

1 Introduction
The study of transition-metal σ-alkynyl complexes can be traced back to 1960s [1,2]. Since the mid-1980s, this area has attracted much more intense interests as evidenced by increasing numbers of research papers from approximately 200 to over 20,000 [3]. Compared with the pure organic conjugated systems [4,5], introduction of metal units into the polymeric or oligomeric systems may result in a broad range of improved properties including electronic communication (molecular wires) [6,7], nonlinear optical effects [8], luminescence and photoconductivity [9,10].
In the research of utilizing metal σ-alkynyl complexes as building blocks for molecular electronic devices, two types of metal alkynyl wires were proposed (Chart 1) [6,11]. Polyyn-diyls (C2n) capped by two metal termini (type I in Chart 1) are very attractive prototypes of “organometallic molecular wires”. Many of the pioneering efforts focused on the design of type I wires featuring facile charge transfer across the sp carbon chain (polyyn-diyls), with [M] as CpFe(P–P) [12], CpRe(P)NO [13–15], CpRu(P)2 [16], and Mn(P–P)2I [17] where P and P–P denote mono- and bidentate phosphines, respectively. Mono-disperse oligomers (type II in Chart 1) could be better alternatives than dimers (I) on the aspects of controlling length and connecting to the outside world, from the practical applications point of view. However, some limitations preclude the possibility of many metal complexes as candidates for type II wires. To form oligomers, the metal units should be capable of forming bis-alkynyl complexes, and in addition, the selection of the metal center [M] is also critical to avoid insulating behavior [6,18]. Examples of well-characterized type II oligomers are rare, and those based on Ru2(DPhF)4-butadiynyl [19] and Ru(P–P)2–σ-arylacetylide monomers [20,21] are noteworthy.
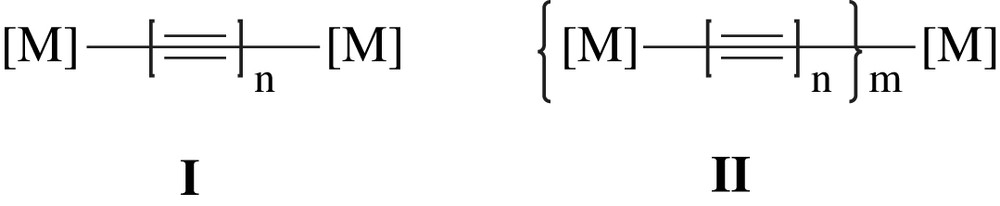
Dimeric (I) and metallayne (II) wires.
Our effort in developing organometallic molecular wires focuses on using diruthenium paddlewheel species as [M] [22–25], due to their attractive features including the controlled syntheses of the mono- and bis-alkynyl complexes, the stability toward ambient atmosphere and heat, the redox flexibility, and very small HOMO–LUMO gaps (1.2–1.5 eV) [26–34]. As excellent chromophores and electrophores, the diruthenium alkynyl complexes have been studied as promising building blocks for novel (opto)electronic materials [23].
2 Ru2 polyyn-diyls
Our investigation of diruthenium alkynyl compounds of a paddlewheel motif demonstrated that σ-alkynyl compounds of [Ru2L4] have good charge mobility along the axial direction. Ru2(II,III) based wire-like compounds (type I) with n up to 10 have been prepared with various supporting ligands (L). Compounds of long polyyn-diyl segments are desired for better understanding of the electron coupling within the type I molecules and the distance dependence of Had (adiabatic electronic coupling elements).
The N,N′-bidentate bridging ligands (L) utilized in supporting the Ru2 core include 2-anilinopyridinate (ap) and its anilino-substituted derivatives, 2-(3,5-dimethoxyanilino)pyridinate (DiMeOap), and 2-(i-butoxyanilino)pyridinate (iBuOap) (Chart 2). Using ap as the auxiliary ligand, type I compounds with n up to 6 were prepared and studied [35,36]. Both of the modified ap ligands afford significantly improved solubility in common organic solvents [37,38], and facilitate the preparation of type I compounds with n up to 10, and the access to compounds of odd n as well.
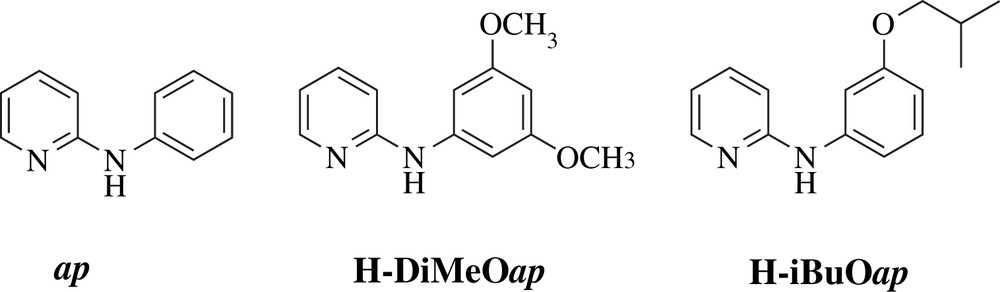
N,N′-bidentate ligands supporting the Ru2 core.
2.1 Monomers and dimers
Wire-like molecules [Ru2L4]2(μ-C2), [Ru2L4]2(μ-C4) and [Ru2L4]2(μ-C6) (L = DiMeOap, compounds A1–A3; L = ap, compounds A1′–A3′) were synthesized through anion metathesis between [Ru2L4]–Cl and LiC2nLi [35,36]. However, this technique became impractical with longer polyyn-diyl ligands, due to the instability of LiC2nLi with n ≥ 4. Hence, the longer type I compounds (n ≥ 4) were prepared through the Glaser oxidative coupling [39] of their corresponding “half” molecules which are the monomer type in Chart 3.
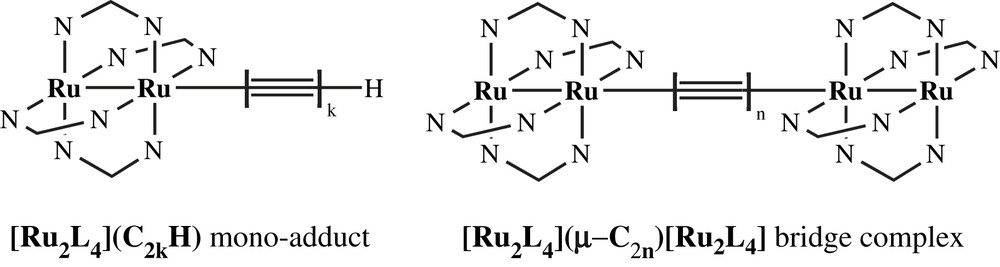
Diruthenium mono-alkynyl building block and wire-like bridge compound.
The mono-adduct with k = 2 was prepared from a reaction between [Ru2L4]–Cl and LiC4SiMe3. After the removal of terminal protection group –SiMe3 (K2CO3, THF/MeOH), a two-carbon sp chain extension reaction was achieved by the Glaser oxidative coupling [39] of [Ru2L4]–C4H and excess HCCSiMe3 (Hay conditions: O2, cat. CuCl, TMEDA and acetone) to give [Ru2L4]–C6SiMe3 (monomer with k = 3). However, extending this sequence to longer monomers (k = 4 and 5) was unsuccessful with the problem of increasing tendency for homo-coupling of [Ru2L4]–C2kH during the reaction. The Cadiot–Chodkiewicz (Cadiot for short) coupling method [39] was successfully employed by Gladysz and coworkers to extend Re–(CC)n– chains [40], which involved the formation of a copper alkynyl complex intermediate. As shown in Scheme 1, the extension of Ru2–(CC)k– monomers (k > 2) under Cadiot conditions was achieved with significantly increased yields compared to those under Hay conditions.

Extension of carbon chain for monomers through Cadiot coupling.
The longer dimers, namely the [Ru2L4](μ-C2n)[Ru2L4] (n ≥ 4) type compounds (Chart 3), were synthesized through either oxidative homo-coupling of corresponding monomers (for dimers with even-numbered n (C8, C12, C16 and C20)) or cross coupling of two different monomers (for type I dimers with odd n (C10, C14 and C18)) (Scheme 2), under Hay conditions in moderate to good yields. During the preparation of the latter type compounds, symmetric dimers (Ru2–μ-C2n–Ru2 and Ru2′–μ-C2n–Ru2′) were generated as the byproducts and tedious chromatography, typically preparative TLC, was necessitated for separation.

Homo/cross-coupling of mono-adduct to yield dimers.
Type I dimers are generally difficult to crystallize and only molecules containing C4 (short), C6 (short), and C8 (medium) bridges have been structurally characterized in our diruthenium system (Fig. 1) [23,35,36].
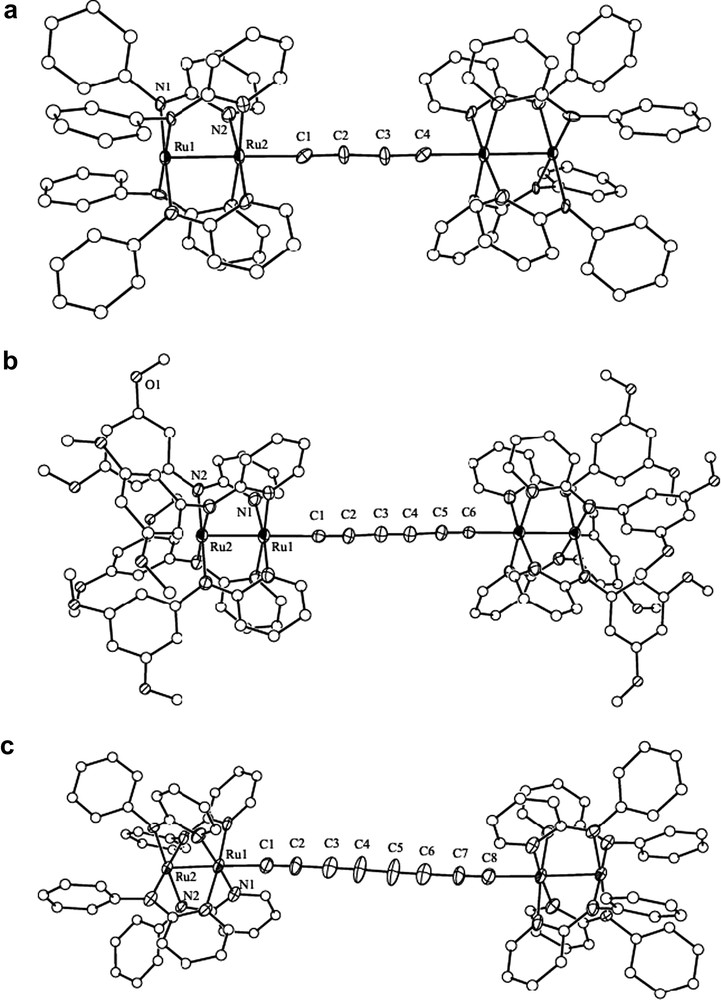
Structural plots of (a) [Ru2(ap)4]2(μ-C4), (b) [Ru2(DiMeOap)4]2(μ-C6), (c) [Ru2(ap)4]2(μ-C8).
Clearly, the coordination environment of the [Ru2L4] cores in dimers is identical to that of monomer type compounds (i.e. [Ru2L4]–C2Y). The N,N′-bidentate bridging ligands in both monomers and dimers adopt the (4,0) arrangement, namely that all anilino N-centers coordinate to the Ru center (4 site) and all pyridine N-centers coordinate to the other Ru center (0 site). Compared to its corresponding monomer, there is no significant change of Ru–Ru distance in the dimer (∼2.33 Å), while Ru–Cα bond became shorter (∼2.02 Å) due to the strengthened Ru–Cα interaction on the formation of polyyn-diyls systems.
2.2 Characterizations
2.2.1 Redox properties
In the type I compounds, exploration of the electron mobility across the polyyn-diyl chains is related to the investigation of the electronic coupling between two redox-active [Ru2L4] moieties, which was assessed on the basis of the comparison of their voltammetric behavior (CV/DPV) with that of the “half” molecule (monomer). For instance, the cyclic voltammogram of monomer [Ru2]–C4TMS ([Ru2] = [Ru2(DiMeOap)4]) exhibits two reversible one-electron processes at ca. 0.5 V (oxidation) and ca. −0.7 V (reduction) [38]. On the other hand, the dimer molecule, [Ru2]2(μ-C6) (A3) clearly exhibits more complex redox features including multiple reversible one-electron couples within the same potential window (Fig. 2): two stepwise oxidations (2+/1+) and (+1/0) at potentials close to 0.5 V and two stepwise reductions (0/−1) and (−1/−2) at potentials close to −0.7 V. Both oxidation couples and the first (least cathodic) two reduction couples are Ru2 based. The pairwise appearance of one-electron couples instead of a two-electron wave is an indicative of significant electronic coupling between two [Ru2] units. Moreover, the potential splitting within each pair (ΔE1/2) is proportional to the coupling strength and can serve as a qualitative indictor, as well known in the mixed-valence chemistry [41–43]. In addition, the potential differences within the oxidations (ΔE1/2(+1)) and reductions (ΔE1/2(−1)) allow the estimation of comproportionation constants for the corresponding mixed-valence cation (Kcom(+1))and anion (Kcom(−1)), respectively [41–44], which are summarized in Table 1. Finally, [Ru2L4] monomers with L as both ap and DiMeOap exhibit almost identical redox potentials (differences within 30 mV), which indicates minimal effect of ligand L on the electronic structures of Ru2 core [38,45].

Cyclic voltammograms of [Ru2]–C4TMS (monomer) and A3 (dimer) recorded in 0.20 M THF solution of Bu4NPF6 at a scan rate of 0.10 V/s.
Electrode potentials (V) for dimers A3–A9 from DPV measurementa
| Compounds | +2/+1 | +1/0 | 0/−1 | −1/−2 | −2/−3 | −3/−4 | Kcom(−1)b |
| A3 | 0.54 | 0.44 | −0.65 | −0.83 | −1.81 | −2.80 | 1300 |
| A4 | 0.54 (2e−) | −0.57 | −0.72 | −1.68 | −1.94 | 370 | |
| A5 | 0.56 (2e−) | −0.52 | −0.64 | −1.55 | −1.80 | 130 | |
| A6 | 0.57 (2e−) | −0.45 | −0.57 | −1.42 | −1.69 | 110 | |
| A7 | 0.58 (2e−) | −0.43 | −0.54 | −1.31 | −1.60 | 70 | |
| A8 | 0.58 (2e−) | −0.39 | −0.49 | −1.19 | −1.51 | 60 | |
| A9 | 0.59 (2e−) | −0.36 | −0.45 | −1.07 | −1.43 | 40 |
a Unpublished results.
b Kcom(−1) = exp (ΔE1/2/0.0257) at 25 °C.
The voltammetric plots (differential pulse voltammetry) in Fig. 3 and electrode potential data in Table 1 reveal similar redox behaviors within [Ru2L4]2(μ-C2n) type compounds and some noteworthy trends. First, for each dimer, the difference between E1/2(0/−1) and E1/2(−1/−2) (ΔE1/2(−1)) is much larger than the difference between E1/2(+2/+1) and E1/2(+1/0) (ΔE1/2(+1)), which suggests that the [Ru2L4]2(μ-C2n) type compounds are much better electron carrier than hole carrier, partly due to the electron-deficient nature of both the diruthenium core and polyyn-diyl bridge [36]. Second, as the polyyn-diyl chain elongates, both ΔE1/2(+1) and ΔE1/2(−1) gradually decrease (from resolved one-electron couples to merged pseudo two-electron waves) indicating the decrease of electronic coupling between two Ru2 centers. This trend is consistent with the consensus that the degree of coupling between a pair of donor–acceptor centers decays exponentially as the distance increases [41,43,46]. Third, potentials for all redox couples display a significant anodic shift as the polyyne chain elongates, which is attributed to the electron-deficient nature of the added acetylene units. Additionally, the second pair of one-electron reductions at more negative potentials was tentatively assigned as polyyn-diyl based [36].

Differential pulse voltammograms of dimers A3–A9 recorded in 0.20 M THF solution of Bu4NPF6 at a scan rate of 0.10 V/s.
2.2.2 Spectroelectrochemistry
As established in the mixed-valence chemistry, the splitting in potentials of oxidation/reduction pairs (ΔE1/2) and the resultant comproportionation constant Kcom usually serve as qualitative indicators of the nature of mixed-valency. These parameters generated from the electrochemical measurements significantly depend on the nature of solvents and electrolytes [47], and hence may be inappropriate as quantitative measures. A more suitable approach is to examine spectroscopic characteristics of the mixed-valence species, namely the intervalence transitions between donor and acceptor Ru2 moieties as mediated by the polyyn-diyl bridge, through spectroelectrochemical studies [41,43,44,48]. Fig. 4 shows electronic absorption spectra derived from spectroelectrochemical studies of [Ru2(ap)4]2(μ-C4) A2′ and its reference compound Ru2(ap)4(C2TMS) [36]. Upon either one-electron reduction or one-electron oxidation, intervalence charge transfer (IVCT) bands appeared and half-widths (bandwidths at half-peak height) were estimated from spectral deconvolution assuming Gaussian profiles (data are summarized in Table 2). The band at ca. 8450 cm−1 is clearly attributed to the CT between two Ru2 moieties of {[Ru2(ap)4]2(μ-C4)}−1. The lower energy band (5670 cm−1) is likely due to the mixing of LMCT and IVCT states in such a strongly delocalized system, and a definitive answer awaits a high level computational study. Based on the comparison of the estimated half-widths with those predicted from the Hush model, which describes weakly coupled mixed-valence species (Robin-Day class II) [49,50], the mixed-valence cation and anion of compound A2′ are strongly coupled species due to the narrowness of the observed IVCT bands [36,44]. They may be regarded as the class III species close to the borderline of class II–III using the Γ parameter analysis [51].
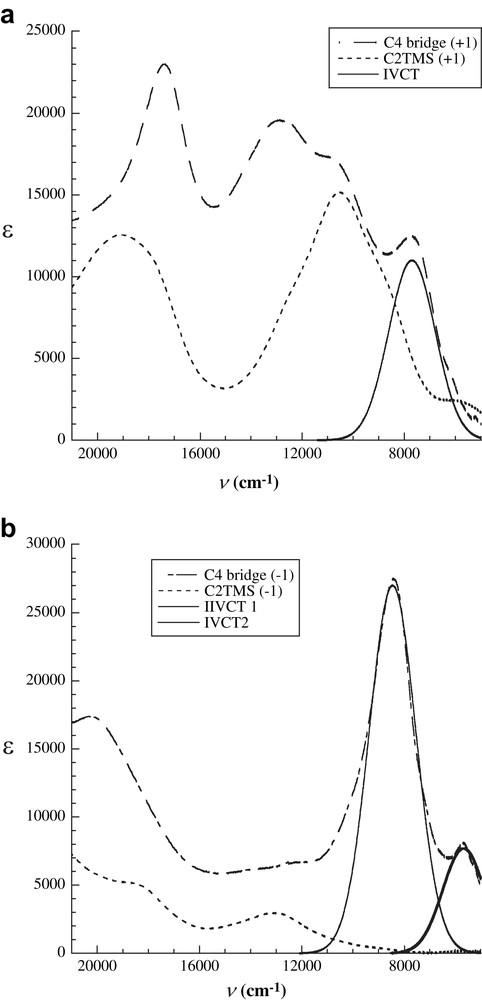
Spectroelectrochemistry results of A2′ and its “half” molecule Ru2(ap)4(C2TMS): (a) spectra with one deconvoluted IVCT band upon one-electron oxidation (b) spectra with two deconvoluted IVCT bands upon one-electron reduction.
Spectroelectrochemistry resultsa of mixed-valence ions of [Ru2(ap)4]2(μ-C4).
| Mixed-valence species | EIVCT | ɛ | Δν1/2 | Δν1/2(Hush)b | Γc |
| (A2′)1− | 8450 | 27000 | 1820 | 4420 | 0.59 |
| 5670 | 7700 | 1630 | 3620 | 0.55 | |
| (A2′)1+ | 7700 | 11000 | 1820 | 4220 | 0.57 |
a EIVCT, Δν1/2 and Δν1/2(Hush) in cm−1 and extinction coefficient (ɛ) in M−1 cm−1.
b Δν1/2(Hush) = (2310νmax)1/2.
c Γ = 1 − Δν1/2/Δν1/2(Hush).
The IVCT bands are also useful in estimating the adiabatic electronic coupling element Had for the series of [Ru2L4]2(μ-C2n) through Had = (ΔGrEIVCT)1/2, where ΔGr is the free energy of resonance exchange and EIVCT is related to the energy of IVCT band [52]. Furthermore, the electronic coupling attenuation constant γ, an efficient measure of the medium's ability to mediate electronic coupling, can be extracted using the equation Had = H0 exp(−γR), where Had and H0 are donor and acceptor wave function resonance exchanges at distance R and van der Waals contact distance, respectively, and R is the separation between the donor and acceptor from van der Waals contacts [41,43,44]. The success of extending the polyyn-diyl bridge to C20 provides the possibility of probing the electronic coupling over a long distance. For the reduced mixed-valence ions of the [Ru2L4]2(μ-C2n) compounds, two estimated γ were derived from a ln(Had) versus plot1 (Fig. 5): γ1 = 0.100 Å−1 for n = 1–3 and γ2 = 0.015 Å−1 for n = 4–10, which are comparable to that found for oligoene-bridged pentaamineruthenium(II,III) donor–acceptor pairs (γ = 0.070 Å−1) [53] and Fc–Fc + donor–acceptor pairs (γ = 0.087 Å−1) [54]. The appearance of dual attenuation parameters (bimodal) within a homologous series of molecules is relative rare [55], but has been frequently documented in electron transfer mediated by peptide chains [56]. The bimodal behavior in the latter case has been interpreted as superexchange (tunneling) at shorter distances and hopping at longer distances, although the precise mechanisms await more detailed studies.

Distance-dependence plot of ln(Had) (Had in cm−1) versus (in Å).
3 Ru2 bis-alkynyls capped by Fc termini
To realize the molecular wire as proposed in Chart 1, not only the charge transfer process on linear conjugated polyyn-diyl chains should be investigated, but also the role of the Ru2 core in mediating electron mobility needs to be addressed [57,58]. This is realized by covalently attaching a pair of ferrocenyl units at the opposite ends of the Ru2 fragment [Fc(CC)n–Ru2–(CC)mFc] and using both the comproportionation constant Kcom = exp(ΔEc/0.0257) (at room temperature, and ΔEc is defined by the equation below) [59] and spectroscopic characteristics of the mixed-valence ions to study the electron delocalization across the Ru2 core [49].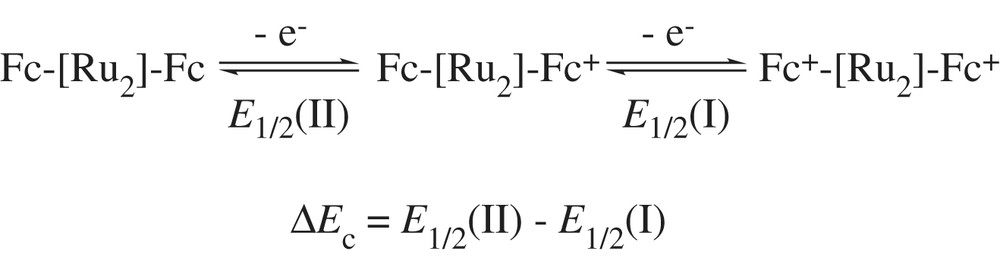
In this diruthenium bis-alkynyl series, the N,N′-bidentate bridging ligands (L) utilized in supporting the Ru2 core are N,N′-dimethylbenzamidinate (DMBA) and its phenyl-substituted derivative, N,N′-dimethyl-3-methoxy-benzamidinate (MeODMBA) (Chart 4). The modified DMBA ligand also has the advantage of significantly improved solubility of bis-compounds in common organic solvents [60].
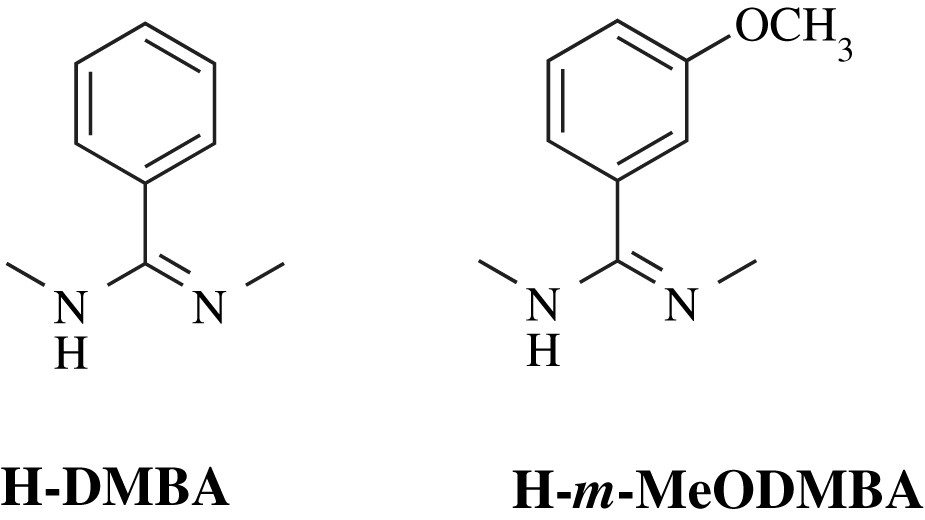
N,N′-dimethylbenzamidinate and its modified ligand.
3.1 Alkynylation protocols
A series of diruthenium bis-alkynyl compounds which contain two Fc termini trans-(FcC2n)[Ru2(Y-DMBA)4](C2mFc) (Chart 5) were successfully synthesized through either anion metathesis [57,58] route or the weak-base protocol (Scheme 3) [23,60–63].

trans-(FcC2n)[Ru2(Y-DMBA)4](C2mFc) series.
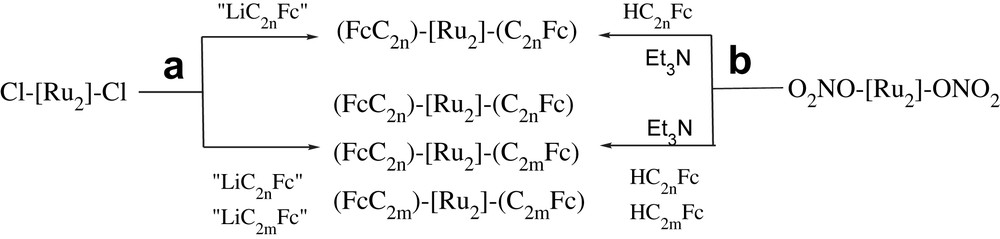
Synthetic routes (a) anion metathesis (b) weak-base protocol to the Fc-capped symmetric and unsymmetric compounds ([Ru2] = “Ru2(DMBA)” or “Ru2(MeODMBA)”).
Weak-base protocol is superior to anion metathesis for the preparation of compounds containing functional groups (i.e. –NO2, –CN) sensitive to organolithium agents [61]. In addition, anaerobic or anhydrous conditions are not required, which facilitates the reaction monitoring and optimization (choice of base and stoichiometry of reactions) [23,63].
Symmetric compounds [Ru2(Y-DMBA)4](C2nFc)2 were prepared in excellent yields, while unsymmetric compounds (FcC2n)[Ru2(Y-DMBA)4](C2mFc) were produced at lower yields after chromatographic separation from the symmetric compounds [57,58]. It is notable that compounds [Ru2(Y-DMBA)4](C6Fc)2 and [Ru2(Y-DMBA)4](C8Fc)2 cannot be synthesized through reactions between Ru2(Y-DMBA)4Cl2 and Li(CC)nFc (n = 3 and 4), due to the instability and difficult isolation of H(CC)nFc (n ≥ 3) [64].
Crystallization of the series of bis-alkynyl compounds was successful on less soluble Ru2(DMBA)4-based complexes [58]. Fig. 6 shows structural plots of compounds B1–B3 determined via single-crystal X-ray diffraction and reveals similar structures under successive extensions by one acetylene unit [57,58]. Other features are: (1) two ferrocenyl groups occupy the opposite axial positions, and the longest edge–edge distance between two Fc centers (Cω…Cω′) is calculated as 21.6 Å for the symmetric compound B4; (2) the relative orientations between two Fc units are either almost trans to each other in B1 (162°) or close to orthogonal in B2 (104°) [58]; (3) In compounds B1 and B2, Ru–N bond lengths vary and Ru′–Ru–C angles deviate from linearity. Such distortions from an idealized D4h geometry have been documented in many bis-alkynyl compounds of a Ru2(III,III) core and were addressed by a second-order Jahn–Teller effect [31,32,60,61].

Structural plots of (a) [Ru2(DMBA)4](C2Fc)2 (B1), (b) [Ru2(DMBA)4](C2Fc)(C4Fc) (B2), (c) [Ru2(DMBA)4](C4Fc)2 (B3).
3.2 Characterizations
3.2.1 Redox properties
Both the cyclic and differential pulse voltammograms of more soluble Ru2(m-MeODMBA)4-based compounds recorded in THF are shown in Fig. 7. For instance, compound B1′ exhibits three sequential one-electron oxidations (between +0.4 V and +1.0 V) and one one-electron reduction at ∼−1.2 V in the potential window of −1.5–+1.5 V (vs. Ag/AgCl). Spectroelectrochemical studies (details discussed below) identified the reduction and first oxidation as Ru2 based, while the second and third oxidations as Fc based. While the butadiynyl spaced di-ferrocene (Fc(CC)4Fc) to a pseudo two-electron oxidation [65], compound B1′ displays a remarkably large potential splitting of 310 mV, indicating a strong coupling between two ferrocenyl units (Kcom ≈ 173 000). As the Fc…Fc distance increases, similar redox behaviors to those illustrated in the [Ru2]–(μ-C2n)–[Ru2] are observed: (1) all redox couples are positively shifted, which is attributed to the strong electron-withdrawing nature of the added acetylene units; (2) Ru2 oxidation couples drastically shift and overlap with the first Fc oxidation couples as n is increased to generate a two-electron wave.

Electrochemical results of trans-(FcC2n)[Ru2(MeODMBA)4](C2mFc) series (compounds B1′–B5′) recorded in 0.20 M THF solution of Bu4NPF6 at a scan rate of 0.10 V/s with a glassy carbon working electrode.
3.2.2 Spectroelectrochemistry
Since the reduction couple is Ru2 based, spectroelectrochemical measurements focused on oxidations only [57,58]. Spectroelectrochemical study of symmetric compound B1′ provided absorption spectra of its oxidation products (B1′)+, (B1′)2+ and (B1′)3+, which show clean isosbestic points with good reversibility (>95% recovery) (Fig. 8). The first oxidation to (B1′)+ resulted in appearance of a very broad low energy band (band I) in the spectrum extending from the near-IR to IR region, with the maximum absorption out of the spectral range of study. The spectrum of dication (B1′)2+ displays a new higher energy band (band II) around 6000 cm−1 in addition to band I. Upon third oxidation to (B1′)3+, both bands disappeared. Bands I and II are assigned to IVCT bands; which vanished in (B1′)3+ since both Fe are in the 3+ state. Compared to the IVCT bands observed for biferrocene [65], band II has approximately the same energy and hence is assigned as Fc–[Ru2]+–Fc+ based (Fc to Fc+ transition). The other two oxidized forms are Fc–[Ru2]+–Fc ((B1′)+, mixed-valent, Fc to Ru27+ transition) and Fc+–[Ru2]+–Fc+ ((B1′)3+, no transitions), respectively. Unsymmetrical compound B2′ and symmetric compound B3′ exhibit similar spectroelectrochemical behaviors and data related to their mixed-valent species are listed in Table 3. Bandwidths of Fc to Fc+ IVCT are generally narrower than those predicted by Hush model, even across a distance of 16.6 Å [23,49,57,58]. Obviously, the characteristics of the IVCT band are consistent with the magnitude of the comproportionation constants, indicating a valence delocalized state. The extent of delocaliztion is sufficient to classify (B1′)2+, (B2′)2+ and (B3′)2+ as Robin-Day class III, class II–III and II–III species, respectively [50,51]. In contrast, other Fc–X–Fc+ systems (with X = none, acetylene, butadiyne or metal-containing units) exhibit very weak electronic communication despite short Fc–Fc+ distances [65–67]. Thus, all these results evidence the proficiency of [Ru2] core in mediating superexchange electronic coupling (especially hole transfer) over a long distance. The mechanism of [Ru2] acting as an efficient charge transfer bridge is under investigation.
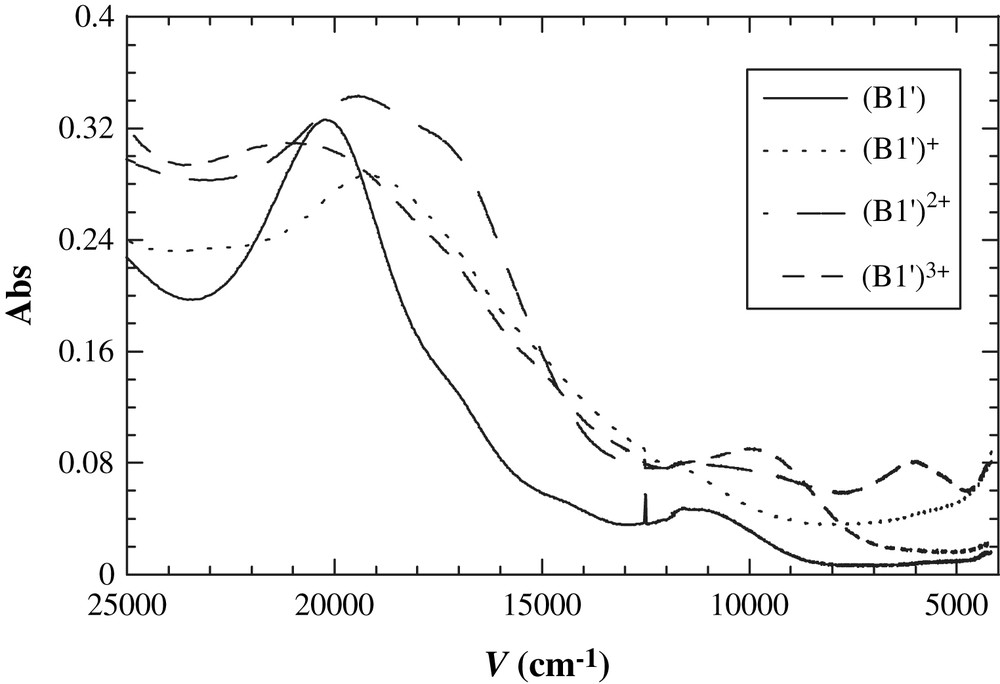
Spectroelectrochemistry results of B1′ upon sequential one-electron oxidation.
Electrochemical and spectroelectrochemical results of Fc–X–Fc+ type species.
| Fc–X–Fc+ | Cω…Cω′a | Kcom | EIVCTa | Δν1/2 | Δν1/2(Hush)b | Γ | ref |
| None | 1.45 | 821 000 | 5000 | 3700 | 3400 | – | [57] |
| –C2– | 4.0 | 7700 | 6410 | 5000 | 3850 | – | [56] |
| –C4– | 6.8 | 50 | 8470 | 5000 | 4420 | – | [56] |
| –C2Ru(dppm)2C2– | 9.36 | 5200 | 4770 | 3300 | 3320 | – | [58] |
| B1′ | 11.58 | 173 000 | 6000 | 800 | 3720 | 0.78 | [50,51] |
| B2′ | 13.84 | 3500 | 6230 | 1520 | 3790 | 0.60 | [51] |
| B3′ | 16.60 | – | 6680 | 1780 | 3930 | 0.55 | [51] |
a Cω…Cω′ (in Å) is the edge–edge distance between two Fc centers, EIVCT, Δν1/2 and Δν1/2(Hush) in cm−1.
b Δν1/2(Hush) = (2310νmax)1/2.
4 Conclusions and outlook
Reviewed herein is our exploration in the chemistry of wire-like diruthenium σ-alkynyl species (Chart 1), including their syntheses, structural characterization, bulk solution voltammetry and spectroelectrochemistry. Facile electron transfer across the polyyn-diyl chains was demonstrated and very small electronic coupling attenuation constants were determined, which favor the realization of the proposed molecule wires [68,69]. Also illustrated is the exceptional ability of diruthenium unit in enhancing charge mobility (especially hole mobility) on the basis of the studies of the trans-(FcC2n)[Ru2(Y-DMBA)4](C2mFc) series. Nevertheless, several pertinent issues remain unresolved: (1) charge transfer mechanisms resulting in two different attenuation constants in one system [Ru2]–(μ-C2n)–[Ru2] and (2) mechanism of [Ru2] acting as an efficient charge transfer bridge between two ferrocenes. Also, the access to type I compounds with n beyond 10 is a challenging task, although one may find comfort in the recent breakthrough of Gladysz in preparing diplatinum compounds of n up to 14 [70,71]. Other related future efforts include seeking the in-depth knowledge of the ground-state electronic structures and the spectroscopic characteristics of these carbon-rich systems, controlling the direction of charge transfer (rectification) through donor/acceptor functionalization [63], and studying the distance dependence of the spin–spin interaction between two [Ru2] units [23,58].
Acknowledgement
Financial support from the National Science Foundation (CHE 0715404) and Purdue University is gratefully acknowledged.
1 The energies of IVCT for most of the members in this series are currently unavailable and that of A2′ was used instead.