

1 Introduction
The camptothecins constitute one of the most important classes of antitumor agents in use and development today [1]. The parent camptothecin 1 was isolated from the leaves of the Camptotheca acuminata tree by Wani and Wall (Fig. 1), but development was stopped because of problems with solubility and toxicity. Nonetheless, camptothecin retains more than historical importance because it is commercially available at reasonable prices. Second-generation camptothecins were extensively produced by both semi-synthesis and total synthesis during the late 1980s and early 1990s. This work culminated in the introduction of two commercial drugs, topotecan 2 (trade name, Hycamtin) and irinotecan 3a (trade name, Camptosar). Irinotecan, also commonly known as CPT-11, is a pro-drug whose active component is the 10-hydroxy analog, SN-38 3b. These drugs have shown both promise and problems in the clinic, and that combination has spurred an ongoing search for third-generation derivatives. A substantial pipeline of such derivatives now exists [1].
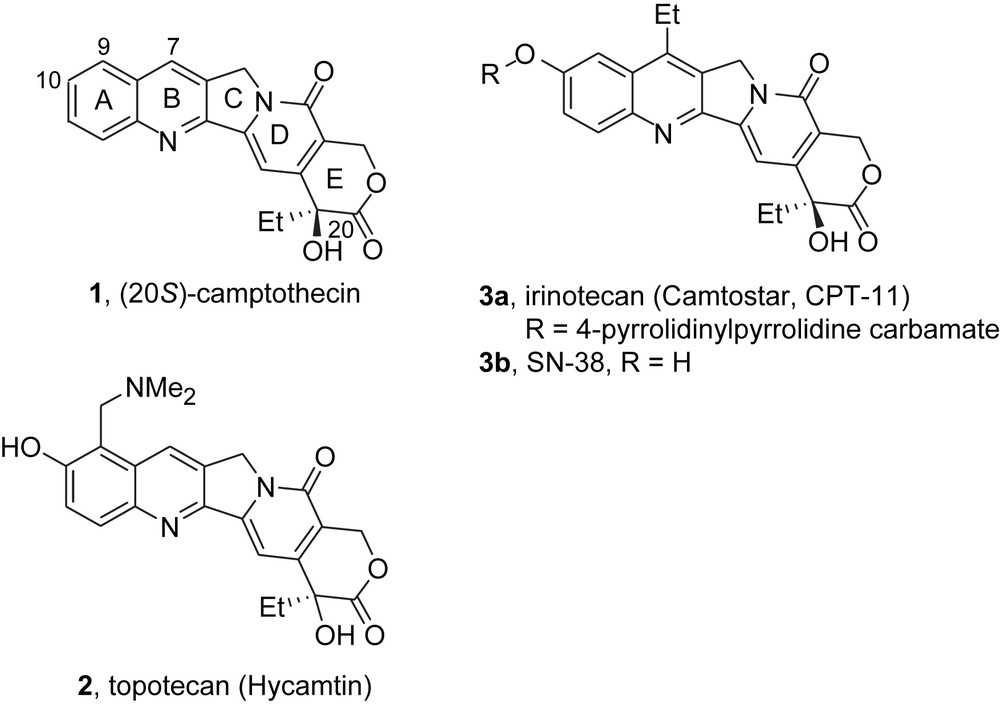
Structures of camptothecin, topotecan, irinotecan (CPT-11) and SN-38.
The molecular mechanism of action of the camptothecin class of anti-cancer agents is now relatively well understood [2]. The compounds are classed as “topoisomerase 1 inhibitors” or “topoisomerase 1 poisons”. The latter name might be better since the camptothecins do not competitively inhibit topoisomerase 1, a key enzyme that helps to unwind DNA during cell replication. Instead, they bind to a covalent intermediate of the enzyme topoisomerase 1 and DNA. The resulting termolecular complex is stable, DNA replication is blocked, and cell death eventually ensues.
The pharmacodynamics of the camptothecins are fundamentally different from most other drugs. Generally, drugs are single small molecules with associated dynamic features such as bond rotations, ring inversions, etc. These single molecules are then gradually metabolized to other molecules. Camptothecin, in contrast, is a chemically dynamic drug. Under physiological conditions, it exists in relatively rapid chemical equilibrium with its open chain hydroxy acid salt 4 [3] (Fig. 2). This hydroxy acid is not thought to be an active anti-cancer agent. The relative concentration of the two forms depends on many features, some of which (pH, lipophilicity, presence of serum albumin, among others) are now understood. Thus, unlike most drugs, which are gradually metabolized after administration, camptothecins are subject to chemical equilibration both before and during metabolism. This added level of complexity provides opportunities and poses problems at the same time.

The lactone/hydroxy acid equilibrium.
Because of the potential problems posed by the dynamic nature of the camptothecins, it would be of interest to have camptothecin analogs that maintain biological activity but that lack the signature equilibrium. For a long time, the search for biologically active E-ring analogs of camptothecin did not bear fruit, and some even speculated that equilibrium and activity were linked. However, in recent years, a few E-ring analogs of camptothecins have shown activity either in cell assays or in vitro in topoisomerase assays (Fig. 3). These include the unusual ether 5 (which shows rather low activity) [4], and the cyclopentanone 6 [5] and the natural product luotonin 7 [6], which show significantly higher levels of activity.
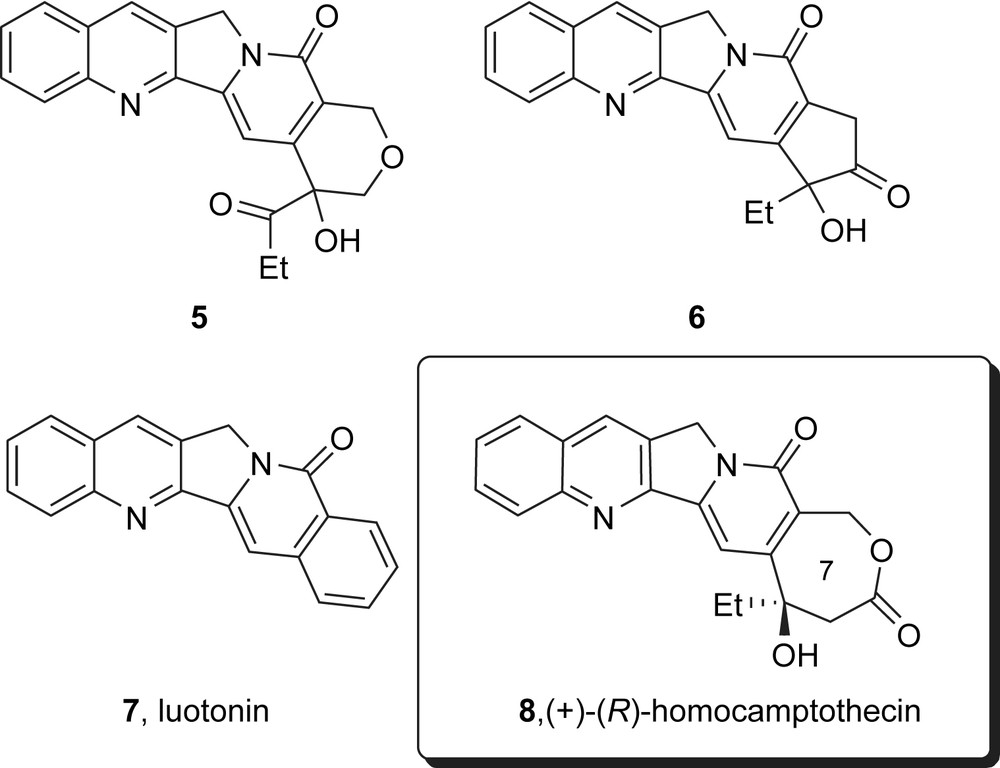
Selected E-ring Analogs of camptothecin with biological activity, including homocamptothecin.
However, the most important class of E-ring analogs to emerge to date has been the homocamptothecins. This short review focuses on the synthesis of homocamptothecins while at the same time including key biological and medicinal chemistry information to illustrate why the class is unique and important. The structure of (R)-homocamptothecin 8 is shown in Fig. 3.1
2 Discovery of homocamptothecin
Homocamptothecin 8 was first described by a multidisciplinary team led by Lavergne and Bigg of the Institut Henri Beaufour [7]. To make the initial sample of the racemate (Scheme 1), the lactone ring of (S)-camptothecin 1 was first disassembled by reduction to the hydroxylactol and oxidative cleavage to give achiral ketoformate 9 [8]. This effectively excised one carbon (C21) from the ring. Next, a Reformatsky reaction on the liberated ketone added back two carbons to give ester 10 in 31% yield. Finally, acid treatment removed the formate and closed the seven-membered ring lactone of racemic hydroxycamptothecin 8 in 55% yield. Despite the low yield of the Reformatsky reaction, the sequence is short, so substantial quantities of (rac)-8 were produced.

Semi-synthesis of racemic homocamptothecin from camptothecin.
Racemic homocamptothecin showed remarkable properties [9]. Despite being a β-hydroxy lactone rather than an α-hydroxy lactone, it exhibited potent antiproliferative activity in assorted cell lines. It also cleaved DNA in the presence of toposiomerase 1 in the signature assay for “camptothecin-like” molecules, although with a different sequence selectivity from camptothecin. However, unlike camptothecin, it was quite stable in plasma and other fluids, opening slowly but irreversibly to the derived β-hydroxy carboxylate 11.
Both enantiomers of homocamptothecin were then produced by chemical resolution of the derived hydroxy acid, and the activity was shown to reside in the dextrorotatory (+)- enantiomer. This suggested that (+)-homocamptothecin had the R configuration, a suggestion that was later confirmed by asymmetric total synthesis (see below). Because of a change in Cahn–Ingold–Prelog (CIP) priorities of the attached groups, (S)-camptothecin is analogous to (R)-homocamptothecin.
These preliminary biological and pharmacological studies spurred significant interest in the synthesis of homocamptothecin and its analogs.
3 The “Lavergne–Comins route” to homocamptothecins
The group from Institut Henri Beaufour prepared one analog of homocamptothecin (the 7-ethyl derivative) by the semi-synthetic route from the analogously substituted camptothecin. But recognizing the limitations of the semi-synthesis for structure–activity studies, they also put in place a total synthesis of racemic homocamptothecin and several interesting analogs in the first paper [10]. This was based on the powerful Comins route to camptothecin, whose key penultimate and ultimate steps are summarized in Scheme 2. N-Alkylation unites AB fragment 12 with DE fragment 13 to give 14. Subsequent Heck reaction closes the C ring to give camptothecin 1.

Key steps in the Comins route to camptothecin and adaptation to homocamptothecins.
In the adaptation to the analog 10,11-difluorohomocamptothecin 18 (diflomotecan), linking of the two fragments 15 and 16 by a Mitsunobu reaction gave 17 in 35% yield. Then Heck cyclization of 17 provided racemic 18 in 18% yield. Later, the resolution of the hydroxy acid derived by hydrolysis of 16 was described, and this then provided access to enantioenriched intermediates for the preparation of the active R enantiomers [11]. Yields for the key coupling and cyclization steps were also improved.
The synthesis of the requisite DE fragment for making homocamptothecin is summarized in Scheme 3. Conversion of 19 to a ketal followed by displacement of the chloride by methoxide provided 20 in 80% yield. Ortholithiation and in situ formylation followed by reduction provided alcohol 21 (62%). Protection of the alcohol followed by removal of the acetal provided a ketone 22 (70%) ready for the same sequence of reactions that was used in the semi-synthesis. Reformatsky reaction of 22 gave β-hydroxyester 23 in 95% yield. In the end game, hydrogenolytic removal of the benzyl group, acid-promoted lactonization and demethylate provided the target lactone pyridone 16 in 40% overall yield.
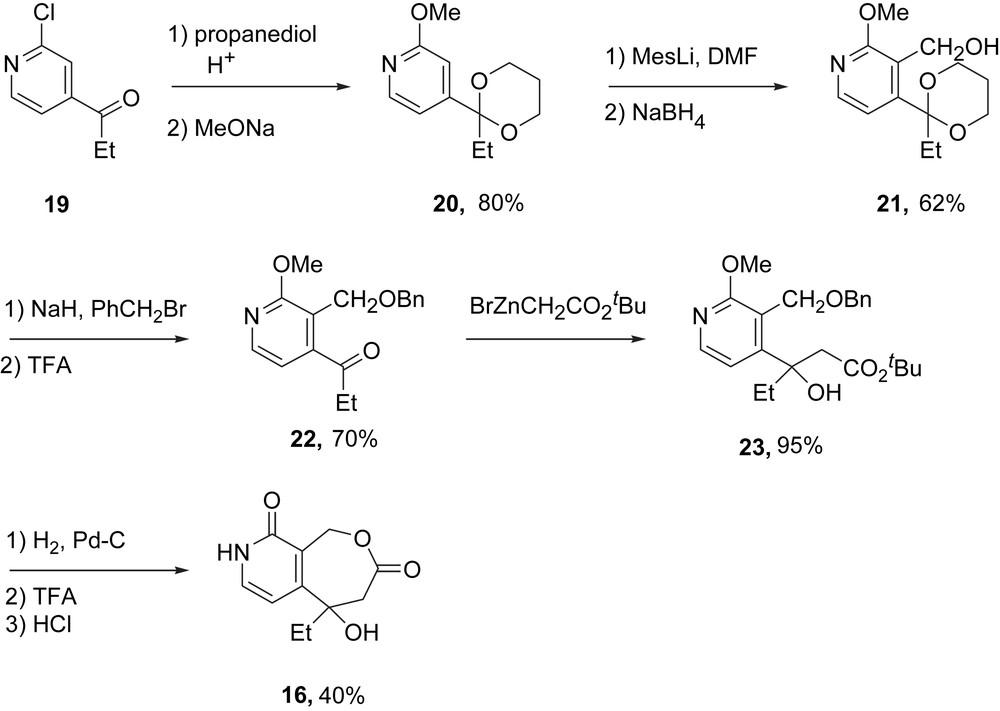
Initial synthesis of the racemic DE fragment 16.
Assorted derivatives of homocamptothecin with one or two substituents on the A ring were also made by this route. The precursors are made starting from appropriately substituted anilides by Vilsmeier-type double formylation and reduction of the resulting alcohol. Especially important among these analogs are fluorinated derivatives, including BN80915 18 (10,11-difluorohomocamptothecin), which is a clinical candidate for cancer chemotherapy called diflomotecan [12]. The synthesis of the requisite AB fragment 15 for BN80915 from 3,4-difluoroacetanilide 24 is shown in Scheme 4.
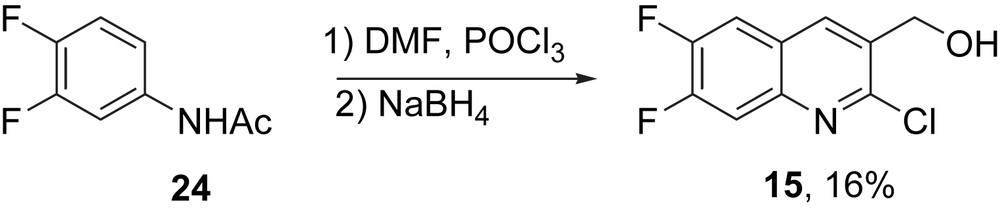
Synthesis of the AB fragment illustrated with 15.
Practical asymmetric routes to BN80915 based on asymmetric aldol reactions have recently been reported, as summarized in Scheme 5 [13]. Streamlined routes to benzyl- and TBS-protected ketones 22 and 25 were first developed. Then these intermediates were subjected to asymmetric aldol reactions to give target R-isomers 26 and 27 in ratios of about 87/13. These aldol products were readily converted to lactone (R)-16 in comparable enantiomeric ratios. The moderately selective aldol reactions were rendered very practical by the property of (R)-16 to crystallize in very high ee (>99.9%) from the enriched mixtures.
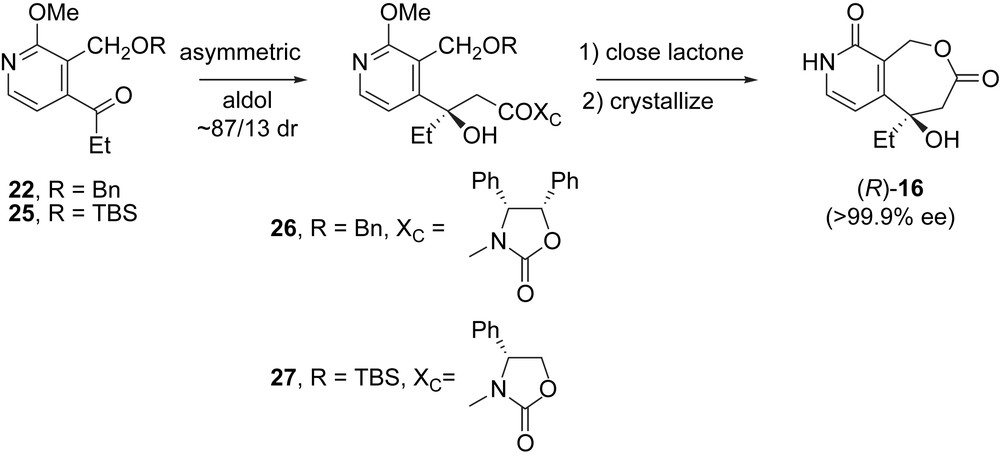
Asymmetric aldol routes to the DE fragment (R)-16.
Even more recently, a completely different “de novo pyridone” route to enantiopure (R)-16 has been put into place [14]. The R-stereocenter at C20 in this route ultimately derives from lactic acid through a self-reproduction of chirality. The authors concluded that this approach could serve as the basis for a technical synthesis of diflomotecan.
Though useful for introducing A-ring substituents, the Lavergne–Comins route is not readily amenable to direct introduction of B-ring substituents a C7. Such substituents have proved very important in the medicinal chemistry of the classical α-hydroxylactones of the camptothecin class.
4 The cascade radical annulation approach to homocamptothecins
During the 1990s, we developed a route to substituted camptothecins that was notable for its brevity and generality [15]. In the penultimate step, lactone pyridone 28 is N-propargylated to give 29. Finally, cascade radical annulation of 29 and an A-ring isonitrile 30 unites the two building blocks and directly forms rings B and C to give the analogous camptothecin 31 (Fig. 4). Dozens of known and new analogs of camptothecin, including the clinical candidate AR-67 [16] (formerly DB-67), were prepared by this route. AR-67 is a member of a large family of 7-silylcamptothecins that go by the name of silatecans [17].

The cascade radical annulation route to camptothecins.
Although the cascade reaction is usually run in a radical mode with a tin (or sometimes silicon) reagent, a palladium-mediated variant is also available that starts from the same precursors and gives the same products [18]. The tin- and silicon-mediated reactions involve radicals. Presumably, organopalladium intermediates are involved in the palladium variant, whose mechanism has not been studied in detail.
Within a few months of the report of homocamptothecin activity, we were able to retool the cascade annulation route to make racemic homosilaecan analogs (Scheme 6) [19]. Starting from readily available enol ether 32, dihydroxylation and ozonolysis provided the requisite ketoformate 33. Standard Reformatsky reaction and ring closure, followed by iododesilylation and demethylation, provided the key lactone pyridone 34, again in racemic form. Now, N-propargylation provided the finished DE fragments 35a,b, ready for the radical annulation.
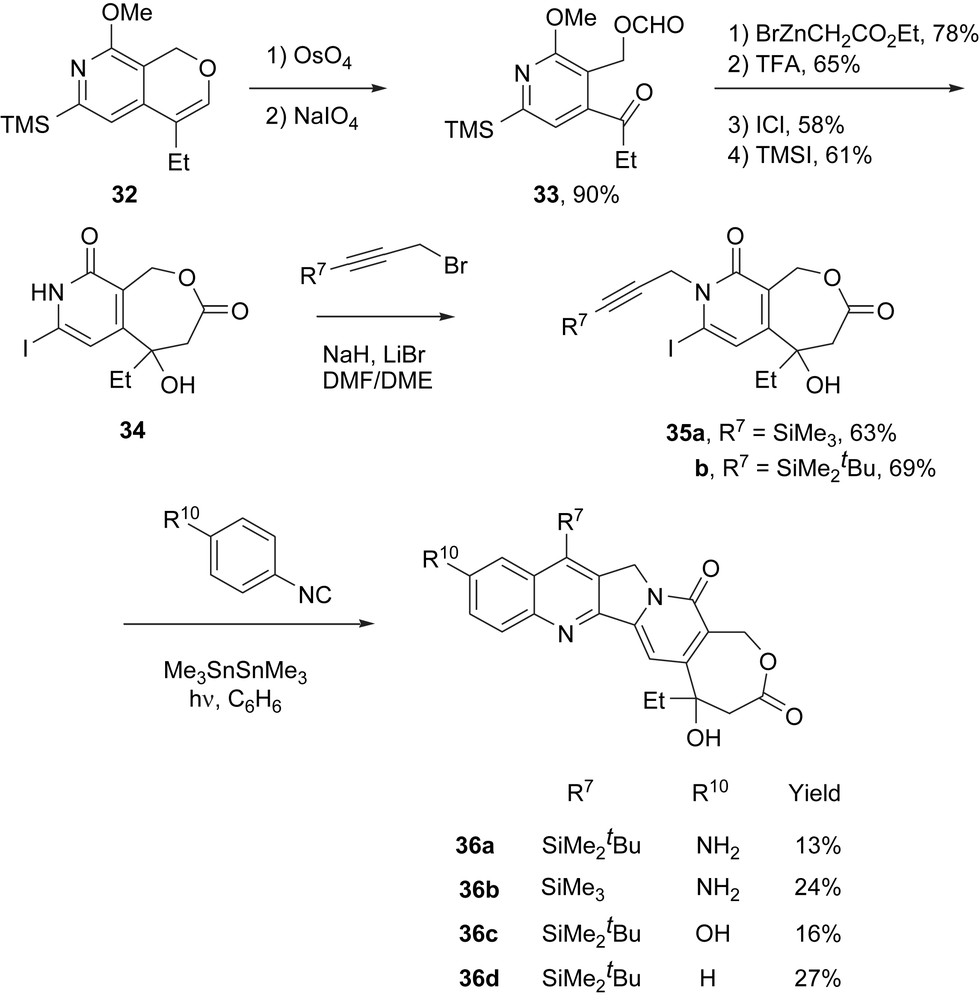
Racemic homosilatecans made by co-opting camptothecin intermediate 32.
In a preliminary illustration of the usefulness of this approach for making analogs, we prepared four 10-substituted-7-silylhomosilatecans 36a–d from the appropriate propargyl bromide and isonitrile. These homosilatecans are triply ring-modified (A, D and E) camptothecins that are highly biologically active and are considerably more stable in blood than homocamptothecin (which in turn is already very stable compared to standard camptothecins) [20]. That they can be made so easily is a testament to the power and generality of the cascade radical annulation approach.
After further shortening of the synthesis of 34, the route in Scheme 6 was parallelized and 115 analogs of homosilatecans were made in individual, pure form on 1–5 mg scale [21]. Simple manual techniques for parallel reactions were coupled with automated purifications (SPE, HPLC) to give high-purity final products. The speed and simplicity of the automated purification protocol more than compensated for yield losses in the synthesis of some analogs relative to traditional flash chromatographic purifications. The ready adaptability of the cascade radical annulation route to parallel synthesis is yet another testament of its generality. We failed to isolate only one of the designed library products. The entire library was made by a single student over several months.
Recognizing the need for an asymmetric synthesis of the DE fragment of homocamptothecins for both the Lavergne–Comins and cascade radical annulation approaches, we put in place the sequence in Scheme 7 based on a classical Sharpless asymmetric epoxidation (SAE). Stille coupling of ready available iodide 37a (R = TMS) and 38 followed by ester reduction provided 39 as a single alkene stereoisomer. Following the SAE reaction, epoxide 40 was isolated in 79% yield and 93% ee. A standard three-step sequence of reactions converted this intermediate into lactone 41, which in turn was converted to 42 by acid-promoted lactonization and iododesilylation. Finally, demethylation provided (R)-34 in >90% ee. This synthesis again borrows the starting material 32 and the last several steps from the original camptothecin synthesis. An analogous route starting from the 37b (now lacking the silyl group, R = H) provided intermediate (R)-16 for the Lavergne–Comins route in high ee.
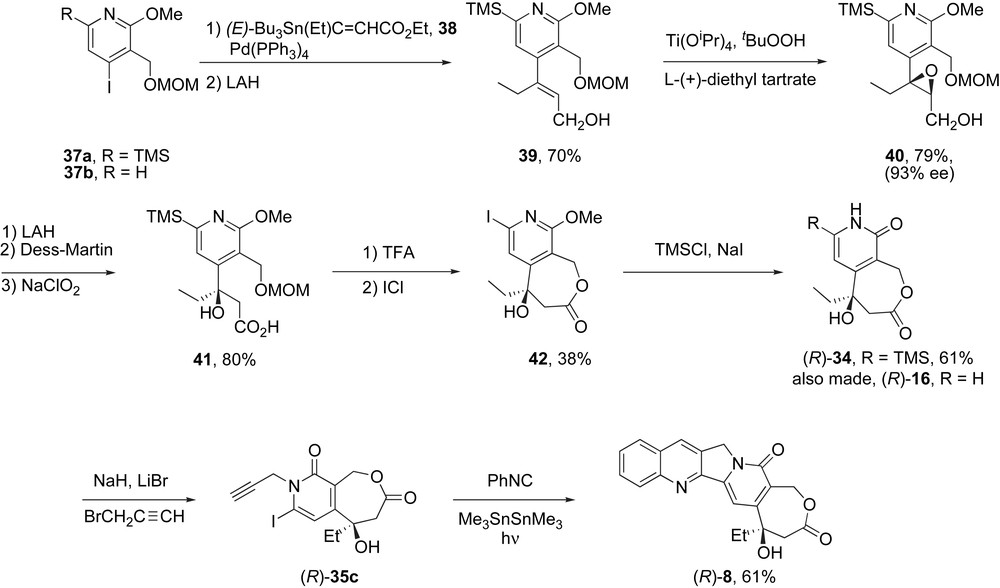
Asymmetric synthesis of both cascade radical (34) and Heck (16) DE fragments.
The conversion of the defined intermediate (R)-34 to (R)-35c and then onward to (+)-homocamptothecin (R)-8 provided a chemical confirmation of the absolute configuration of homocamptothecin that validated the earlier biological assignment.
Finally, in a further demonstration of the versatility of this approach, we synthesized a series of unsaturated analogs of homocamptothecins [22]. The synthesis of the parent, dehydrohomocamptothecin 44 from its DE precursor 43 is typical, as summarized in Scheme 8. These compounds had significantly reduced biological activity compared to the analogous homocamptothecins.
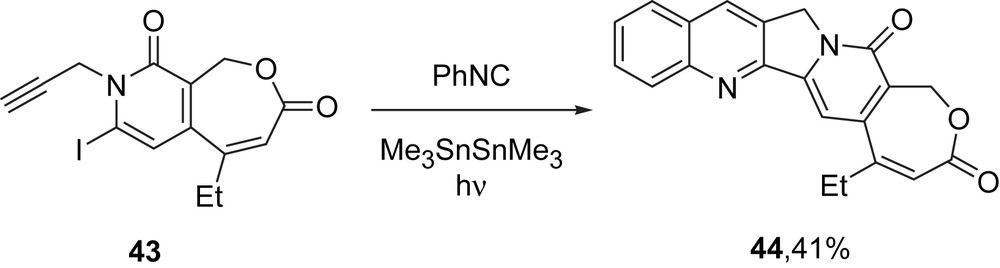
Synthesis of dehydrohomocamptothecin.
5 The Friedlander route to homocamptothecins
The Lavergne–Comins and cascade radical annulation routes to camptothecins are generally considered second-generation syntheses because they were developed during the revival of camptothecins as anti-cancer agents. From among the first-generation syntheses [23], the Friedlander approach has commonly been used for preparing analogs for medicinal chemistry purposes. Summarized in Scheme 9, this involves the Friedlander condensation of tricyclic keto pyridone 46 with analino aldehyde 45 (or an equivalent) to provide camptothecin 1.
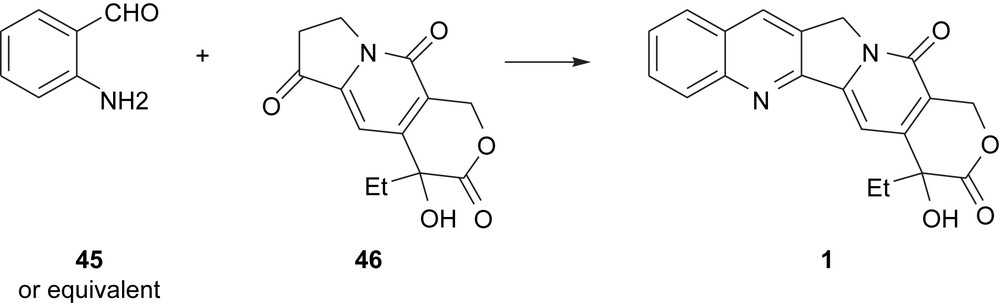
The Friedlander approach to camptothecins.
Recently, Miao and coworkers adapted this route to make homocamptothecins (Scheme 10). Ketal 47, the immediate precursor of 46, was ring expanded by a series of reactions similar to those above to provide tricyclic ketone 48, now with the seven-membered lactone, in excellent overall yield. Friedlander condensation of this lactone with 49 provided homocamptothecin 8. The route was generalized by producing a number of A- and B-ring analogs starting from suitably substituted anilino aldehydes or ketones. To date, only racemic homocamptothecins have been produced by this route.
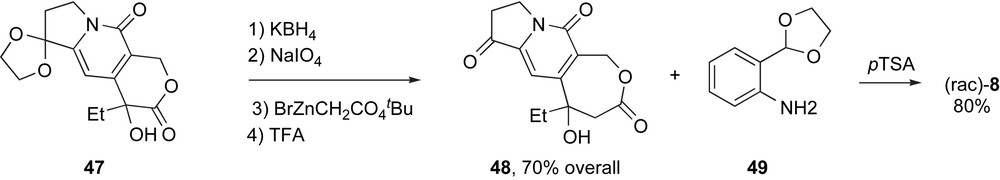
The Friedlander approach to homocamptothecins.
6 Conclusions
Homocamptothecin and its analogs are proving to be an especially interesting class of camptothecins that merits an independent classification because the lactones of these analogs open slowly and irreversibly, rather than rapidly and reversibly. The discovery of this class of molecules provided unexpected new opportunities to leverage lessons learned during the synthesis of the camptothecins.
Three powerful and general routes—the Lavergne–Comins route, that cascade radical annulation route, and the Friedlander route—have been put into place to make homocamptothecins. While homocamptothecin was initially made in racemic form from camptothecin, substituted homocamptothecins are increasingly remote from the parent, especially considering that the sole stereocenter of camptothecin is destroyed in the semi-synthesis. Thus, total synthesis efforts have dominated the medicinal chemistry development of the homosilatecans. Likewise, both the preclinical and clinical developments of diflomotecan have been driven by increasingly efficient variants of the Lavergne–Comins route.
Acknowledgements
I thank David Bom, Ana Gabarda, Thomas ISARNO, Wu Du and Raghuram Tangirala for all their experimental and intellectual contributions on this project, and I am grateful to the National Institutes of Health, National Institute of General Medical Sciences, for funding.
1 Camptothecin numbering is used for the homocamptothecins in this review to facilitate comparison.