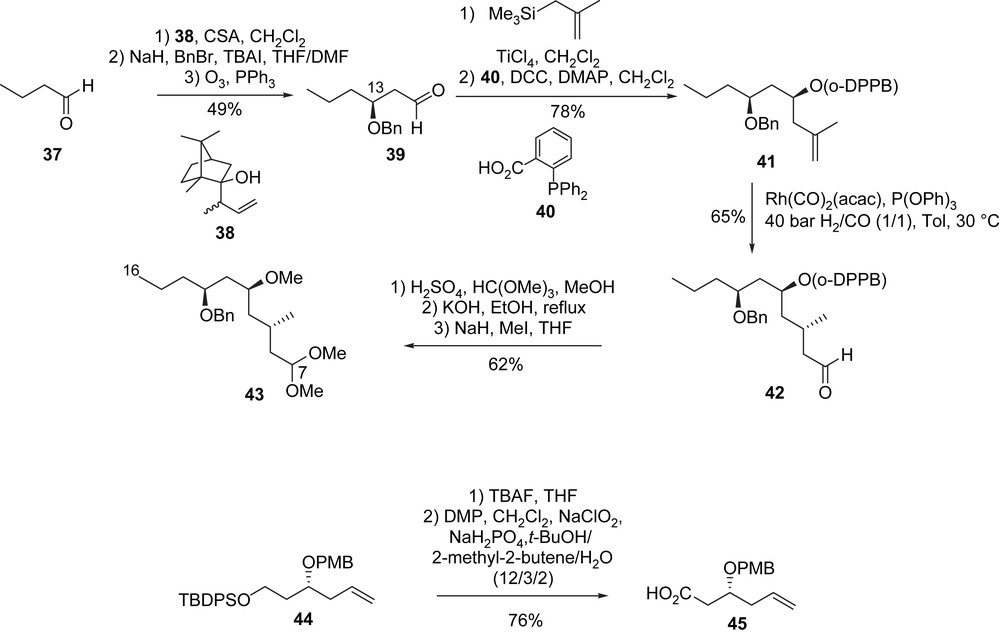
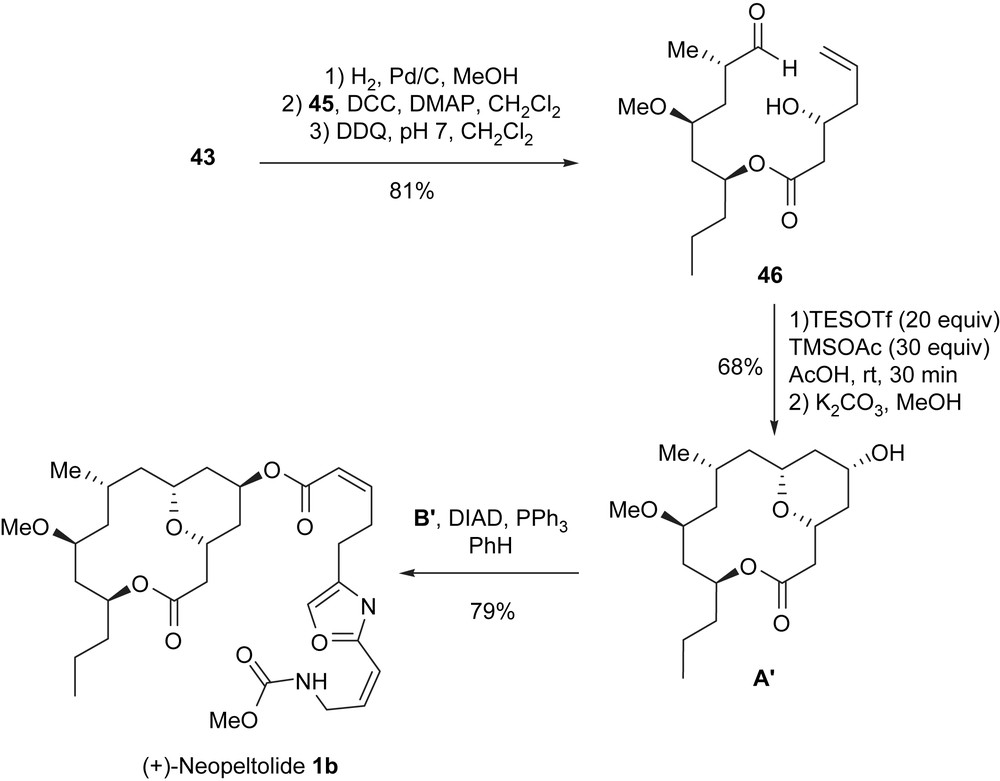
The cleavage of the benzyl ether in 43 led to a secondary alcohol at C13 which was acylated with acid 45 to furnish, after prolongated exposure to DDQ, the aldehydic homoallylic alcohol 46 (81%), which is the substrate of the Prins intramolecular cyclization. Compound 46 was treated under the Prins conditions [(TESOTf (20 equiv), TMSOAc (30 equiv), AcOH)] and then subjected to a methanolysis (K2CO3, MeOH) to give bicyclic macrolide A′ in good yield (68%); this bicyclic system was constructed efficiently with complete stereocontrol of the two newly created stereocenters. Finally, a Mitsunobu reaction of 47 with carboxylic acid B′ produced (+)-neopeltolide.
In this approach, (+)-neopeltolide was synthesized in 15 steps for the longest linear sequence from butyraldehyde 37 with an overall yield of 6.7% (Scheme 12).
4.5 Kozmin et al. racemic total synthesis [4a]
Recently, Kozmin et al. reported the synthesis of racemic neopeltolide. In their approach, they applied a Prins desymmetrization in order to establish the highly functionalized THP moiety [38]. They previously applied the same strategy to the racemic synthesis of leucascandrolide.
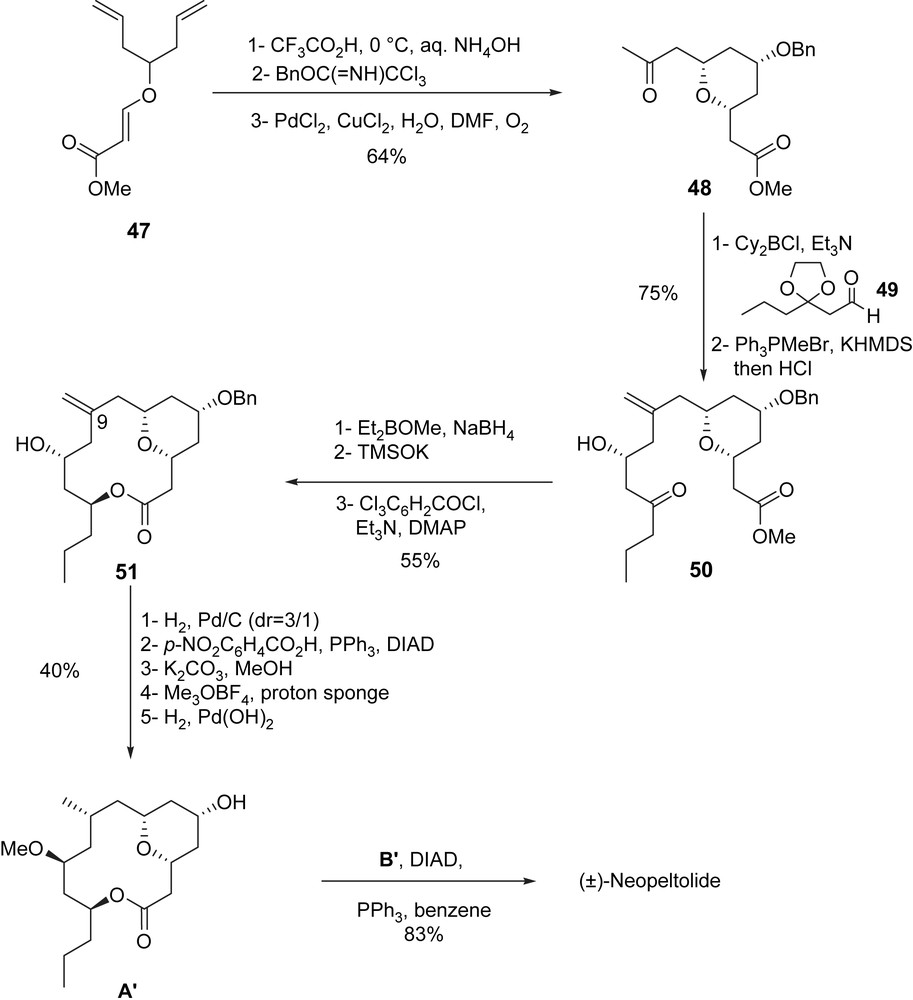
The racemic synthesis of neopeltolide started with the Prins desymmetrization [38] of diene 47 (CF3COOH, NH4OH) followed by the protection of the generated hydroxyl group at C5 (BnOC(NH)CCl3, TfOH) and a Wacker alkene oxidation to produce ketone 48. An aldol condensation [39] between a boron-enolate generated from ketone 48 and aldehyde 49, gave the expected aldol product (dr > 98/2) which was then transformed to 50 by using a Wittig methylenation (Ph3PMeBr, KHMDS) followed by acidic treatment in order to remove the dioxolane (HCl) (75% yield, 2 steps). After a diastereoselective reduction of the hydroxyketone 50 (Et2BOMe, NaBH4) followed by a saponification (TMSOK, Et2O) and a Yamaguchi macrolactonization, macrolide 51 was obtained (55% yield).
Five steps were then necessary to transform macro-lactone 51 to A′. After a diastereoselective hydrogenation of the olefin at C9 (Pd/C, H2), an inversion of the alcohol at C11 using a Mitsunobu reaction, followed by the methanolysis of the formed p-nitrophenylbenzoate, a subsequent methylation of the generated alcohol and, after hydrogenolysis of the benzyl ether, lactone A′ was obtained in 40% yield from 51. The final coupling step between macrolactone A′ and carboxylic acid B′ under standard Mitsunobu conditions provided (±)-neopeltolide.
(±)-Neopeltolide was efficiently synthesized in 14 steps for the longest linear sequence from diene 47 with an overall yield of 8.7% (Scheme 13).
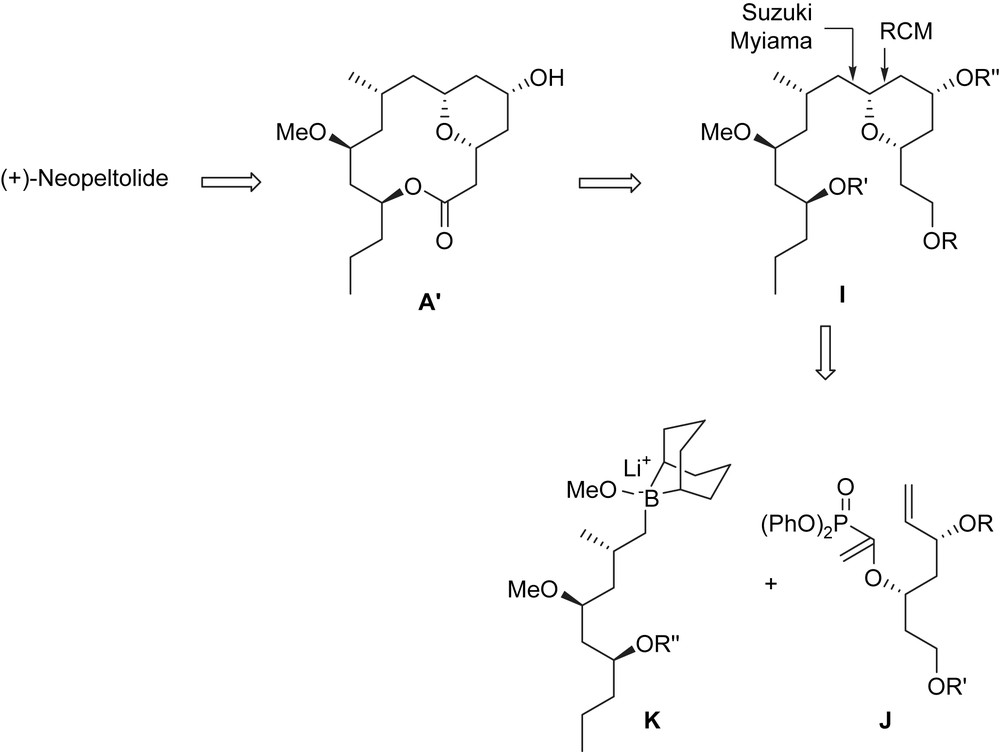
4.6 Fuwa–Sasaki et al. total synthesis [40]
As in most syntheses of (+)-neopeltolide, the introduction of the oxazole-containing side chain was achieved by using a Mitsunobu reaction between macrolactone A′ and carboxylic acid B′. The macrocyclic lactone A′ was prepared from tetrahydropyran I. To construct this latter, contrary to the previous syntheses employing a Prins reaction, a Suzuki–Miyaura coupling of the enol phosphate J and alkylborane K was used, followed by a RCM of the obtained diene to build I (Scheme 14).
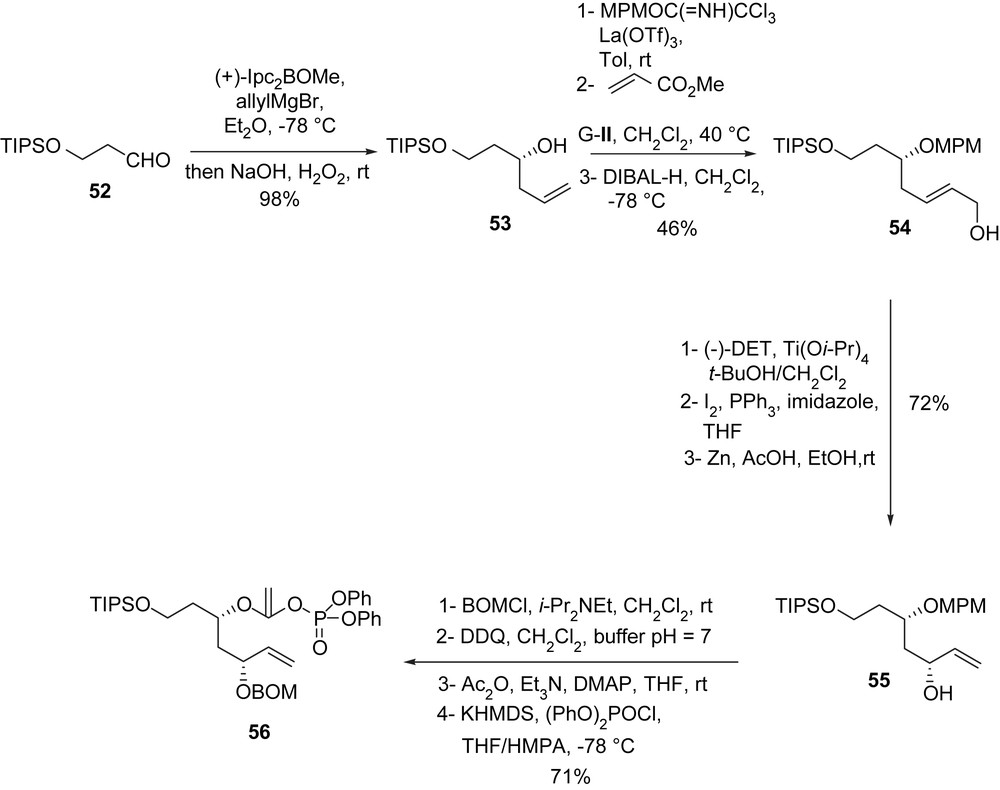
The synthesis of enol phosphate of type J, compound 56, started with the asymmetric allylation [41] of aldehyde 52, furnishing alcohol 53 [(+)-Ipc2BOMe, allylMgBr, then NaOH, H2O2, 98% yield]. After protection of the hydroxyl group (MPMOC(NH)CCl3, La(OTf)3, toluene, rt) followed by a cross-metathesis with methyl acrylate (Grubbs II = G-II) and reduction of the obtained α,β-unsaturated ester (DIBAL-H, CH2Cl2), homoallylic alcohol 54 was isolated. A Sharpless epoxidation [(−)-DET, Ti(Oi-Pr)4, t-BuOH, CH2Cl2, 97%] was followed with a reductive iodination (I2, PPh3, imidazole; Zn, AcOH, EtOH, 72.5%) to open the epoxide, allowing the isolation of the vinylic alcohol 55. This latter compound was then converted to the enol phosphate 56 in four steps. At first, alcohol 55 was protected (BOMCl, i-Pr2NEt), and then the MPM ether was cleaved (DDQ, CH2Cl2, buffer pH = 7). A subsequent acetylation (Ac2O, Et3N, DMAP, 99%) led to the corresponding acetate which, after enolization with KHMDS in the presence of (PhO)2P(O)Cl, furnished enol phosphate 56 (Scheme 15).
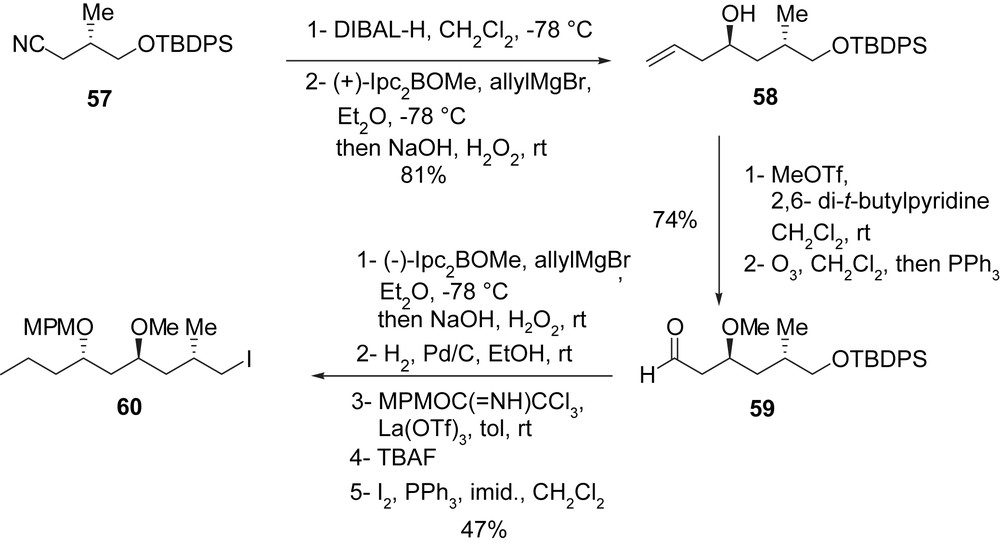
The synthesis of the precursor of borate of type K, iodide 60, commenced with nitrile 57 [42]. After reduction with DIBAL-H, followed by asymmetric allylation [41] of the obtained aldehyde [(+)-Ipc2BOMe, allylMgBr then NaOH, H2O2, 87%], allylic alcohol 58 was isolated. The methylation of alcohol 58 (MeOTf, 2,6-di-t-butylpyridine, CH2Cl2) followed by oxidative cleavage of the double bond (O3 then PPh3, 85%), afforded aldehyde 59. A second asymmetric allylation [41] [(−)-IpcBOMe, allylMgBr then NaOH, H2O2, 96%] and a hydrogenation of the obtained homoallylic alcohol (H2, Pd/C, EtOAc, 100%) allowed the introduction of the terminal alkyl chain. Finally, protection of the remaining hydroxyl group (MPMOC(NH)CCl3, La(OTf)3, 75%) followed by desilylation (TBAF, 87%), afforded the corresponding alcohol which was transformed to iodide 60 (47% yield) under standard conditions (Scheme 16).
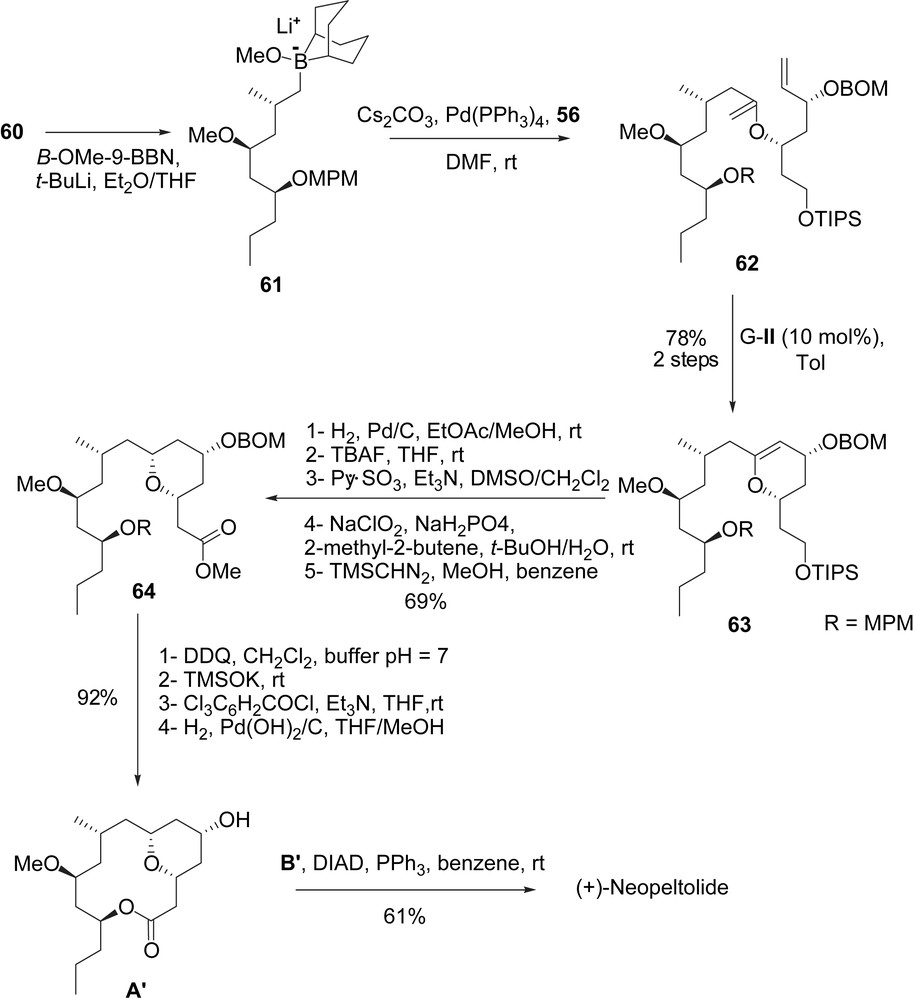
Assembly of enol phosphate 56 and iodide 60 was realized via the formation of an alkylborate intermediate generated in situ. For this coupling, lithiation of iodide 60 with t-BuLi in the presence of B-OMe-9-BBN was achieved and furnished alkylborate intermediate 61, which was treated in situ with enol phosphate 56 (Cs2CO3, Pd(PPh3)4, DMF) to give the acyclic enol ether 62. Subsequent RCM of 62 (Grubbs II, toluene) led to the cyclic enol ether 63 (78% yield). The stereoselective hydrogenation of the double bond (H2, Pd/C, EtOAc/MeOH) formed the cis-tetrahydropyran as a single diastereomer. The TIPS protecting group was then removed (TBAF, THF, 99%), and an oxidation/esterification sequence (Py·SO3, Et3N, DMSO/CH2Cl2 then NaClO2, NaH2PO4 and then TMSCHN2, 89%) furnished ester 64. The completion of the synthesis of macrolactone A′ was achieved in four steps. At first the cleavage of the MPM ether (DDQ, 100%) followed by saponification of the methyl ester (TMSOK, 100%) led to a seco-acid, which was treated under the Yamaguchi conditions to form the macrolactone core. A final hydrogenolysis of the BOM protecting group (H2, Pd(OH)2/C, 100%) gave intermediate A′. The Mitsunobu reaction with carboxylic acid B′ provided (+)-neopeltolide (61%).
Even if, compared to the other syntheses, this latter one is longer (25 steps for the longest linear sequence), the excellent overall yield of 8.3% shows the efficiency of the employed reactions (Scheme 17).
5 Conclusion
Since the discovery of (+)-neopeltolide by Wright et al. in 2007, six syntheses (one formal synthesis, five total syntheses) have been reported. The most efficient syntheses of (+)-neopeltolide were achieved by using intra- or intermolecular Prins reactions as these reactions allow the formation of the cis-trisubstituted tetra-hydropyrans with the control of two new stereogenic centers. The recent investigations on the mechanism of action of neopeltolide [4] will probably stimulate the synthesis of neopeltolide analogues for SAR studies.
Acknowledgments
We thank Pr. K.-H. Altmann and Pr. S. Kozmin for generously sending us their article related to neopeltolide in Nat. Chem. Biol. (Ref. [4]).
1 Carboxylic acid B′ was prepared according to refs. [6a,b].