

1 Introduction
Metal catalyzed homo- and cross-coupling reactions are among the most effective tools for organic and medicinal chemistry [1,2]. Ru and Mo catalyzed olefin metathesis reactions have become an indispensable arsenal for the synthesis of delicate molecular targets of pharmaceutical and materials interests [3]. Conventional C–C bond formation (between sp/sp2 carbons) methodologies have also experienced a renaissance during the last decade [4,5]. In comparison, applications of coupling methodologies to coordination/organometallic compounds have remained largely unexplored and limited. Recent efforts have focused on the utility of the Sonogashira [6] and Suzuki [7] reactions in derivatizing metallo-porphyrins, metal bi-/ter-pyridine complexes and metal-alkynyl compounds.
During the course of developing organometallic molecular wires, we noticed that Ru2-alkynyl compounds are excellent chromophores and electrophores, and hence promising building blocks for novel (opto)electronic materials [8]. Further exploration of the materials' potential of Ru2-alkynyl species necessitates controlled assemblies of these building blocks without significantly altering their (opto)electronic properties. Cross-coupling reactions at the periphery of Ru2-alkynyls are appealing tools for such assemblies owing to the formation of robust C–C bonds and chemical selectivity therein. Described in this short review are our recent efforts in both the development of appropriate Ru2-precursors and execution of ensuing coupling reactions including the Sonogashira, Suzuki, Heck and Negishi types.
2 Ru2-species as cross-coupling substrates
Aryl halides, especially aryl iodides, are the most common substrates for cross-coupling reactions. Hence, iodo substituents were introduced via the preparation of Ru2L4−xL′x (x = 1 or 2) type compounds, where auxiliary ligand L is a diarylformamidinate (DArF), and L′ is an iodo-containing ligand, either N,N′-dimethyl-4-iodobenzamidinate (DMBA-I) or N,N′-di(4-iodophenyl)formamidinate (D(4-IPh)F) (Chart 1). The selective formation of Ru2L4−xL′x relies on the high yield syntheses of both the Ru2(DArF)3(OAc)Cl and Ru2(DArF)2(OAc)2Cl type compounds. DArFs are used as ancillary ligands due to their inertness toward substitution and good solubility of the resultant Ru2 complexes in common organic solvents. The first examples of such compounds were reported in 1999 [9], and further explorations by several groups [10–13] demonstrated that Ru2(DArF)3(OAc)Cl can be prepared on a multi-gram scale. As shown in Scheme 1, a typical preparation of Ru2(DArF)3(OAc)Cl involves refluxing Ru2(OAc)4Cl with 3 equiv of HDArF in THF, and subsequent recrystallization or column purification results in analytically pure material. This simple route works well with a range of DArF ligands such as N,N′-di(3-methoxyphenyl)formamidinate (DmAniF) [11,12] and N,N′-di(3,5-dichlorophenyl)formamidinate (D(3,5-Cl2Ph)F) [13]. Preparation of Ru2(DArF)2(OAc)2Cl type compounds (Scheme 1) is similar to that of Ru2(DArF)3(OAc)Cl, but requires lower reaction temperatures [12,13]. Both the Ru2(DArF)3(OAc)Cl and Ru2(DArF)2(OAc)2Cl type compounds are paramagnetic S = 3/2 species and their structures were established through single crystal X-ray diffraction studies (Fig. 1), which revealed the cis-configuration of the Ru2(DArF)2(OAc)2Cl type compounds.
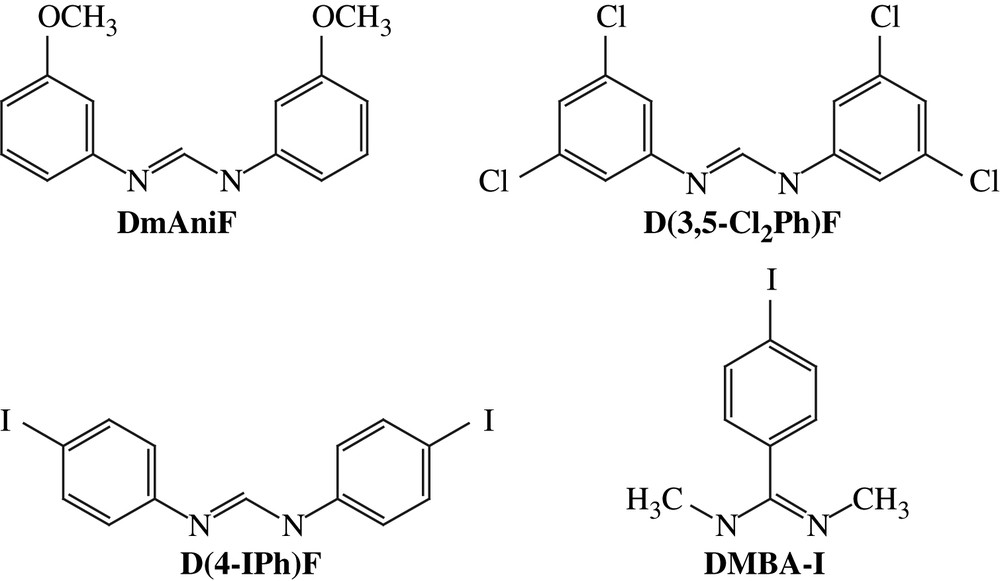
Structures of ligands L and L′.

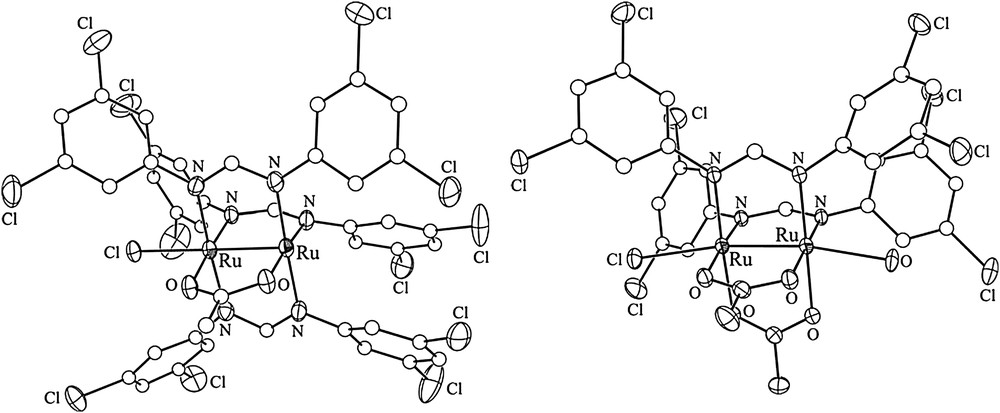
Structural plots of Ru2(D(3,5-Cl2Ph)F)3(OAc)Cl (left) and cis-Ru2(D(3,5-Cl2Ph)F)2(OAc)2Cl(H2O).
Also shown in Scheme 1, the Ru2(DArF)4−x(OAc)xCl (x = 1 and 2) type compounds undergo smooth ligand displacement reactions with a different N,N′-bidentate ligand, L′, to yield the Ru2(DArF)4−x(L′)xCl type compounds. L′ can be either DMBA-I or D(4-IPh)F, and contains one or two peripheral iodo substituents that enables further modification. The Ru2(DArF)4−x(L′)xCl type compounds also undergo reactions with LiC2R (R as Ph or C2SiMe3, in excess) to afford the corresponding axial alkynyl derivatives trans-[Ru2(DArF)4−x(L′)x](C2R)2. As shown in Fig. 2, structural studies of the derivatized Ru2(L)2(L′)2 type compounds revealed the retention of cis-configuration, confirming the substitution inertness of the DArF ligand in Ru2(DArF)4−x(OAc)xCl.

Structural plots of Ru2(D(3,5-Cl2Ph)F)3(D(4-I–Ph)F)Cl (left) and cis-Ru2(DmAniF)2(DMBA-I)2(CCPh)2.
3 Peripheral cross-coupling reactions
Common C–C bond formation reactions via cross-coupling pathway include the Heck, Suzuki, Sonogashira, Negishi, Kumada, Stille, and Hiyama types [1], and the first four types have been successfully executed at the periphery of diruthenium species. Pd catalysts commonly used in organic cross-coupling reactions are employed here, and all observations are consistent with the oxidative addition and reductive elimination pathway. Although transition metals other than Pd are also known to facilitate cross-coupling reactions [1], our scope is limited to the Pd-based catalysts.
3.1 Sonogashira coupling
In a typical Sonogashira reaction [5,14] an iodoarene and a terminal alkyne are coupled in the presence of trans-Pd(PPh3)2Cl2, CuI and a weak base to afford the corresponding arylacetylene, and the reaction conditions have not been modified much since the publication of the original report [14]. Two types of Sonogashira reactions with a Ru2 substrate are presented in Scheme 2. Our initial attempt was based on Ru2(DmAniF)3(DMBA-I)Cl, which cross coupled with HCCY (Y = SiMe3 and Fc) to afford the corresponding Ru2(DmAniF)3(DMBA–CCY)Cl in modest yields, as shown in Table 1 [11]. Upon reacting with LiC4SiMe3 or LiC2Ph, the Ru2(DmAniF)3(DMBA–CCY)Cl type compounds were converted to the axially alkynylated derivative trans-(RCC)2[Ru2(DmAniF)3(DMBA–CCY)]. Concurrent Sonogashira couplings on Ru2(DmAniF)2(DMBA-I)2Cl resulted in Ru2(DmAniF)2(DMBA–CCFc)2Cl (Fig. 3) in good yield [12]. Similarly, both Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl and Ru2(D(3,5-Cl2Ph)F)2(DMBA-I)2Cl underwent Sonogashira coupling to afford, respectively, Ru2(D(3,5-Cl2Ph)F)3(DMBA–CCY)Cl and Ru2(D(3,5-Cl2Ph)F)2(DMBA–CCY)2Cl with yields better than those of DmAniF auxiliary ligands [13]. The improved yields are attributed to the electron deficiency of D(3,5-Cl2Ph)F ligand, which renders Ru2(D(3,5-Cl2Ph)F)4−x(DMBA-I)xCl type compounds better substrates for cross-coupling reactions. Ru2(DArF)4−x(D(4-IPh)F)xCl (x = 1 and 2) type compounds also underwent Sonogashira coupling reaction under similar conditions. Sonogashira coupling reactions using trans-(RCC)2[Ru2(DmAniF)3(DMBA-I)] as the Ru2 substrate were also attempted but failed to yield the expected products. Instead, Ru2-species free of axial alkynyl ligands were detected as the main by products [11]. In initiating the Sonagashira coupling, Cu(I) ion activates terminal alkyne through a η2 coordination [19], which probably also induces the cleavage of Ru–CC bond in our experiments. The copper-free Sonogashira coupling was demonstrated recently [20], and it will be interesting to see whether trans-(RCC)2[Ru2(DmAniF)3(DMBA-I)] can be similarly derivatized.
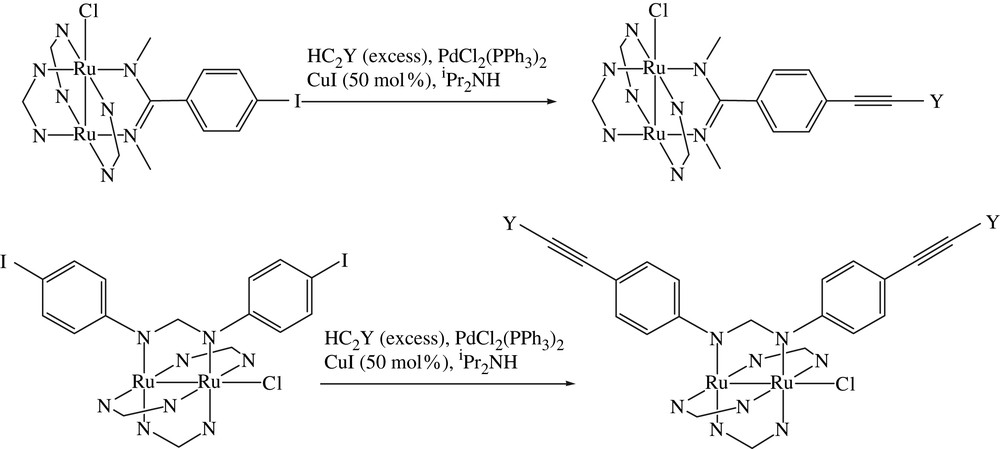
Sonogashira couplings of Ru2 substrates.
Peripheral cross-coupling reactions
| Ya | Product | Yieldb (%) | Ref. | |
| Sonogashira | ||||
| Ru2(DmAniF)3(DMBA-I)Cl | SiiPr3 | Ru2(DmAniF)3(DMBA–CCSiiPr3)Cl | 41 | [11] |
| Fc | Ru2(DmAniF)3(DMBA–CCFc)Cl | 39 | ||
| Ru2(DmAniF)2(DMBA-I)2Cl | Fc | Ru2(DmAniF)2(DMBA–CCFc)2Cl | 52 | [12] |
| Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl | SiMe3 | Ru2(D(3,5-Cl2Ph)F)3(DMBA–CCSiMe3)Cl | 75 | [13] |
| Ph | Ru2(D(3,5-Cl2Ph)F)3(DMBA–CCPh)Cl | 84 | ||
| Ru2(D(3,5-Cl2Ph)F)2(DMBA-I)2Cl | SiMe3 | Ru2(D(3,5-Cl2Ph)F)2(DMBA–CCSiMe3)2Cl | 78 | [13] |
| Fc | Ru2(D(3,5-Cl2Ph)F)2(DMBA–CCFc)2Cl | 46 | ||
| Ru2(DmAniF)3(D(4-IPh)F)Cl | SiMe3 | Ru2(DmAniF)3(D(4-CCSiMe3Ph)F)Cl | 90 | [15] |
| [Ru2(DmAniF)3(DMBA-I)](σ-C4SiMe3)2 | SiiPr3 | No product isolated | – | [11] |
| Suzuki | ||||
| Ru2(DmAniF)3(D(4-IPh)F)Cl | H | Ru2(DmAniF)3(D(4-Ph–Ph)F)Cl | 50 | [16] |
| Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl | H | Ru2(D(3,5-Cl2Ph)F)3(D(4-Ph–Ph)F)Cl | 41 | [16] |
| CH3C(O)– | Ru2(D(3,5-Cl2Ph)F)3(D(4-CH3C(O)Ph–Ph)F)Cl | 20 | ||
| Ru2(DmAniF)3(D(4-IPh)F)(σ-CCPh) | H | Ru2(DmAniF)3(D(4-Ph–Ph)F)(σ-CCPh) | 49 | [16] |
| Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)(σ-CCPh) | H | Ru2(D(3,5-Cl2Ph)F)3(D(4-Ph–Ph)F)(σ-CCPh) | 55 | [16] |
| CH3C(O)– | Ru2(D(3,5-Cl2Ph)F)3(D(4-CH3C(O)Ph–Ph)F)(σ-CCPh) | 17 | ||
| Negishi | ||||
| Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl | H | NR | [17] | |
| CH3O | ||||
| CF3 | ||||
| Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl | H | Ru2(D(3,5-Cl2Ph)F)3(D(4-Ph–Ph)F)Cl | 79 | [17] |
| CH3O | Ru2(D(3,5-Cl2Ph)F)3(D(4-MeOPh–Ph)F)Cl | 85 | ||
| CF3 | Ru2(D(3,5-Cl2Ph)F)3(D(4-CF3Ph–Ph)F)Cl | 92 | ||
| Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)(σ-CCPh)2 | H | Ru2(D(3,5-Cl2Ph)F)3(DMBA-4-Ph)(σ-CCPh)2 | 47 | [17] |
| CH3O | Ru2(D(3,5-Cl2Ph)F)3(DMBA-4-(4′-MeOPh))(σ-CCPh)2 | 75 | ||
| CF3 | Ru2(D(3,5-Cl2Ph)F)3(DMBA-4-(4′-CF3Ph))(σ-CCPh)2 | 79 | ||
| Heck | ||||
| Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl | Ph | Ru2(D(3,5-Cl2Ph)F)3(D(4-PhCHCHPhF)Cl | 52 | [18] |
| 3-NO2Ph | Ru2(D(3,5-Cl2Ph)F)3(D(4-(3′-NO2Ph)CHCHPh)F)Cl | 65 | ||
| C(O)OCH3 | Ru2(D(3,5-Cl2Ph)F)3(D(4-(MeC(O)OCHCHPh)F)Cl | 70 | ||
| Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl | Ph | Ru2(D(3,5-Cl2Ph)F)3(DMBA–CHCHPh)Cl | 68 | [18] |
| SiMe3 | Ru2(D(3,5-Cl2Ph)F)3(DMBA–CHCHSiMe3)Cl | 78 | ||
| Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)(σ-CCPh)2 | Ph | Ru2(D(3,5-Cl2Ph)F)3(DMBA–CHCHPh)(σ-CCPh)2 | 60 | [18] |
| C(O)OCH3 | Ru2(D(3,5-Cl2Ph)F)3(DMBA–CHCHC(O)OMe)(σ-CCPh)2 | 52 |
a The definition of Y is given in Schemes 2–5 for Sonogashira, Suzuki, Negishi and Heck reactions, respectively.
b All reported yields are based on column purified products.

Structural plots of Ru2(D(3,5-Cl2Ph)F)3(DMBA–CCSiMe3)Cl (left) and [cis-Ru2(DmAniF)2(DMBA-4-CCFc)2](σ-CCPh)2.
3.2 Suzuki and Negishi couplings
Suzuki (also known as Suzuki–Miyaura) coupling has been one of the most frequently practiced C–C bond formation reactions in medicinal and materials chemistry [21], and reaction conditions are more demanding on the substrate than that of Sonogashira due to the requirement of refluxing at elevated temperature. The initial attempt of the reaction between Ru2(DmAniF)3(D(4-IPh)F)Cl and PhB(OH)2 in the presence of Pd(OAc)2, K2CO3 and refluxing THF(aq) resulted in the degradation of diruthenium species. After trials of several sets of conditions, the combination of trans-PdCl2(PPh3)2, tBuOK and THF solvent resulted in the expected Suzuki derivative Ru2(DmAniF)3(D(4-Ph–Ph)F)Cl (Scheme 3) [16]. The optimized conditions were also successfully applied to the reactions with Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl as the Ru2 substrate (see Table 1 and Fig. 4). In contrast to the experience of Sonogashira coupling, the Ru2(DArF)3(D(4-IPh)F)(σ-CCPh) type compounds also underwent Suzuki coupling to yield the corresponding derivatives in satisfactory yields. It is noteworthy that free ligand HD(4-IPh)F either decomposed or became the dehalogenated derivative under various Suzuki coupling conditions, reaffirming the necessity of the peripheral modification reaction.
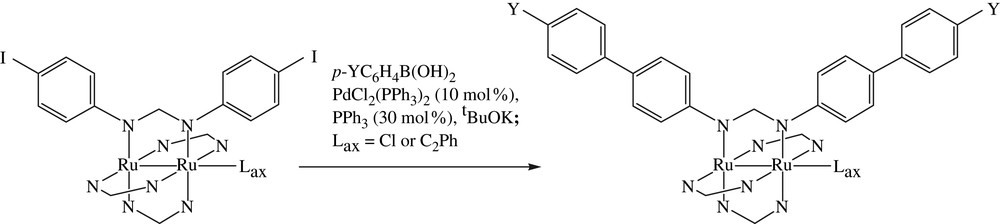
Suzuki couplings of Ru2 substrates.

Structural plots of Ru2(D(3,5-Cl2Ph)F)3(D(4-Ph–Ph)F)Cl (left) and Ru2(D(3,5-Cl2Ph)F)3(D(4-CH3C(O)Ph–Ph)F)(σ-CCPh).
The Negishi coupling method encompasses the cross-coupling reactions between organic halides and organometals including Zn, Al and Zr [22]. Our foray has been limited to the Pd catalyzed reactions between Ru2 substrates and organozinc reagent that result in the formation of biphenyl. Our initial efforts were based on Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl starting material, but all attempts resulted in the decomposition of the starting materials. The use of Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl as the Ru2 substrate resulted in immediate successes: cross-coupling with BrZnPh (10 fold excess) in the presence of 10% trans-Pd(DPEPhos)Cl2 (Scheme 4) [23] yielded the expected product Ru2(D(3,5-Cl2Ph)F)3(D(4-Ph–Ph)F)Cl in 80% yield. The biphenyl derivative was previously prepared from the Suzuki coupling in much lower yield (see Table 1) [16]. The biphenyl formation reaction under Negishi conditions was successfully extended to BrZnC6H4-4-Y with Y as MeO and CF3 in excellent yields. Further demonstrating the advantage of Negishi coupling over Suzuki coupling, the biphenyl formation reaction was successfully performed on Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)(σ-CCPh)2, a substrate that is prone to the cleavage of axial CCPh ligand under extensive heating.
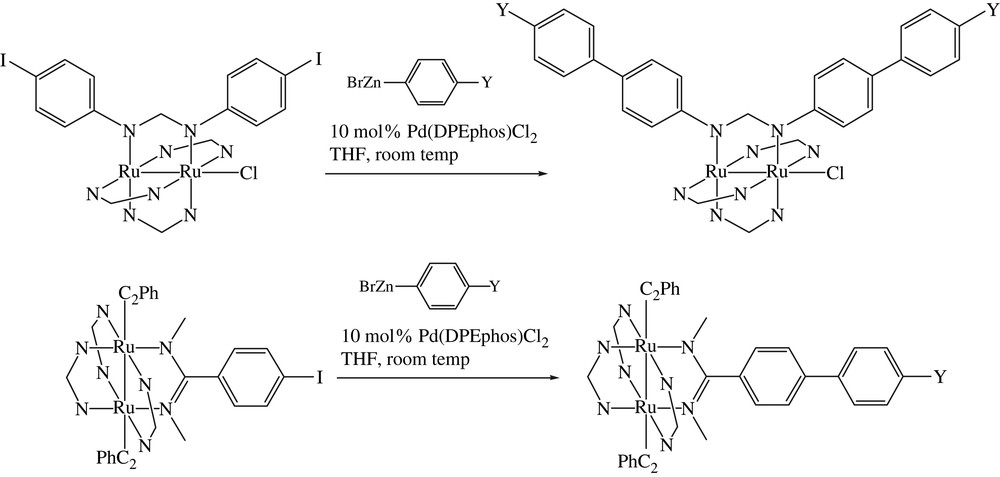
Negishi couplings of Ru2 substrates.
3.3 Heck coupling
The Heck reaction refers to the cross-coupling between an organic halide and a terminal olefin [24], and often requires prolonged reaction at elevated temperatures similar to the Suzuki reaction. Depending on substrates, organic Heck reactions vary vastly in reaction conditions such as Pd catalyst, the nature of the weak base and solvent. Similar dependence on conditions is encountered in Heck reactions with Ru2 substrates. With Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl as the substrate, the reaction was most efficient with Pd(OAc)2 as the catalyst and Et3N as both the base and solvent [18]. With Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl, the combination of Pd(OAc)2 and nBu4NOAc, the so called Jeffrey conditions [25], was found to be the most effective. Diruthenium species bearing axial phenylacetylide, i.e., Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)(σ-CCPh)2, was successfully cross coupled with various olefins in the presence of Pd(dba)2, KF, and nBu4NCl at room temperature.
4 Properties of cross-coupling products
The identities of several cross-coupling products have been confirmed with the X-ray structures shown in Figs. 3 and 4. In addition, the comparison of these structures with those of their parent compounds (Fig. 2) reveals that the coordination geometry around the Ru2-core is not altered upon peripheral modification. Careful comparisons, given in the original publications [11–13,15,16,18], further indicate that the key geometrical parameters such as Ru–Ru, Ru–N, and Ru–Lax (Lax is either Cl or CCY axial ligand) are nearly identical before and after peripheral modifications.
One of the objectives of peripheral chemistry is the retention of electrophore and chromophore characteristics upon the modification, which can be examined using simple UV–vis spectroscopic and voltammetric techniques. Shown in Fig. 5 is a collection of visible absorption spectra of Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl and its cross-coupling surrogates [15,16,18]. Although not identical, all spectra feature an intense peak around 490 nm, and a weak peak/shoulder around 620 nm except the Heck product. The spectral similarity indicates that the distribution of frontier orbitals remains relatively invariant before and after modifications.

Visible absorption spectra of Ru2(D(3,5-Cl2Ph)F)3(D(4-IPh)F)Cl and its derivatives via Suzuki (Y = H), Heck (Y = Ph) and Sonogashira (Y = H) couplings recorded in THF.
Cyclic voltammograms of both Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl and its Heck and Sonogashira derivatives are shown in Fig. 6. Each compound displays three Ru2-based one electron processes: a reversible oxidation (A), a quasi-reversible reduction (B), and a reversible reduction (C). The electrode potentials of the first oxidation and reduction correspond to the ionization potential (EHOMO) and electron affinity (ELUMO) of the solvated molecule, respectively. Hence, the perfect alignment of both the A and B peak positions in Fig. 6 reveals that the absolute HOMO/LUMO energies of the Ru2-species are not affected by peripheral modifications.
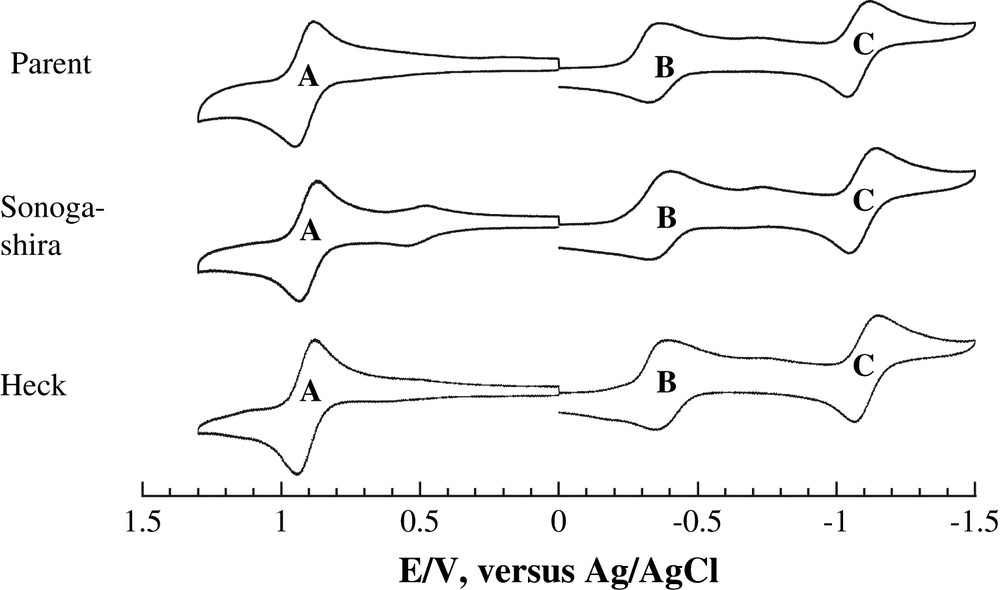
Cyclic voltammograms of Ru2(D(3,5-Cl2Ph)F)3(DMBA-I)Cl (parent) and its derives via Heck (Y = SiMe3) and Sonogashira (Y = SiMe3) couplings recorded in 0.20 M THF solution of Bu4NPF6 at a scan rate of 0.10 V/s with a glassy carbon working electrode.
5 Conclusions and outlook
Using diruthenium species as the working examples, research from our laboratory demonstrated programmatically that a variety of cross-coupling reactions can be performed at the periphery of inorganic and organometallic species. The merit of peripheral cross-couplings goes beyond novelty: it was noted on several occasions that free ligands do not undergo the same type of cross-coupling reactions at all. These reactions are also remarkable to ponder about should one take into account that the entire diruthenium species functions as a very bulky substrate of cross-coupling processes – being oxidatively added to the Pd center and then reductively eliminated from.
In general, peripheral modifications do not significantly alter molecular and electronic structures of the Ru2-core. Furthermore, certain functional groups introduced through cross-coupling reactions, such as olefins and alkynes, may undergo further covalent bond formation reactions such as olefin metathesis, “click”, and oxidative homocoupling reactions [15,26]. Currently, we are exploring the application of these reactions in hierarchical assemblies of Ru2-species.
Acknowledgement
Financial support from the National Science Foundation (CHE 0242623/0553564 and CHE 0715404) and Purdue University is gratefully acknowledged. This review was made possible through hardworking of several co-workers, especially Drs. W.-Z. Chen, G.-L. Xu, and L. Zhang. The author is also indebted to Ms. Antoinette S. Cordova for tireless proofreading.