

1 Introduction
Odorant Binding Proteins (OBPs) belong to the family of lipocalins [1]. They are small carrier proteins, thought to participate in the first step of the perception of smell [2,3]. OBPs are considered as non-specific binders and are thought to participate in the olfaction process by carrying hydrophobic odorant molecules to the olfactory receptors to trigger the olfaction process [4,5]. Although various types of OBP can be found in some species (such as the rat, for example [6]) only one OBP is present in the human mucus [7].
At the molecular level, OBPs share the common lipocalins structural motifs: a β-barrel structure, made up of eight strands (noted A–H), linked together by seven Loops (L1–L7) and connected to an α-helix. The β-barrel encloses an internal binding site, dedicated to small hydrophobic ligands such as odorants. The ligand is located within the hydrophobic cavity of the protein and the large majority of the binding energy is driven by van der Waals interactions [8,9].
Many experimental data are now known concerning these systems, particularly on rat, porcine, and human OBPs [7,10–12]. In a general manner, such hydrophobic systems are devoid of any strong specificity, with binding free energies in the range −8 to −10 kcal/mol. These proteins do indeed not show a particular specificity for a given odorant, except in rat [10] or human [7]. In human OBP, a single lysine residue has been shown to drive a larger affinity for aldehydes [7]. The dynamic analysis of this interaction should shed light on this intriguing fact. The presence of a single type of OBP would have rather suggested that it is totally non-selective towards any odorant family. Atomic-level analysis has already been shown to help in describing some important structural features, then nailing down crucial residues for ligand recognition [9,12–15].
In this article, we have examined the dynamic behavior of the free human OBP, called hOBPIIa and its complex with both citral and undecanal, as depicted in Scheme 1. These ligands are particularly sensitive to the role of the lysine 127 residue located within the binding cavity, as shown in Ref. [7]. The role of this lysine residue (potentially responsible for the selectivity towards aldehydes) is more particularly examined.
Chemical structures of undecanal and citral.
2 Methods
The human OBPIIa has no available X-ray structure. The structure, made up of 170 residues has been built based on homology sequences, considering the experimental structures of selected lipocalins: major horse Equ c1 (pdb id: 1EW3) [16], bovine lipocalin allergen Bos D2 (pdb id: 1BJ7) [17], mouse MUP (pdb id: 1MUP) [18], porcine OBP (pdb id: 1DZK) [19] and hamster aphrodisin (pdb id: 1E5P) [20] with Modeller, forcing the typical OBP cysteine bridge (74–166 in the sequence) to be conserved. Energies were minimized by 1000 steps of conjugate-gradient with the AMBER 9 [21] package and models were assessed with PROCHECK. More detailed data and algorithm references are provided as Supplementary data.
The ligands' parameters were obtained from the Generalized Amber Force Field (GAFF). The charges were computed with the “antechamber” module of AMBER 9 using the “bcc” charges on structures optimized at the B3LYP/6-31G∗ level of theory using Gaussian 03 [22].
The flexible ligands were docked in the binding cavity with Autodock, where a systematic contact between the ligands' aldehyde group and Lysine127 was found.
2.1 Molecular dynamics
The molecular dynamics simulations were performed using the AMBER 9 program at 310 K. Simulations were carried out with an implicit Generalized-Born (GB) solvation scheme, using the PMEMD module and SHAKE algorithm on bonds involving hydrogen atoms. A time step of 2 fs was applied. Complexes were minimized by 1000 steps of steepest descent, followed by 2000 steps of conjugate-gradient protocols. Then, 1000 steps of minimisation and 10 ps of Molecular Dynamics (MD) simulation using a restraint of 20 kcal/mol/Å2 on the solute atoms were performed, followed by four rounds of 1000 steps minimisation reducing the restraints by 5 kcal/mol/Å2 at each round, with 10 ps MD simulation. Further, the systems were slowly heated to 310 K over a period of 20 ps. Equilibration runs were continued over 100 ps before production simulations of 2 ns. Notice that the use of such a GB calculation allows a much more efficient relaxation of the protein structure due to the neglect of the molecular nature of the solvent [23]. A factor of 100 has even been suggested concerning the increase of the sampling speed.
2.2 MM–GBSA
The MM–GBSA approach is an end-point method, where only the initial and the final state energies are evaluated [24]. This method is based on analysis of configurations from equilibrated MD simulations, treating the solvent as a dielectric continuum. In this approach, if we consider a receptor/ligand complex, the binding free energy can be computed according to the following equation, where 〈X〉 corresponds to an average of a given value X over snapshots taken from the complex MD trajectory.
3 Results
Throughout the simulations, the three systems keep the same fold, with the β-barrel enclosing the hydrophobic binding cavity. The protein fold is also identical with or without the presence of a ligand within the binding site. A typical snapshot of a complex (here with citral) is shown in Fig. 1. The structural analysis is quantified by the calculation of the Cα root mean square deviation with respect to the starting structure. It never exceeds 4 Å throughout the simulations, revealing that the homology-built structures are kept in a conformational attractor. This rather large rmsd is more particularly due to the long and unstructured N and C-terminal tails of the protein. If this part of the protein is not considered for the rmsd calculations (considering only residues 21–155), the latters lower to values systematically below 3 Å.

Typical snapshot of the human OBPIIa bounded to citral. The general structural features of the free OBP and of the complex with undecanal are highly similar.
The residues lining the binding cavity are in concordance with those found while building the structures on the basis of human tear lipocalin [7]. The build structure is very similar to all other known mammals' Odorant Binding Proteins. The residues considered to act as the protein door [13] (Tyr93 and Pro46) are located at the same positions to those found for example in pig OBP [11], rat OBP-1F (T. Borysik, personal communication) or MUP [18]. The cavity is mainly made up of hydrophobic residues, although some hydrophilic ones can also be found, such as Lysines (Lys 97 and Lys 127, numbered 82 and 112, respectively in [7]). Notice also that on the basis of sequence alignment, rat OBP2 is also supposed to share a lysine residue at the same position and could behave similarly considering aldehyde binding [7].
The flexibility of each residue is quantified by the evaluation of its root mean square fluctuation throughout the molecular dynamics. The three rmsf are shown in Fig. 2. One can see a typical rmsf plot, where more flexible parts are found on residues belonging to the loops between the β-barrel secondary structures. These loops have a poorly defined shape and can appear more or less structured depending on the initial structure. The residues directly in interaction with the ligands show a weaker mobility in the bound systems. It is notably the case for Lys 127 that makes a regular hydrogen bond interaction with both citral and undecanal. Other residues belonging to the binding cavity (residues 80–100 and 127–140) also show a weaker mobility since they also undergo some contacts with the ligands, although they are of hydrophobic nature.
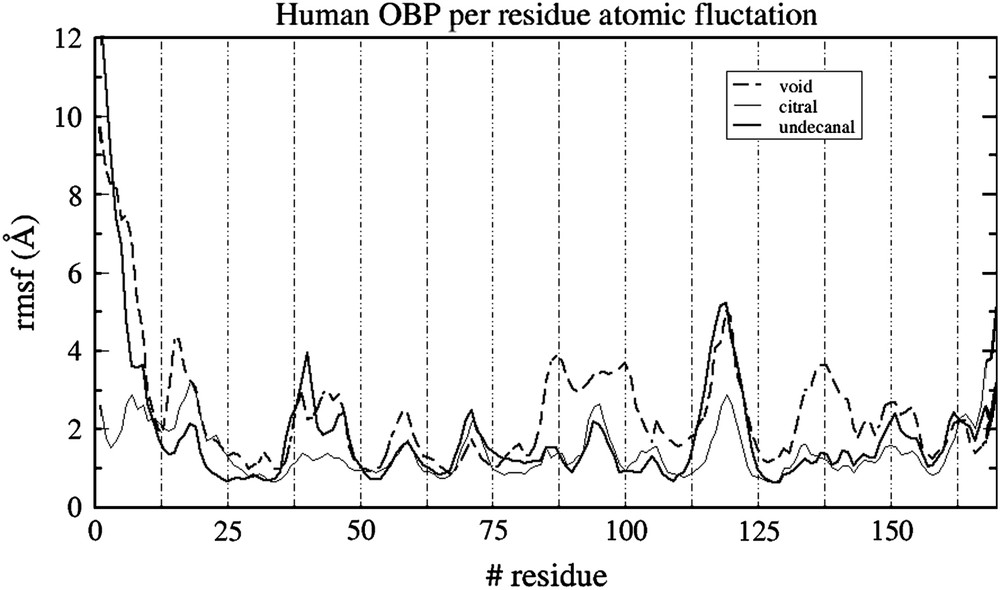
Residue atomic fluctuation for the three systems throughout the simulations.
In the complexes, there is a regular interaction between the ligands and lysine 127 throughout the simulations. Figs. 3 and 4 report the hydrogen bond distance between both citral and undecanal and this lysine 127, a residue belonging to the OBP cavity and considered to be responsible for the large affinity for aldehydes [7].
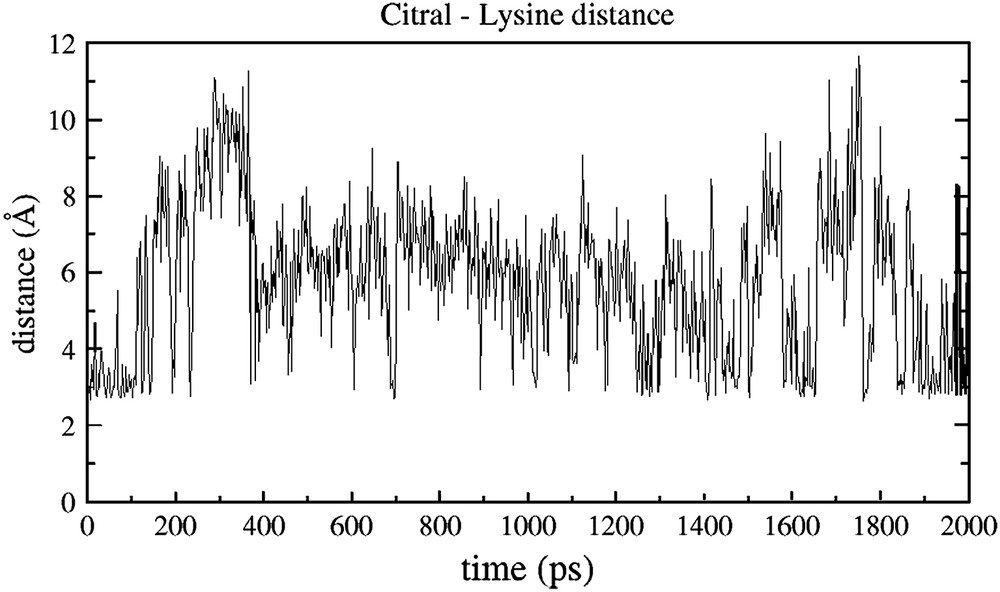
Time evolution of the distance between the aldehyde oxygen atom of citral and the nitrogen atom of the lysine 127 of human OBPIIa.

Time evolution of the distance between the aldehyde oxygen atom of undecanal and the nitrogen atom of the lysine 127 of human OBPIIa.
From Fig. 3, it appears clearly that Lys 127 undergoes a strong interaction with citral. The hydrogen bond is regularly sampled all along the simulation, with an average value of 5.8 Å during these 2 ns. On the contrary, the interaction of citral with other residues of the binding cavity is never as strong as that found with Lys 127. It is notably the case for Lys 97, for which the distance with the aldehyde function never gets closer than 9 Å (data not shown).
Concerning undecanal, the distance measurements are in relative accordance with a higher experimental affinity with hOBPIIa (1.1 kcal/mol with respect to citral, on the basis of measured kdiss) [7]. The interaction with Lys 127 also goes back and forth throughout the simulation, but can sample a close interaction with a longer residence time (between 1.3 ns and 1.7 ns, for example). The resulting average value is similar to that of citral (6.05 Å during the 2 ns). From a structural point of view, this Lys 127 seems to play an important role for both ligand recognitions.
The results are in good accordance with previous experimental results. Undecanal has the strongest affinity for hOBPIIa and the mutation of the lysine 127 residue originates the strongest difference in the affinity amongst all the tested ligands with respect to the wild-type protein [7]. This clearly demonstrates that the affinity of undecanal is more strongly dependent on the lysine 127 residue than that of citral. These distance measurements provide the atomic-level explanation of this experimental fact: undecanal is more regularly bound to this lysine.
A good way to get an idea of the validity of a model is to compare computed values with experiment. Although little is known concerning the atomic-level structural feature of the system, experiments have been performed by means of fluorescence binding assays, to estimate the relative binding free energies between ligands [7]. For the purpose of a comparison with experiment, we have performed an MM–GBSA analysis on both the hOBP/citral and hOBP/undecanal systems. This so-called end-point approach allows one to estimate the binding energetics of the considered systems. Notice, however, that this is mainly used as a semi-quantitative way to compute free energy difference between structurally related systems, as already performed by us on OBP–odorants complexes [15]. In this case, the entropic correction has not been computed since one can consider that it will be roughly identical for the citral and the undecanal complexes.
The binding free energy is found to be −19.0 (with a standard deviation of 2.5 kcal/mol) and −21.9 kcal/mol (with a standard deviation of 2.6 kcal/mol) for citral and undecanal, respectively. A detail of the computed data is provided in Supplementary data. The preference of 2.9 kcal/mol predicted for undecanal is in good accordance with fluorescence experiments, since these differential binding measurements also reported a preference for undecanal by a free energy difference of 1.1 kcal/mol [7]. Such a good predictive calculation allows us to consider our computed model with a higher confidence.
4 Discussion
Human OBPIIa, like all other Odorant Binding Proteins in mammals is thought to be responsible for the binding and transport of odorant from the nasal mucus to the Olfactory Receptors located at the neuron membrane surface. This protein shares the global structure of typical lipocalins, with a β-barrel calyx occluded from water and dedicated to bind hydrophobic ligands. A lysine residue within this binding cavity appears to interact directly with odorants belonging to the family of aldehydes through a hydrogen bond with the ligand aldehyde group. The structural deviation upon odorant binding is deemed rather minor, with small structural difference between the bonded and the free protein. The MM–GBSA approach allowed us to compute the difference of binding free energy between the citral and the undecanal complexes on the basis of our model and recovered the experimental higher affinity for undecanal. The role of this lysine residue is clearly emphasized and a few interesting comments can be put forward.
The hydrogen bond interaction occurs between the aldehyde function of the odorant and the ɛ-amino group of the lysine amino-acid. Although OBPs are not considered to make chemical bonding with odorants, such an interaction between a lysine and an aldehyde is nonetheless typical of the early step of the formation of a Schiff base, as shown in Scheme 2.

Typical equilibrated reaction between a Lysine residue and an aldehyde functional group, leading to a Schiff base and H3O+.
Although this Schiff base formation cannot be observed in our simulation (it would have needed the use of a quantum chemical potential), the strength of the interaction is connected to the first step of the reaction. Indeed, the reaction requires that the reactants are both in a good position and orientation prior to the reaction to occuring. The previous hydrogen bond analysis suggests that both ligands are regularly in contact with the lysine side-chain and are thus correctly positioned for the reaction to proceed.
OBPs are, however, only considered as odorants' carriers and are not supposed to undergo chemical reaction with odorants during the early steps of the perception of smell [5]. This chemical reaction is however equilibrated and is likely to proceed backward when the system is largely hydrated. In the absence of any other protagonist than the odorant and the OBP, the odorant is within the cavity and is thus totally occluded from water. The system is likely to be found as a Schiff base. However, to activate the Olfactory Receptor at the neuron membrane surface, the odorant has to be recovered in its initial form (the aldehyde). With the opening of the binding pocket, as the protein reaches near the Olfactory Receptor, the filling of the cavity with water molecules would recover the aldehyde function. The odorant would then be properly delivered for triggering the receptor activation.
The solvent would then play not only a solvation role but would also act as a real chemical protagonist, inducing the formation of the Schiff base when it is absent (within the hydrophobic pocket) or inducing the recovery of the aldehyde when present (during the opening of the cavity near the neuron membrane).
A deeper examination of the Schiff Base formation between the ligands and Lys 127 would require the use of quantum chemical approaches, eventually through hybrid Quantum Mechanical–Molecular Mechanics calculations taking into account for the protein environment and the water phase explicitly. Human OBP and eventually rat OBP2 (since a lysine seems to be aligned with Lys 127 in the sequence) could be unique transport proteins, able to originate a chemical bond with odorants.
Acknowledgments
Sylvain Antoniotti is acknowledged for the discussion about the Schiff Base formation. The CINES (Centre informatique national de l'enseignement supérieur) in Montpellier and the CMIM (Centre d'imagerie et de modélisation moléculaire) of the University of Nice Sophia Antipolis have provided the computer time for this study.