

1 Introduction
α-Aminonitriles have gained an increasing interest in recent years thanks to their versatile utility as precursors and intermediates in the preparation of numerous biologically-active compounds [1–9]. In particular, the synthesis of optically-active α-aminonitriles constitutes an area of considerable interest in asymmetric organic synthesis. The preparation of chiral N-monosubstituted and N-unsubstituted α-aminonitriles is very well documented. These compounds can be obtained by resolution of racemates with tartaric acid [10], through enantioselective enzymatic transformation [11], via catalytic enantioselective Strecker reaction [12,13] or by dehydration of optically-active amides derived from amino acids [14–17]. However, the literature is bereft of reports on the preparation of N,N-disubstituted α-aminonitriles in an enantiomerically enriched form. This is also significantly contrasted with the vast number of examples of non-racemic aminonitriles having two or more stereogenic centers [18,19]. Indeed, we are aware of only three such reports: (R)-2-alkyl-2-(1-piperidinyl)alkanenitrile (ee 6–84%) obtained via dehydration of the corresponding commercially carboxamides using various dehydrating agents [20], (S)-1-benzyl-α-cyanopiperidine (ee 91%) prepared in several steps from non-racemic cyanohydrin [21] and N-allyl-N-trifluoroacetyl-α-aminonitriles (ee 37–95%) obtained via addition of hydrogen cyanide to imines, catalyzed by a chiral (Salen)Al(III) complex and followed by the trifluoroacetylation of the N-monosubstituted α-aminonitriles [22].
Herein, we report the synthesis of enantiomerically pure N-methyl-N-arylsulfonyl-α-aminonitriles from the corresponding (l)- and (d)-α-amino acids by means of N-methyl-N-arylsulfonyl-α-amino amides. These compounds are new and represent the first example of enantiomerically pure N,N-disubstituted α-aminonitriles.
2 Results and discussion
The synthetic route to target compounds 4a–k is outlined in Schemes 1 and 2. The commercially available (D)- and (L)-amino acids, used as starting materials, were converted to the corresponding N-arylsulfonyl-α-amino acids 1a–k according to the procedure described in the literature [23]. Compounds 1a–i were converted to N-methyl-N-arylsulfonyl-α-amino acids 2a–i in two steps, following a similar method reported by Freindinger et al. [24–26] The treatment of 2a–i and 1j,k with thionyl chloride followed by treatment with aq. NH3 led to the corresponding α-amino amides 3a–k. The last step is the dehydration of aminoamides 3a–k with thionyl chloride which led to the corresponding α-aminonitriles 4a–k.
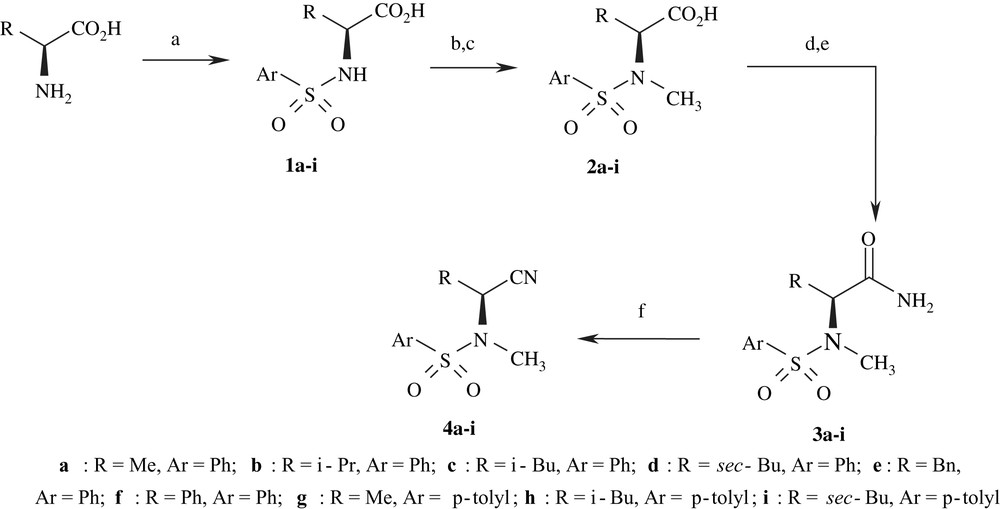
Synthesis of N-methyl-N-arylsulfonyl-α-aminonitriles from the corresponding amino acids. Reaction conditions: (a) ArSO2Cl/NaOH/EtN(i-Pr)2; (b) (CH2)n/p-toluene sulfonic acid; (c) Et3SiH/CF3CO2H; (d) SOCl2; (e) aq. NH3; (f) SOCl2 reflux, 2 h.

Synthesis of the substituted α-aminonitrile from (l)-proline.
α-Aminonitriles 4a–k, prepared by dehydration of the corresponding amides 3a–k, were obtained with excellent yields. Moreover, the reaction occurred without a significant racemization of the stereogenic center. Indeed, we have analyzed the enantiomeric ratio of compounds 4a–c,f prepared from both optically pure and racemic amino acids, by chiral HPLC analysis. We have found that racemization did not occur for compound 4a–c,f prepared from optically pure amino acids.
In this work, thionyl chloride is used as a dehydrating agent to convert amides into the corresponding aminonitriles. This agent appeared to be convenient to provide α-aminonitriles without a significant racemization of the α-bearing carbon. However, as described in the literature, the use of other dehydrating agents such as POCl3/Py, TsCl/PyTf2O/Et3N and Burgess' salt [20] involved racemization of the N,N-disubstituted α-aminonitriles. Within this work, we have found that the use of POCl3 instead of SOCl2, to convert the amide 3a into the corresponding nitrile 4a, occurred with total racemization of the α-bearing carbon. As described by Sheldon et al. [10] in α-aminonitriles, the α-proton is somewhat acidic due to the electron-withdrawing effect of the cyano group. Then, the presence of a basic site would catalyze the racemization process. Accordingly, we think that the use of SOCl2, which frees HCl (g) and SO2 (g) during the reaction, is convenient to avoid the racemization, since there is no possible acid–base interaction between the acidic α-proton of nitriles and these compounds.
3 Conclusion
To conclude, the preparation of a single-stereocentre series of enantiomerically pure N-methyl-N-arylsulfonyl-α-aminonitriles from their corresponding α-amino acids has been achieved for the first time in the combined terms of quantitative yields and enantiomeric purity. We analyzed the enantiomeric ratio of compounds 4a–k by chiral HPLC analysis. The study of the synthetic features of these products to prepare new chiral heterocyclic compounds is under investigation in our laboratory.
4 Experimental section
TLC was performed on Merck 60F-254 silica gel plates (layer thickness 0.25 mm). Column chromatography was performed on silica gel (70e230 mesh) using ethylacetate and cyclohexane mixture as eluents. CH2Cl2 was distilled over CaH2. Melting points were determined on a Electrothermal 9002 apparatus and are uncorrected. NMR spectra were recorded on Bruker AC-300 apparatus (1H at 300 MHz and 13C at 75 MHz). All chemical shifts are reported as δ values (ppm) relative to internal tetramethylsilane. IR spectra were recorded on BIO-RAD FTS-6000 apparatus. Mass spectra (EI) were recorded on Hewlett Packard 5897. Elemental analyses were carried out by ‘Service de microanalyse’ of ‘Institut national de recherche et d'analyse physico-chimique de Tunis’. HPLC analyses were conducted on a heptane/isopropanol system with a UV detector at 254 nm, using a Chirobiotic V column (250 × 46 mm) and a flow rate of 0.5 mL/min. Compounds 2a–i were prepared as described [25,26].
4.1 Typical procedure for the preparation of N-methyl-N-arylsulfonyl-2-amino amides
A mixture of N-methyl-N-sulfonyl-2-amino acid (10 mmol), SOCl2 (40 mmol) and three drops of dry DMF in anhydrous dichloromethane was stirred overnight at room temperature under N2. After removal of the excess of SOCl2 under vacuum, the residue was added slowly to a cold solution of aqueous ammonium hydroxide and was allowed to warm slowly to room temperature with stirring. The mixture was filtered and the resultant solid was dissolved in boiling ethanol. The solution was, then, filtered while hot and water was added dropwise to the cloud point. After cooling, the pure N-methyl-N-arylsulfonyl-2-amino amide was collected.
4.1.1 (2S)-N-methyl-N-phenylsulfonyl-2-aminopropanamide 3a
Yield = 88%; mp 144–143 °C; IR (cm−1): νCO = 1716,
4.1.2 (2S)-N-Methyl-N-phenylsulfonyl-2-amino-3-methylbutaneamide 3b
Yield = 87%; mp 122–123 °C; IR (cm−1): νCO = 1715,
4.1.3 (2S)-N-Methyl-N-phenylsulfonyl-2-amino-4-methylpentaneamide 3c
Yield = 89%; mp 137–138 °C; IR (cm−1): νCO = 1717,
4.1.4 (2S,3S)N-Methyl-N-phenylsulfonyl-2-amino-3-methylpentaneamide 3d
Yield = 90%; mp 127–128 °C; IR (cm−1): νCO = 1719,
4.1.5 (2S)-N-Methyl-N-phenylsulfonyl-2-amino-3-phenylpropaneamide 3e
Yield = 85%; mp 157–158 °C; IR (cm−1): νCO = 1715,
4.1.6 (2R)-N-Methyl-N-phenylsulfonyl-2-amino-2-phenylethaneamide 3f
Yield = 84%; mp 165–166 °C; IR (cm−1): νCO = 1712,
4.1.7 (2S)-N-Methyl-N-tolylsulfonyl-2-aminopropaneamide 3g
Yield = 89%; mp 133–134 °C; IR (cm−1): νCO = 1717,
4.1.8 (2S)-N-Methyl-N-tolylsulfonyl-2-amino-4-methylpentaneamide 3h
Yield = 90%; mp 140–141 °C; IR (cm−1): νCO = 1714,
4.1.9 (2S,3S)-N-Methyl-N-tolylsulfonyl-2-amino-3-methylpentaneamide 3i
Yield = 88%; mp 147–148 °C; IR (cm−1): νCO = 1715,
4.1.10 (2S)-N-Phenylsulfonyl-2-carbamoylpyrrolidine 3j
Yield = 86%; mp = 150–151 °C; IR (cm−1): νCO = 1716,
4.1.11 (2S)-N-Tolylsulfonyl-2-carbamoylpyrrolidine 3k
Yield = 91%; mp = 160–161 °C; IR (cm−1): νCO = 1714,
4.2 Typical procedure for the dehydration of N-methyl-N-arylsulfonyl-2-amino amides
N-methyl-N-arylsulfonyl-2-amino amide (8 mmol) was dissolved in 5 mL of freshly distilled thionyl chloride. The mixture was refluxed for 2 h until the amide was consumed (TLC). The excess of thionyl chloride was removed under vacuum using a rotary evaporator, and the reaction residue was, then, mixed with crushed ice and extracted with ethylacetate (3 × 50 mL). The organic layers were combined, washed sequentially with a saturated aqueous NaHCO3 solution and water and dried with anhydrous MgSO4. After that, the solvent was removed under vacuum and the crude product was purified by column chromatography on silica gel, using [cyclohexane:ethylacetate (8:2)], to afford pure N-methyl-N-arylsulfonyl-2-aminonitrile 4a–k.
4.2.1 (2S)-N-Methyl-N-phenylsulfonyl-2-aminopropanenitrile 4a
Yield = 93%; mp: 78–79 °C; [α]D −120, (c0.5, CHCl3); ee > 99.5% [HPLC analysis using a Chirobiotic V column 4.6 mm × 250 mm under the following conditions: heptane/isopropanol (95:5) as mobile phase, rt, λ = 254 nm, flow rate = 0.5 mL/min. Retention times: (S)-4a, 10.45 min; (R)-4a, 12.86 min]; IR (cm−1): νCN = 2245; 1H NMR (300 MHz, CDCl3): δ: 1.57 (d, 3H, J = 7.2 Hz), 2.84 (s, 3H), 5.04 (q, 1H, J = 7.2 Hz), 7.56-7.83 (m, 5H); 13C NMR (75 MHz, CDCl3), δ: 18.52, 30.25, 44.70, 116.1 (CN), 127.61, 129.54, 133.79, 136.35; MS/EI m/z = 224 (M+); Anal. Calcd for C10H12N2O2S, 224.24: C 53.55, H 5.39, N 12.49. Found: C 53.43, H 5.22, N 12.31.
4.2.2 (2S)-N-Methyl-N-phenylsulfonyl-2-amino-3-methylbutanenitrile 4b
Yield = 94%; mp 75–77 °C; [α]D −80, (c 0.5, CHCl3); ee > 99.5% [HPLC analysis using a Chirobiotic V column 4.6 mm × 250 mm under the following conditions: heptane/isopropanol (95:5) as mobile phase, rt, λ = 254 nm, flow rate = 0.6 mL/min. Retention times: (S)-4b, 25.95 min; (R)-4b, 31.97 min]; IR (cm−1): νCN = 2246; 1H NMR (300 MHz, CDCl3): δ: 0.90 (d, 3H, J = 6.6 Hz), 0.96 (d, 3H, J = 6.6 Hz), 2.04 (m, 1H), 2.83 (s, 3H), 4.88 (d, 1H, J = 10.5 Hz), 7.66–7.83 (m, 5H); 13C NMR (75 MHz, CDCl3): δ: 19.52, 19.62, 26.93, 30.15, 51.70, 117.4 (CN), 127.5, 129.25, 135.81, 139.84; MS/EI m/z = 252 (M+); Anal. Calcd for C12H16N2O2S, 252.34: C 57.12, H 6.39, N 11.10. Found: C 56.98, H 6.27, N 11.03.
4.2.3 (2S)-N-Methyl-N-phenylsulfonyl-2-amino-4-methylpentanenitrile 4c
Yield = 95%; mp = 73–75 °C; [α]D −70 (c 0.5, CHCl3); ee > 99.5% [HPLC analysis using a Chirobiotic V column 4.6 mm × 250 mm under the following conditions: heptane/isopropanol (95:5) as mobile phase, rt, λ = 254 nm, flow rate = 0.6 mL/min. Retention times: (S)-4c, 15.43 min; (R)-4c, 19.58 min]; IR (cm−1): νCN = 2244; 1H NMR (300 MHz, CDCl3) δ: 0.91 (d, 6H J = 5.4 Hz), 1.52–1.77 (m, 3H), 2.73 (s, 3H), 4.84 (dd, 1H, J = 8.7 Hz, J = 7.0 Hz), 7.48–7.78 (m, 5H); 13C NMR (75 MHz, CDCl3) δ: 21.84, 22.62, 24.63, 30.74, 40.67, 48.03, 116.15(CN), 128.00, 129.83, 134.07, 136.75; MS/EI m/z = 266 (M+).
4.2.4 (2S,3S)N-Methyl-N-phenylsulfonyl-2-amino-3-methylpentanenitrile 4d
Yield = 95%; mp = 81–83 °C; [α]D −110, (c 0.5, CHCl3); IR (cm−1): νCN = 2245 (CN); 1H NMR (300 MHz, CDCl3): δ: 0.92 (t, 3H J = 7.5 Hz), 1.13 (d, 3H J = 6.6 Hz), 1.18–1.27 (m, 1H), 1.69–1.85 (m, 2H), 2.79 (s, 3H), 4.47 (d, 1H J = 10.5 Hz), 7.55–7.81 (m, 5H); 13C NMR (75 MHz, CDCl3): δ: 10.17, 15.75, 24.53, 30.91, 35.81, 54.89, 115.37 (CN), 127.59, 129.49, 133.71, 136.42, MS/EI m/z = 266 (M+).
4.2.5 (2S)-N-Methyl-N-phenylsulfonyl-2-amino-3-phenylpropanenitrile 4e
Yield = 93%; mp = 97–99 °C; [α]D −30, (c 0.5, CHCl3); IR (cm−1): νCN = 2241 (CN); 1H NMR (300 MHz, CDCl3): δ: 2.81 (s, 3H), 3.06 (m, 2H), 4.99 (m, 1H), 7.17–7.69 (m, 10H); 13C NMR (75 MHz, CDCl3) δ: 31.33, 39.03, 51.59, 115.60 (CN), 127.90, 128.41, 129.44, 129.67, 129.83, 134.05, 136.95, 136.95, MS/EI m/z = 300 (M+). Anal. Calcd for C16H16N2O2S, 300.38: C 63.98, H 5.37, N 9.33. Found: C 63.86, H 5.35, N 9.40.
4.2.6 (2R)-N-Methyl-N-phenylsulfonyl-2-amino-2-phenylethanenitrile 4f
Yield = 94%; mp = 86–88 °C; [α]D +90; (c 0.5, CHCl3); ee > 99.5% [HPLC analysis using a Chirobiotic V column 4.6 mm × 250 mm under the following conditions: heptane/isopropanol (95:5) as mobile phase, rt, λ = 254 nm, flow rate = 0.5 mL/min. Retention times: (R)-4f, 14.54 min; (S)-4f, 19.73 min]; IR (cm−1): νCN = 2242 (CN); 1H NMR (300 MHz, CDCl3): δ: 2.60 (s, 3H), 5.86 (s, 1H), 7.18–7.33 (m, 10H); 13C NMR (75 MHz, CDCl3): δ: 31.19, 62.77, 117.56 (CN), 127.68, 128.35, 128.99, 129.45, 130.01, 130.78, 132.25, 132.85; MS/EI m/z = 286 (M+).
4.2.7 (2S)-N-Methyl-N-tolylsulfonyl-2-aminopropanenitrile 4g
Yield = 92%; mp = 85–87 °C; [α]D −130, (c 0.5, CHCl3); IR (cm−1): νCN = 2246; 1H NMR (300 MHz, CDCl3): δ: 1.35 (d, 6H, J = 7.2 Hz), 2.37 (s, 3H), 2.75 (s, 3H), 4.94 (h, 1H, J = 7.2 Hz), 7.28 (d, 2H J = 8.1 Hz), 7.64 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ: 18.89, 27.30, 30.53, 45.00, 116.54 (CN), 128.013, 130.45, 133.79, 145.05, MS/EI m/z = 238 (M+); Anal. Calcd for C11H14N2O2S, 238.31: C 55.44, H 5.92, N 11.67. Found: C 55.34, H 5.81, N 11.49.
4.2.8 (2S)-N-Methyl-N-tolylsulfonyl-2-amino-4-methylpentanenitrile 4h
Yield = 93%; mp = 83–85 °C; [α]D −60 (c 0.5, CHCl3); IR (cm−1): νCN = 2243; 1H NMR (300 MHz, CDCl3) δ: 0.91 (d, 6H J = 5.6 Hz), 1.59–1.83 (m, 3H), 2.43 (s, 3H), 2.78 (s, 3H), 4.90 (dd, 1H J = 8.4 Hz, J = 6.75 Hz), 7.36 (d, 2H J = 8.1 Hz), 7.78 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ: 21.48, 21.69, 22.28, 24.28, 30.36, 40.34, 47.65, 115.91 (CN), 127.71, 130.09, 133.38, 144.67; MS/EI m/z = 280 (M+).
4.2.9 (2S,3S)-N-Methyl-N-tolylsulfonyl-2-amino-3-methylpentanenitrile 4i
Yield = 91%; mp = 78–80 °C; [α]D −100, (c 0.5, CHCl3); IR (cm−1): νCN = 2243 (CN); 1H NMR (300 MHz, CDCl3) δ: 0.95(t, 3H J = 7.4 Hz), 1.18 (d, 3H J = 6.9 Hz), 1.21–1.84 (m, 3H), 2.44 (s, 3H), 2.78 (s, 3H), 4.47 (d, 1H J = 10.4 Hz), 7.35 (d, 2H, J = 8.4 Hz), 7.81 (d, 2H, J = 8.4 Hz); 13C NMR (75 MHz, CDCl3) δ: 10.19, 15.76, 21.69, 24.57, 30.88, 35.90, 54,88, 115.47 (CN), 127.64, 130.08, 133.49, 144.61; MS/EI m/z = 280 (M+).
4.2.10 (2S)-N-Phenylsulfonyl-2-cyanopyrrolidine 4j
Yield = 96%; mp = 99–101 °C; [α]D −1040; (c 0.5, CHCl3); IR (cm−1): νCN = 2236 (CN); 1H NMR (300 MHz, CDCl3): δ: 2.02–2.19 (m, 4H), 3.33 (m, 2H), 4.58 (dd, 1H, J = 6.9 Hz, J = 2.7 Hz), 7.53–7.88(m, 5H); 13C NMR (75 MHz, CDCl3): δ: 24.73, 31.97, 47.58, 48.66, 118.03 (CN), 127.55, 129.42, 133.59, 137.29, MS/EI m/z = 236 (M+). Anal. Calcd for C11H12N2O2S, 236.29: C 55.91, H 5.12, N 11.86. Found: C 56.13, H 4.99, N 11.75.
4.2.11 (2S)-N-Tolylsulfonyl-2-cyanopyrrolidine 4k
Yield = 94%; mp = 92–94 °C; [α]D −980, (c 0.5, CHCl3); IR (cm−1): νCN = 2235 (CN); 1H NMR (300 MHz, CDCl3) δ: 1.95–2,17 (m, 4H), 2.36 (s, 3H), 3.39 (m, 2H), 4.51 (dd, 1H, J = 7.5 Hz, J = 2.55 Hz), 7.27 (d, 2H, J = 8.1 Hz), 7.72 (d, 2H, J = 8.1 Hz); 13C NMR (75 MHz, CDCl3) δ: 21.99, 25.03, 32.30, 47.93, 48.99, 118.50 (CN), 127.95, 130.35, 134.61, 144.86; MS/EI m/z = 250 (M+).
Acknowledgements
The authors thank the DGRSRT (Direction générale de la recherche scientifique et de la rénovation technologique) of the Tunisian Ministry of Higher Education and Scientific research and Technology for the financial support of this research.