

1 Introduction
Recently, the synthesis, structure, and physical properties of polyynes and acetylenic compounds have attracted increasing attention [1]. The carbon–carbon triple bond is a crucial building block for the preparation of carbon-rich compounds as well as π-conjugated molecules. Especially, in the field of polymer chemistry, it is an essential unit to construct a rigid rod π-conjugated framework. Among π-conjugated polymers containing carbon–carbon triple bonds in their main chain, poly(p-arylene-ethynylene)s (PAEs) have attracted considerable attention, and they are currently one of the most important classes of conjugated polymers used as emitting, charge-transporting, conducting, and sensing materials [2]. PAEs with relatively high molecular weights can be readily prepared by a palladium-catalyzed coupling reaction [3] and the molybdenum-catalyzed metathesis [4]. PAEs with properties depending on their intended application have been developed by incorporating various aromatic compounds into their backbone.
In the past few years, we have employed [2.2]paracyclophane as the key unit for the construction of through-space conjugated PAEs [5] and aromatic ring-layered PAEs [6] (Chart 1). We evaluated that [2.2]paracyclophane-containing PAEs exhibited an extension of π-conjugation length via a through-bond as well as a through-space interaction; this prompted us to incorporate other cyclophane compounds into PAEs in order to take advantage of their characteristics. Thus, we have focused on dithia[3.3]metaphanes and its derivatives because they are of great interest; these compounds exhibit a unique transannular π–π interaction between two aromatic rings and also exhibit conformational flexibility [7,8]. Particularly, syn–anti ring inversion of two aromatic rings is the most interesting point in dithia[3.3]metaphane chemistry. Depending on the applicability of dithia[3.3]metaphanes, they can be considered to be promising building blocks for the aryl unit of PAEs. On the contrary, the structurally straight and rigid carbon–carbon triple bond is the best way to link dithia[3.3]metaphane units rather than a carbon–carbon double bond and other linkers in order to take advantage of their unique features. This account summarizes our study on PAEs comprising dithia[3.3]metaphane. The synthetic procedures, characterizations, and optical properties of dithia[3.3]metacyclophane- and dithia[3.3](2,6)pyridinophane-containing PAEs [9,10] are introduced.
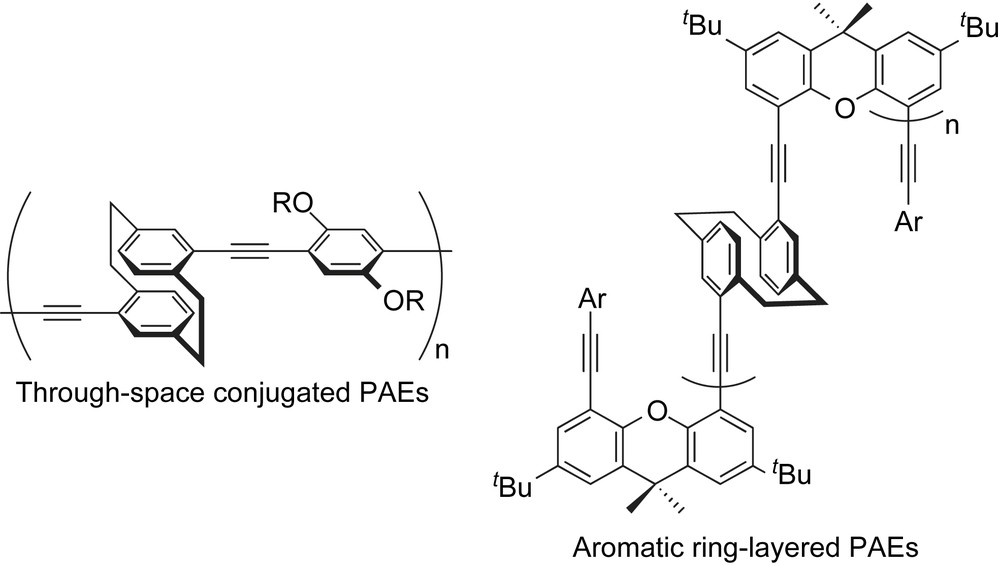
[2.2]Paracyclophane-containing PAEs.
2 PAE containing dithia[3.3]metacyclophane
2.1 Synthesis
A key monomer, 6,15-dibromo-2,11-dithia[3.3]metacyclophane 1, was synthesized by the direct coupling reaction of 5-bromo-1,3-benzenedimethanethiol with 5-bromo-1,3-bis(bromomethyl)benzene in 42% yield [9a,11]. As shown in Scheme 1, the reaction of 1 with diethynylbenzene 2 in the presence of a catalytic amount of Pd(PPh3)4 and CuI yielded a corresponding polymer 3 in 47% isolated yield with a number-average molecular weight of 14,000 (PDI = 1.9 by GPC, polystyrene standards). Polymer 3 was soluble in common organic solvents, and it was thermally and air stable both in solution and in the solid state.

2.2 Characterization and stereochemistry
Model compound 4, which has a dithia[3.3]metacyclophane core, was prepared in order to study the stereochemistry and the optical properties of the polymer [9a]. Fig. 1 shows the 1H NMR spectra (270 MHz) of monomer 1, compound 4, and polymer 3 in CDCl3 at 25 °C. In the 1H NMR spectrum of 1 (Fig. 1A), the singlet signals at 7.05 and 6.96 ppm are assignable to the outer and inner aromatic protons (Ho and Hi, Fig. 1), respectively. It is known that Hi of the anti conformation appears at 5–6 ppm due to a ring current effect [12]. In addition, the bridge methylene protons of 1 appeared at δ = 3.73 ppm as a sharp singlet peak (Fig. 1A). These results imply that the rapid syn–[anti]–syn interconversion of aromatic rings occurs in a solution.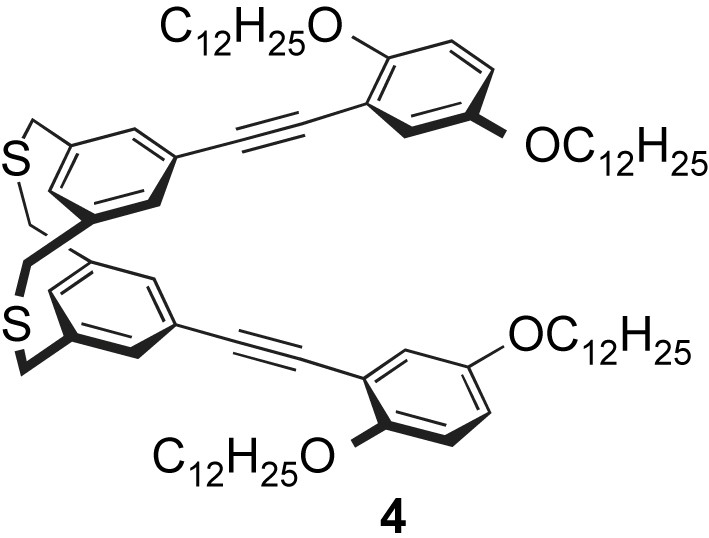

Schematic representation of syn–[anti]–syn isomerization of the dithia[3.3]metacyclophane moiety, and 1H NMR spectra in CDCl3 solution at room temperature; (A) monomer 1, (B) model compound 4, (C) polymer 3, and (D) polymer 5.
The 1H NMR spectrum of compound 4 and polymer 3 also exhibited conformational flexibility, as shown in Fig. 1B and C, respectively. In both cases, the inner protons Hi appeared at normal aromatic range and the bridged methylene protons appeared as a singlet peak at around 3.7 ppm. In dithia[3.3]metacyclophane units, the rapid syn–[anti]–syn interconversion of aromatic groups occurs in a solution, although they are incorporated into the PAE polymer backbone.
2.3 Reactivity
Dithia[3.3]metacyclophanes have been used as synthetic precursors for [2.2]metacyclophanes and [2.2]metacyclophane-dienes [7,13]. Dithia[3.3]metacyclophane-2,2′,11,11′-tetraoxide is an intermediate compound formed during the synthesis of [2.2]metacyclophane, which can be easily prepared by the oxidation reaction of 2,11-dithia[3.3]metacyclophane. According to a method described in the literature, oxidation was carried out by adding an excess amount of m-CPBA to polymer 3 in CHCl3 (Scheme 2) [9b,13,14]. Polymer 3 was easily oxidized to produce polymer 5 in 93% yield, and the 1H, 13C NMR and FTIR spectra revealed that all sulfur atoms were completely converted to sulfones. Additionally, the 1H NMR spectrum of polymer 5 exhibited rapid syn–[anti]–syn isomerization of dithia[3.3]metacyclophane tetraoxide units (Fig. 1D), same as that in polymer 3 [9b].

2.4 Optical properties
The optical properties of polymer 3 and compounds 4 and 6 [5a] were examined. In Fig. 2, the UV–vis absorption spectrum of polymer 3 in a dilute CHCl3 solution at room temperature exhibited absorption peaks at 311 and 372 nm, which agreed with the absorption peak of the PAE backbone. The absorption maxima of compounds 4 and 6 were observed at 331 and 370 nm, respectively, as shown in Fig. 2. The maximum absorption peak of polymer 3 (λmax = 372 nm) was observed at almost the same wavelength as that of 4, and the absorption edge of 3 had a longer wavelength than that of 4. Incidentally, [2.2]paracyclophane-containing PAE (Chart 1, R = C12H25) exhibited a maximum absorption peak at around 385 nm [5a]. Due to the flipping motion of dithia[3.3]metacyclophane units in polymer 3, an ineffective extension of the π-conjugation length of the polymer chain is observed.
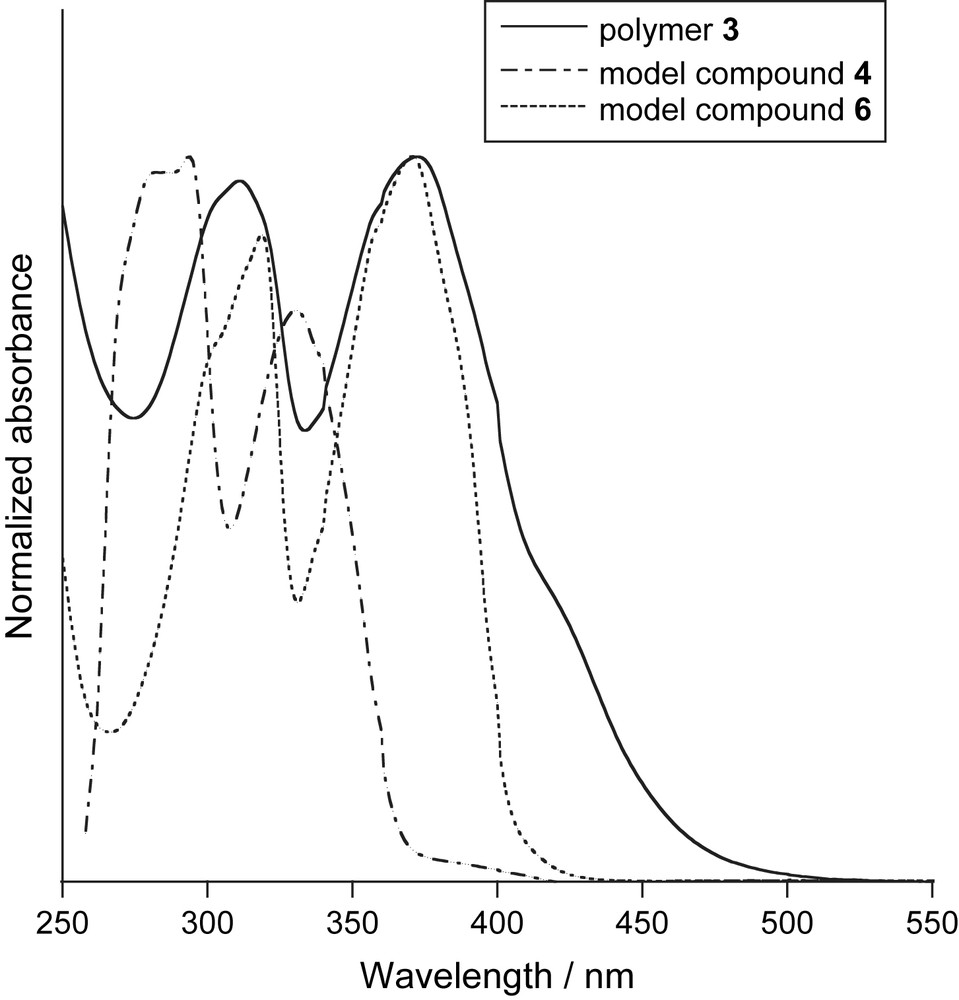
UV–vis absorption spectra of polymer 3 and model compounds 4 and 6 in CHCl3 solution (2.0 × 10−5 M) at room temperature.
In the fluorescence emission spectra of polymer 3 and compound 6 in dilute CHCl3 solution (1.0 × 10−5 M) at room temperature on excitation at absorption maximum, the emission peaks were observed at around 450 and 410 nm in the visible blue region, respectively (Table 1, Fig. 3). A large Stokes shift of polymer 3 was derived from a nonradiative transition caused by molecular movements such as phane-flipping, –CH2–S–CH2– bridge-wobbling. The above-mentioned emission spectra exhibited vibrational structures that were mirror images of the absorption spectra. It is well known that the emission of cyclophane compounds can correspond to two situations, i.e., emission in a monomer (chromophore) state or that in a phane state (excimer-like emission) [15,16]. The emission peak of polymer 3 exhibited a vibrational structure at λ = 405, 440, and around 470 (sh) nm, which was almost the same as that exhibited by compound 6 at λmax = 398 and 415 nm. This result implies that the emission of polymer 3 occurs in the monomer state rather than the phane state. In addition, the fluorescence quantum efficiency of polymer 3 was found to be 0.51 in the CHCl3 solution at room temperature.
Catalytic activity on Heck reaction.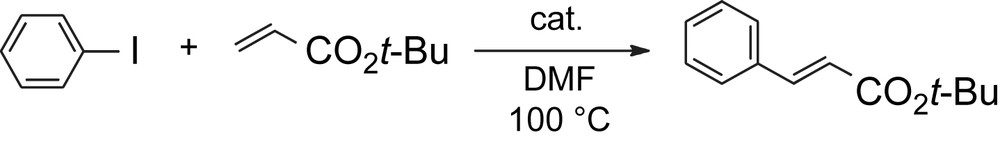
| Entry | Cat. | mol %a | Time/h | Yieldb/% |
| 1 | 10 | 1.0 | 1 | 98 |
| 2 | 10 | 0.05 | 1 | 90 |
| 3 | 10 | 0.05 | 24 | 93 |
| 4c | 10 | 0.05 | 1 | 91 |
| 5d | 10 | 0.05 | 1 | 85 |
| 6e | 10 | 0.05 | 1 | 81 |
| 7 | 10 | 0.01 | 24 | 86 |
| 8 | 11 | 0.05 | 1 | Trace |
| 9 | 11 | 0.05 | 24 | 90 |
| 10 | 12 | 0.05 | 1 | 8 |
| 11 | 12 | 0.05 | 24 | 73 |
a mol % of palladium–pyridinophane units.
b Determined by GLC based on the amount of phenyl iodide charged.
c The reaction was performed in air.
d 2nd cycle.
e 3rd cycle.

Fluorescence emission spectra of polymer 3 and compound 6 in dilute CHCl3 solution (1.0 × 10−5 M) at room temperature.
3 PAE containing dithia[3.3](2,6)pyridinophane
3.1 Synthesis
Dithia[3.3](2,6)pyridinophane, which has two pyridine rings linked by two –CH2–S–CH2– bridges, exhibits a unique property in addition to conformational flexibility and transannular π–π interaction. Moreover, dithia[3.3](2,6)pyridinophane can be used as a bidentate nitrogen ligand for transition metals. As shown in Scheme 3, the polymerization of monomer 7 with 1,4-diethynyl-2,5-dioctyloxybenzene 8 was carried out in toluene-diisopropylamine solution in the presence of a catalytic amount of PdCl2(PPh3)2/PCy3/CuI (Cy = cyclohexyl) to produce the corresponding PAE 9 in 81% yield with Mn = 4200 [10]. Metal species could be removed by washing the polymer solution with aqueous NH3 three times, which was confirmed by the 1H NMR spectrum (vide infra).

3.2 Characterization and stereochemistry
Fig. 4A and B shows the 1H NMR spectra of monomer 7 and polymer 9 in a CDCl3 solution, respectively. The chemical shift of the bridged methylene protons (–CH2–S–CH2–) of monomer 7 appeared as a sharp singlet peak at δ = 3.9 ppm. This indicates that rapid ring syn–anti flipping occurs in the solution of monomer 7. In the 1H NMR spectrum of polymer 9 in CDCl3 (Fig. 5B), the signals of the octyloxy chains were observed at δ = 0.87, δ = 1.2–1.9, and δ = 3.8 ppm as a shoulder peak overlapped with the bridged methylene protons of the dithia[3.3](2,6)pyridinophane unit. Thus, the dithia[3.3](2,6)pyridinophane units in polymer 9 are also flexible, and the rapid syn–anti interconversion of pyridine rings is observed in the solution of polymer 9.
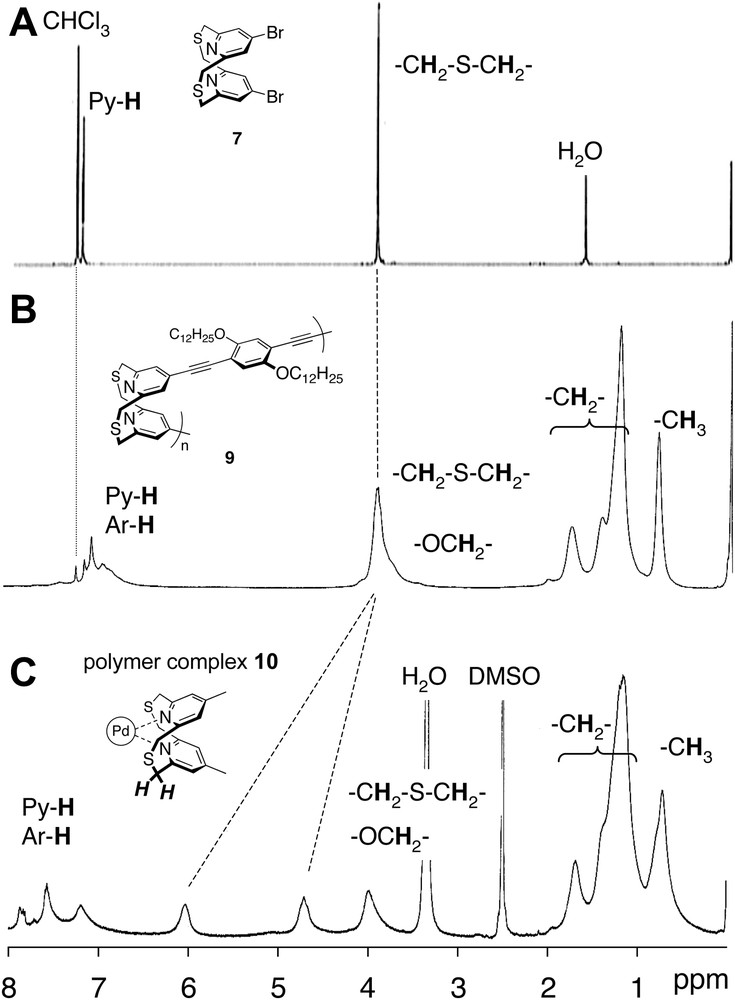
1H NMR spectra at room temperature; (A) monomer 7 in CDCl3, (B) polymer 9 in CDCl3, and (C) polymer complex 10 in DMSO-d6.
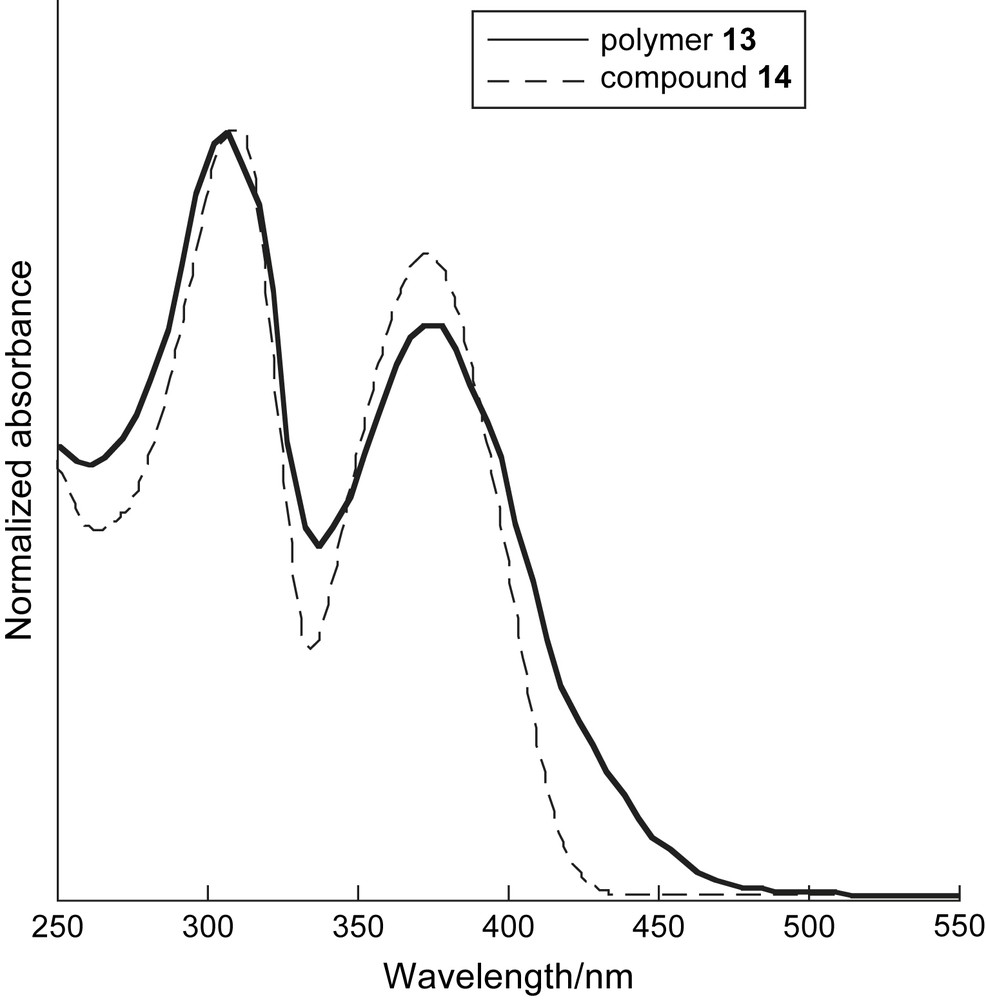
UV–vis absorption spectra of polymer 13 and compound 14 in CHCl3 (2.0 × 10−5 M) at room temperature.
3.3 Complexation behavior
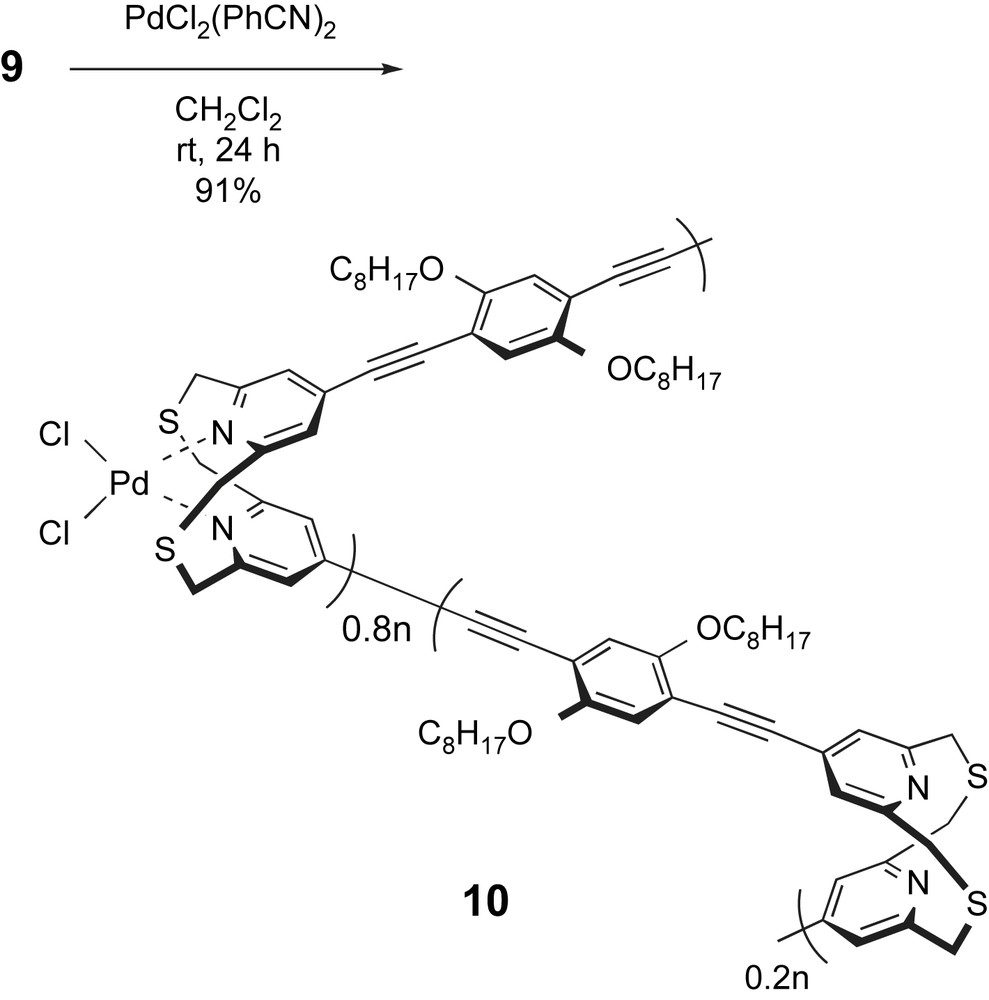
It is reported that the complexation reaction between dithia[3.3](2,6)pyridinophane and the palladium complex PdCl2(PhCN)2 produces PdCl2(dithia[3.3](2,6)pyridinophane) [17]. Therefore, we examined the complexation reaction between polymer 9 and PdCl2(PhCN)2. As shown in Scheme 4, polymer 9 was treated with an equimolar amount of PdCl2(PhCN)2 in CH2Cl2 solution, and a brown-colored polymer complex 10 was precipitated in 91% yield. This polymer complex 10 was insoluble in CH2Cl2, CHCl3, and THF, and it was slightly soluble in aprotic solvents such as DMF and DMSO. Fig. 4C shows the 1H NMR spectrum of polymer complex 10 in DMSO-d6. In Fig. 4C, –CH2–S–CH2– methylene protons of polymer 9 (Fig. 4B) were separated into two signals at δ = 4.7 and 6.0 ppm, which were consistent with those of PdCl2(dithia[3.3](2,6)pyridinophane) (at δ = 4.72 and 6.07 ppm) reported by Tsuge and coworkers [17]. The formation of the palladium complex caused dithia[3.3](2,6)pyridinophane units to fold the syn conformation. The complexation efficiencies were calculated to be 80% by comparing with the integral ratio of separated methylenes of coordinated pyridinophane units (δ = 4.7 and 6.0 ppm) and methylenes of unreacted pyridinophane units (δ = 4.0 ppm). Due to the low solubility of polymer complex 10 in CH2Cl2, it was precipitated from the reaction mixture during the complexation process.
3.4 Catalytic activity of polymer complex 10
The catalytic activity of the obtained polymer complex 10 was examined. In the presence of a catalytic amount of the palladium–pyridinophane units of polymer complex 10, the Heck coupling reaction between phenyl iodide and tert-butyl acrylate was carried out smoothly to produce the corresponding tert-butyl cinnamate with high yields (Table 1). The best result was obtained when 1.0 mol% of palladium–pyridinophane units were used (entry 1, 98% yield), while only 0.01 mol% of palladium–pyridinophane units exhibited a high catalytic activity to give tert-butyl cinnamate in 86% yield (entry 7). The catalytic activity of polymer complex 10 at room temperature also yielded a good result (entry 4). After the reaction, polymer complex 10 was purified by reprecipitation from an excess amount of CHCl3 solution and used for the next reaction. The results of this recycling study are also listed in Table 1, entries 5 and 6. Polymer complex 10 maintained the catalytic activity in the 3rd cycle and yielded tert-butyl cinnamate in 81% yield (entry 6). The catalytic activity of 10 was compared with those of PdCl2(dithia[3.3](2,6)pyridinophane) 11 (entries 8 and 9, Table 1) and PdCl2(2,2′-bipyridine) 12 (entries 10 and 11, Table 1). Complexes 11 and 12 showed good catalytic activities and afforded tert-butyl cinnamate in 90% and 73% yields (entries 9 and 11, Table 1), respectively. However, longer reaction time was required to complete this reaction. It is considered that electron density of palladium in polymer complex 10 is higher than that in 11 and 12 due to the electron-donation from the electron rich alkoxyphenylene units to the pyridinophane units in 10. Thus, this conjugation effect in polymer complex 10 enhances the oxidative addition of aryl halide to palladium.
3.5 Optical properties
The UV–vis absorption spectra of polymer 13, which has dihexyloxyphenylene moieties (Mn = 4200, PDI = 2.3), and model compound 14 [18] are depicted in Fig. 5. Polymer 13 exhibited a maximum absorption peak at 377 nm derived from the typical π–π∗ transition of PAE moieties, and the absorption edge was observed at around 500 nm. Compound 14 had a spectrum similar to that of polymer 13 with the maximum absorption peak at 374 nm and the absorption edge at around 430 nm, as shown in Fig. 5. The through-space interaction of the dithia[3.3](2,6)pyridinophane unit appears to be less effective for the extension of π-conjugation due to conformational flexibility, which is similar to the case of the dithia[3.3]metacyclophane-containing polymer.
The emission spectrum of polymer 13 at 464 nm (fluorescence quantum efficiency of 0.14) in the visible greenish-blue region in dilute CHCl3 solution (1.0 × 10−5 M) is shown in Fig. 6. The emission spectrum of 13 appeared to be featureless and did not have a vibrational structure. However, based on the emission spectrum of the dithia[3.3]metacyclophane-containing polymer 3, and the similar spectra of polymer 13 and compound 14 [18b], it is supposed that the emission spectrum of 13 is the same as that of the pyridine-containing arylene-ethynylene unit possessing an inconspicuous vibrational structure. Additionally, the emission spectrum of polymer 13 would be derived from the monomer state, which is the same as that of polymer 3. The maximum emission peak of polymer 13 appeared at 464 nm, and it was red-shifted as compared to that of polymer 3 (λmax = 440 nm) because of an intramolecular donor–acceptor effect caused by the electron-accepting pyridine ring.

Photoluminescence spectra of polymer 13 in CHCl3 (1.0 × 10−5 M) excited on 377 nm at room temperature, and polymer 13 with 80% of Pd(II) and Pt(II) in CHCl3 (1.0 × 10−5 M) excited on 400 nm at room temperature.
Fig. 6 shows the photoluminescence spectra of polymer complexes with 80% Pd(II) and Pt(II) obtained by the complexation between PdCl2(PhCN)2 and PtCl2(PhCN)2, respectively. The coordination of transition metals such as Pd(II) and Pt(II) affected the fluorescence emission property, and the emission spectra were significantly reduced. This result indicates the possible application of the dithia[3.3](2,6)pyridinophane-containing PAE in a transition metal sensor.
4 Conclusion
In summary, we have synthesized conjugated polymers possessing the dithia[3.3]metacyclophane and dithia[3.3](2,6)pyridinophane units based on the PAE backbone. The polymers were readily synthesized by a palladium-catalyzed coupling reaction. The rapid syn–anti isomerization of dithia[3.3]metaphane in the PAE main chain was observed in solution. The through-space conjugation was ineffective in the ground state because of the flipping motion of the dithia[3.3]metaphane unit, and photoluminescence from the monomer state was observed in the excited state. Dithia[3.3](2,6)pyridinophane-containing PAE exhibited a coordination ability to transition metals such as palladium, and the conformation of the dithia[3.3](2,6)pyridinophane unit was folded in a syn form. This polymer complex acted as a recyclable catalyst for the conventional Heck coupling reaction. The photoluminescence of dithia[3.3](2,6)pyridinophane-containing PAE was considerably reduced by metal coordination, which suggests the possibility of applying this polymer in a heavy metal sensor. These findings will contribute to the design and synthesis of a novel PAE as well as through-space conjugated polymers utilizing their dynamic behavior and/or coordination ability.
Acknowledgments
This work was supported by Grant-in-Aid for Creative Scientific Research of “Invention of Conjugated Electronic Structures and Novel Functions”, No. 16GS0209, from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. Financial support from the Mitsubishi Foundation is also acknowledged. T.I. appreciates Research Fellowships from the Japan Society for the Promotion of Science for Young Scientists.