

1 Introduction
Trifluoromethanesulfonic acid (HOTf, [1493-13-6]) and bis(trifluoromethanesulfonyl)imide (HNTf2, [82113-65-3]) belong to the class of Bronsted superacids, with negative Hammett acidity functions H0 [1]. Various metallic salts formed with their conjugated bases, triflate anion TfO− or triflimidate anion Tf2N− can therefore be considered as Lewis superacids, a terminology introduced to define the strong acidity of some metallic fluoride (SbF5, AsF5, NbF5, …) and popularised by George Olah [2].
We have been interested in providing simple and efficient methods for the preparation of metallic triflates and triflimidates. Initially, an electrochemical method was developed, involving the oxidation of a metal anode in an organic medium containing the corresponding Bronsted superacid, within a monocompartimented cell [3,4]. From this work, a first method was proposed for the synthesis of triflate and triflimidate salts from various metals, obtained as solvates with DMF or nitromethane molecules (1–8), depending on the solvent used, and on the metal cation. Recently, we have reported a straightforward method based on the direct oxidative dissolution of metal powders in DMSO in the presence of the desired superacid [5]. From this method, it was possible to get salts as solvates with DMSO molecules (4–10, depending on the nature of the salt).
Both of these methods present the advantage of providing the salts as solvates with organic molecules instead of the hydrates usually obtained by conventional methods, or commonly supplied when commercially available.
In the present paper, we present a detailed account of the oxidative dissolution method of preparation of metallic triflates and triflimidates published previously as a communication [5] and we compare the catalytic activity of various salts obtained from this oxidative dissolution method in the cycloisomerisation of a model substrate featuring a 1,6-diene moiety.
2 Results
By simply heating the metal powder in DMSO at 100 °C in the presence of stoichiometric amounts of triflic acid or triflimide, the desired metallic salts were obtained quantitatively (Scheme 1 and Table 1). Although an atmosphere of molecular oxygen (1 atm) was used, the reaction could be carried out under normal aerobic conditions simply by preventing the exposure of the reaction medium to air moisture.
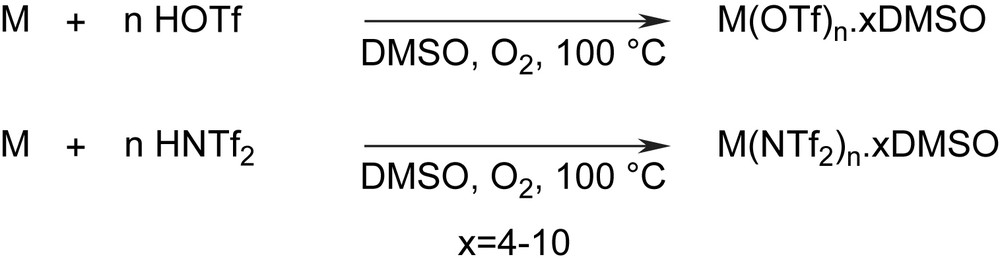
Results for the oxidative dissolution of various metals (taken from Ref. [5]).
| Entry | Metal | E° vs. SHE (V)a | HOTf (equiv.) | HNTf2 (equiv.) | Time (h) | Productb |
| 1 | Cu | 0.34 | 2 | – | 18 | Cu(OTf)2·5DMSO |
| 2 | Sn | −0.14 | 4 | – | 24 | Sn(OTf)4·6.1DMSO |
| 3 | Bi | −0.23 | 3 | – | 24 | Bi(OTf)3·7.8DMSO |
| 4 | In | −0.34 | 3 | – | 22 | In(OTf)3·7DMSO |
| 5 | Fe | −0.44 | 3 | – | 24 | Fe(OTf)3·6.2DMSO |
| 6 | W | −1.07 | 6 | – | 24 | – |
| 7 | Zr | −1.55 | 4 | – | 48 | – |
| 8 | Al | −1.66 | 3 | – | 24 | –c |
| 9 | Sm | −2.01 | 3 | – | 10 | Sm(OTf)3·9.7DMSO |
| 10 | Mg | −2.37 | 2 | – | 17 | Mg(OTf)2·3.9DMSO |
| 11 | Ca | −2.87 | 2 | – | 2 | Ca(OTf)2·6.4DMSO |
| 12 | Cu | 0.34 | – | 2 | 18 | Cu(NTf2)2·4.4DMSO |
| 13 | Sn | −0.14 | – | 4 | 7 | Sn(NTf2)4·7.9DMSO |
| 14 | Bi | −0.23 | – | 3 | 16 | Bi(NTf2)3·10.8DMSO |
| 15 | Al | −1.66 | – | 3 | 48 | –c |
| 16 | Mg | −2.37 | – | 2 | 18 | Mg(NTf2)2·5.0DMSO |
a Standard reduction potentials [7].
b Quantitative yields. Formula determined from 1H and 19F NMR, and elemental analysis.
c Unsuccessful, even with HCl activation of aluminium prior to use.
The reaction conditions were successfully applied to various metals such as copper, tin, bismuth, indium, iron, samarium, magnesium, and calcium, regardless of their standard reduction potential. Tungsten, zirconium and aluminium could not, however, be oxidatively dissolved under these conditions, although a method of preparation of aluminium triflimidate from aluminium dust in water has been reported in a recent patent application [6]. It is worth noting that those three metals have close reduction potentials (−1.07, −1.55, −1.66 V, respectively). From Table 1 and considering the reaction of metal powders with HOTf, one can notice three groups of metals; entries 1–5, those having reduction potentials above −0.44 V, entries 6–8 those having reduction potentials in the interval (−0.44, −1.66 V), and entries 9–11 those having reduction potentials below −1.66 V. We found unreactive under our conditions metals of entries 6–8, while the others were successfully dissolved to the corresponding triflate salts. One explanation on that partition could be found considering two different mechanisms for the formation of metallic triflate. When the metal has itself a reductive character, it could react directly with HOTf to form molecular hydrogen and triflate anion, in a rather fast one-electron process. Such mechanism could be considered for metals of entries 9–11. If the metal has high reduction potential, it gets involved in a rather slow redox reaction with molecular oxygen. It is the case for Cu, Bi, Fe, which are known to form a redox couple with O2 in various processes. Such mechanism could be considered for entries 1–5.
We have used stoichiometric amounts of HOTf or HNTf2 based on the most common highest oxidation state for the considered metals (Cu(II), Sn(IV), Fe(III)) except for bismuth (Bi(III)). Several attempts to form CuOTf or Fe(OTf)2 by using one or two equivalents of HOTf, respectively, were unsuccessful, and a dark brown mixture was obtained in each case leading to the isolation of uncharacterised insoluble material.
For successful reactions, the slow disappearance of the metal powder was observed, except with samarium, which reacted vigorously. The solvent was then evaporated using a bulb-to-bulb oven under reduced pressure. The mixture was not heated beyond the temperature of 100 °C, in order to avoid the putative decomposition of the salt. The solid was collected and washed with cold diethyl ether or dichloromethane. After 1H and 19F NMR analysis in [d6]-acetone, each salt was analysed by elemental analysis for carbon, fluorine, sulphur, hydrogen, and the metal. The salt formulae are proposed on the basis of these analytical data.
We have previously shown that the shielding observed by 1H NMR on the chemical shift of the solvating DMSO molecules was linearly correlated with the Pauling electronegativity of the metal centre considered [5], as an evidence of the tight interaction between the metal cation and DMSO ligands. The influence of the metal centre was also observed on the triflate counter anion. Indeed, the chemical shift of the CF3 group in 19F NMR analysis of each sample of metal triflate M(OTf)n·xDMSO revealed again a correlation with the Pauling's electronegativity of the metal (Fig. 1).
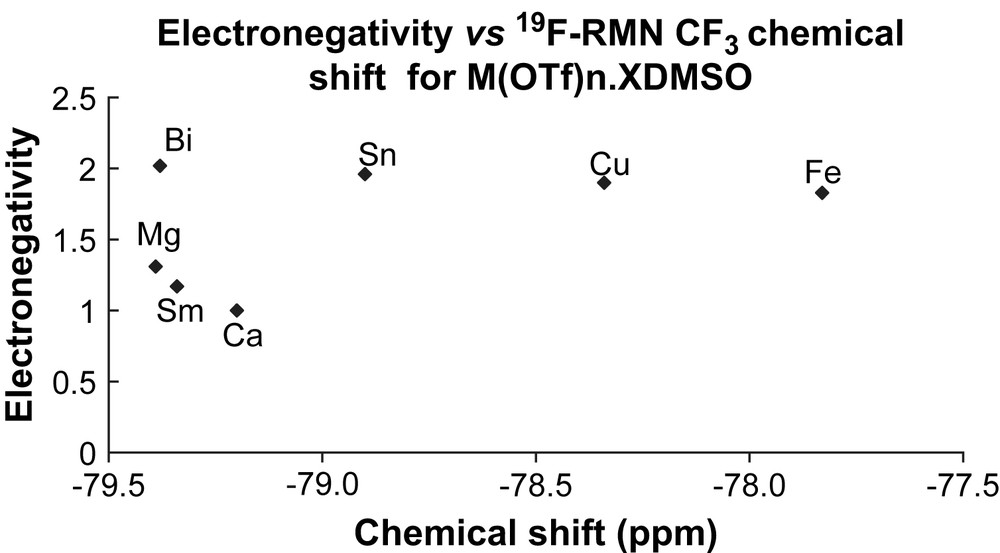
Surprisingly, while salts of bismuth (III), tin (IV), copper (II), and iron (III), displayed chemical shift linearly correlated to their electronegativity, magnesium (II), samarium (III), and calcium (II) were way out of linearity, but presented a rather high shielding, of the order of magnitude of that of bismuth and tin salts. It is worth noting that anhydrous Sn(IV) metal salts have been used as catalysts in many applications in organic synthesis by our group both as tin (IV) triflate and tin (IV) triflimidate [8–10].
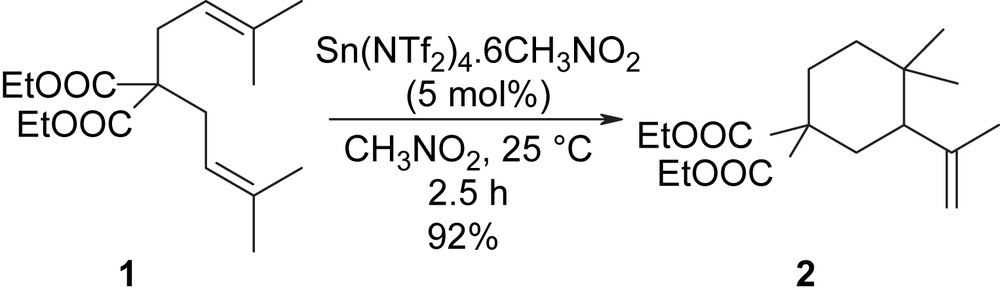
Recently, our interest was focused on the catalytic cycloisomerisation of 1,6-dienes, which is a modern topic of research in chemistry with both fundamental and practical significance [11–13]. Although transition metal-catalysed reactions usually lead to cyclopentene derivatives, our group has recently reported a novel and efficient cycloisomerisation reaction of non-activated 1,6-dienes leading selectively to 6-membered rings [10,14]. In this previous work, the cycloisomerisation of a model substrate, diethyl bis(prenyl)malonate 1, was efficiently carried out with various metallic salts such as chlorides, triflates and triflimidates, among which Sn(NTf2)4·6CH3NO2, prepared by the electrochemical method, was clearly superior (Scheme 2).
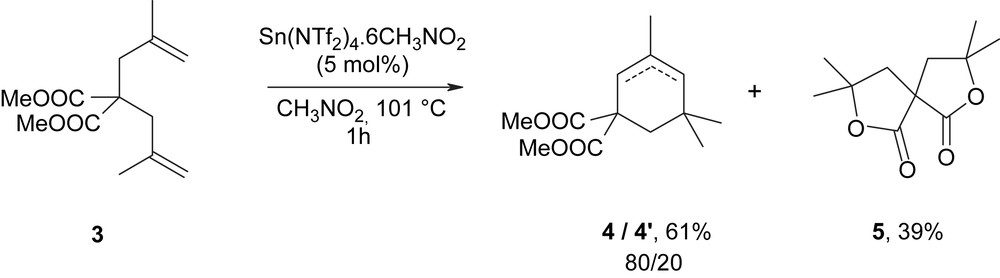
With the same catalyst for dimethyl bis(methallyl)malonate 3 however, heating in refluxing nitromethane was necessary to reach complete conversion in 1 h and the formation of 4 + 4′ in 61% yield, with bis-γ-lactone 5 as side-product (Fig. 2).
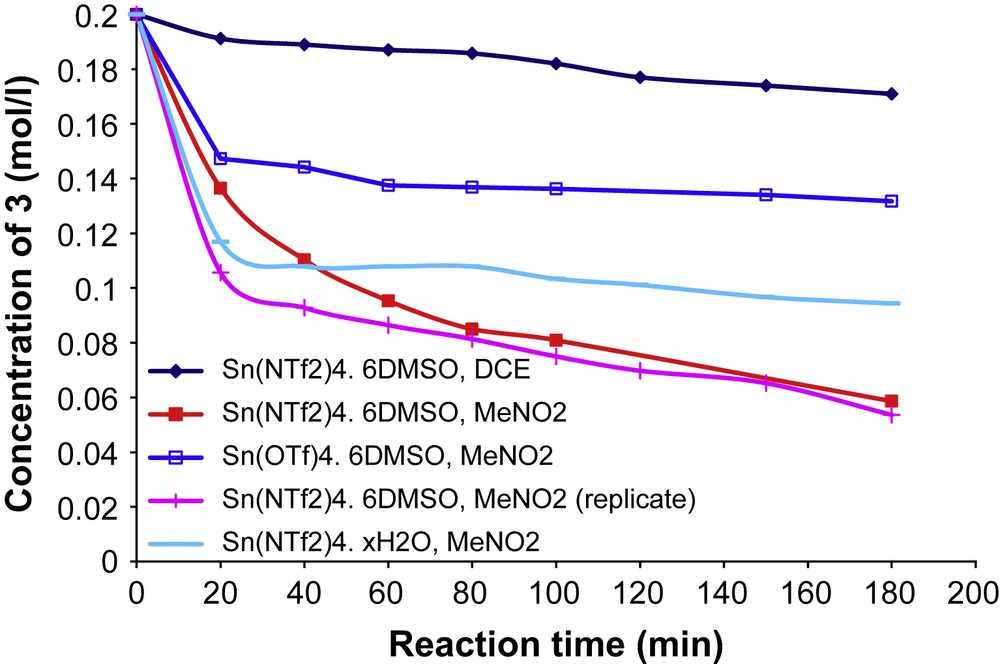
Kinetic plots of the reaction of model substrate 3 under various reaction conditions (in refluxing solvents).
In the continuation of this work, we have been interested in the cycloisomerisation of 3 to the cyclised isomers 4 and 4′, and to use this less reactive substrate as a probe to compare the intrinsic activity and the selectivity of the different catalysts prepared by oxidative dissolution; Sn(OTf)4·6DMSO; Sn(NTf2)4·6DMSO, and the hydrated form, Sn(NTf2)4·xH2O. Good yields of 2 were indeed obtained with substrate 1 and these catalysts under mild conditions, either in dichloroethane or nitromethane as the solvents.
In this regard, various grades of Sn(NTf2)4 and Sn(OTf)4 were prepared according to our procedures [5], leading to Sn(NTf2)4·6DMSO, and Sn(OTf)4·6DMSO. A sample of Sn(NTf2)4·6DMSO was dissolved in acetone/water (9:1) and agitated 3 h to yield after evaporation of the solvent Sn(NTf2)4·xH2O.
In a first attempt, we used Sn(NTf2)4·6DMSO in refluxing dichloroethane for the cycloisomerisation of 3. The reaction was carried in the presence of hexadecane as internal standard for GC-FID quantification. After 3 h, the conversion was only of 18%, leading mainly to degradation product since no compounds 4, 4′, or 5 could be identified. This result was rather unexpected, compared to what could be obtained with Sn(NTf2)4·6CH3NO2 under similar conditions. Various reaction conditions were thus evaluated for the reaction of 3, varying in a first set of experiments only the nature of the catalyst (Sn(NTf2)4·6DMSO, Sn(NTf2)4·xH2O, Sn(OTf)4·6DMSO), and the solvent ((CH2Cl)2 and CH3NO2). Kinetic plots of the conversion of substrate 3 for each system are presented in (Fig. 2).
This set of experiments shows that nitromethane is better suited than dichloroethane as solvent, maybe simply for the reason of having a higher boiling point (101 vs. 81 °C, respectively) (Fig. 2). Comparing the activity of Sn(NTf2)4 with that of Sn(OTf)4 in refluxing nitromethane, it appeared that the metallic triflimidate is superior to the triflate both in terms of reaction rate and conversion. The superiority of metallic triflimidures over metallic triflates has been demonstrated in several examples of Lewis acid-catalysed reactions [10,15–19]. Additionally, the use of hydrated tin triflimidate Sn(NTf2)4·xH2O resulted in an overall moderate conversion, below 50%.
In terms of products, the reactions led to the cyclised products 4 and 4′ in 20–40% selectivities, the main products being the spiro bis-lactone 5 formed with 60–80% selectivities, sometimes with small amounts of monolactone intermediate. With tin (IV) triflimidate under its hydrated form, the undesired bis-γ-lactone 5 was formed with 80% selectivity. These results are to be compared with previous results obtained with Sn(NTf2)4·6CH3NO2, prepared via the electrochemical method, which yielded the cyclised products 4 and 4′ in 61% yield within 1 h, with only 39% of lactone 5 (Scheme 3).
We can conclude that, for the reaction examined, anhydrous tin (IV) triflimidate coordinated only to DMSO molecules is a more active catalyst than the corresponding tin (IV) triflate or than the hydrated forms of tin (IV) triflimidate. Indeed, DMSO solvates, compared to hydrates, are probably more prone to accommodate substrates of low Lewis base character (such as dienes), DMSO being a weak ligand.