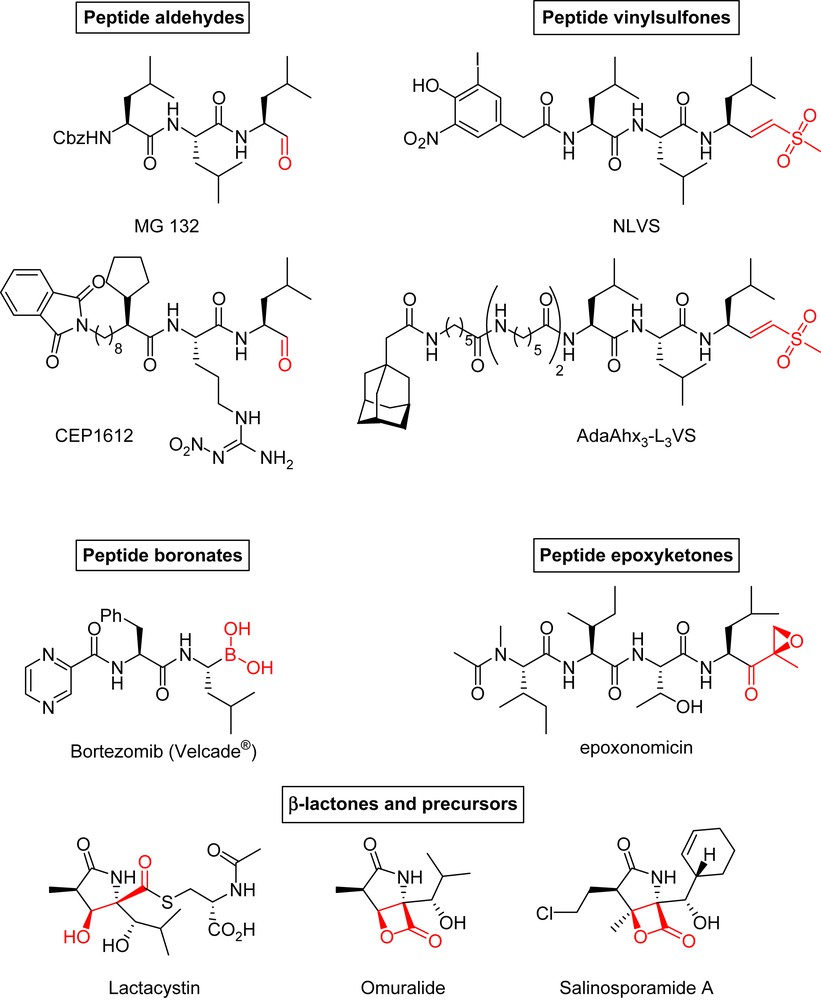
1 Introduction: proteasome inhibition
1.1 Proteasome: a brief overview
Thirty years ago, mammalian cells were shown to contain a highly unusual system for the selective degradation of proteins, now known as the ubiquitin–proteasome pathway [1]. In this pathway, which is the major machinery for protein degradation, proteins are marked by covalent linkage to the protein cofactor ubiquitin [2]. Proteins with a chain of ubiquitin molecules are then rapidly degraded by a very large ATP-dependent proteolytic particle, the 26S proteasome, itself composed of the 20S central core, where proteins are digested, and a 19S regulatory complex (Fig. 1).
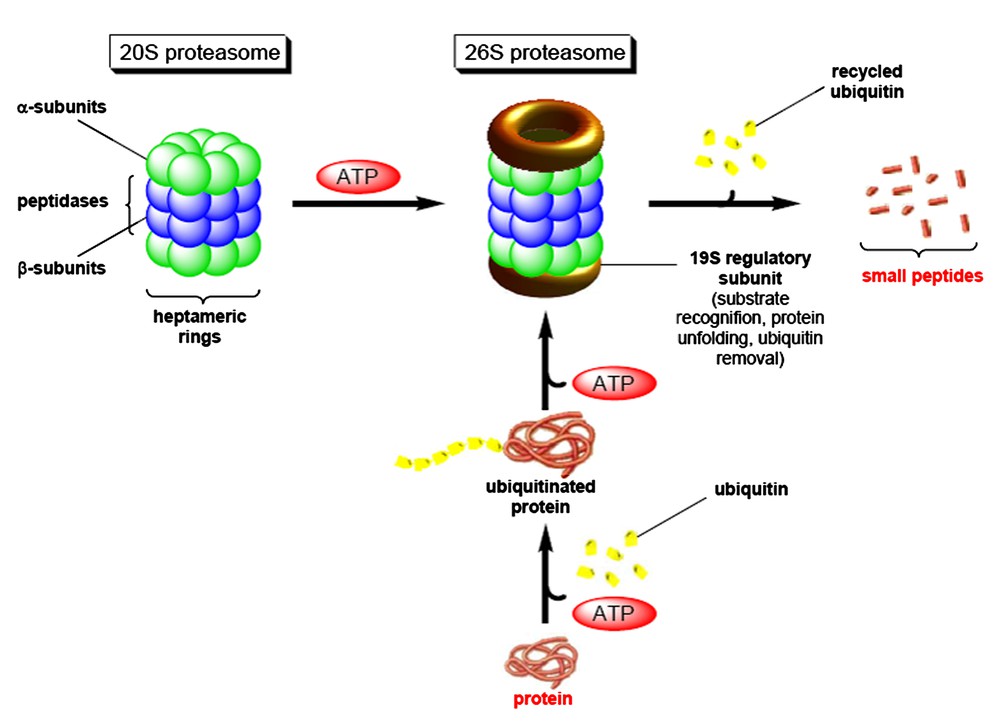
The ubiquitin–proteasome pathway for protein degradation.
The proteolytic sites in the 20S proteasome function by a mechanism distinct from that of other proteases, the initial attack on the peptide bonds being by the hydroxyl group on an N-terminal threonine of the β-subunits which yields to the formation of an acyl–enzyme intermediate. Further hydrolysis releases the carboxylic acid and the free proteasome (Fig. 2).
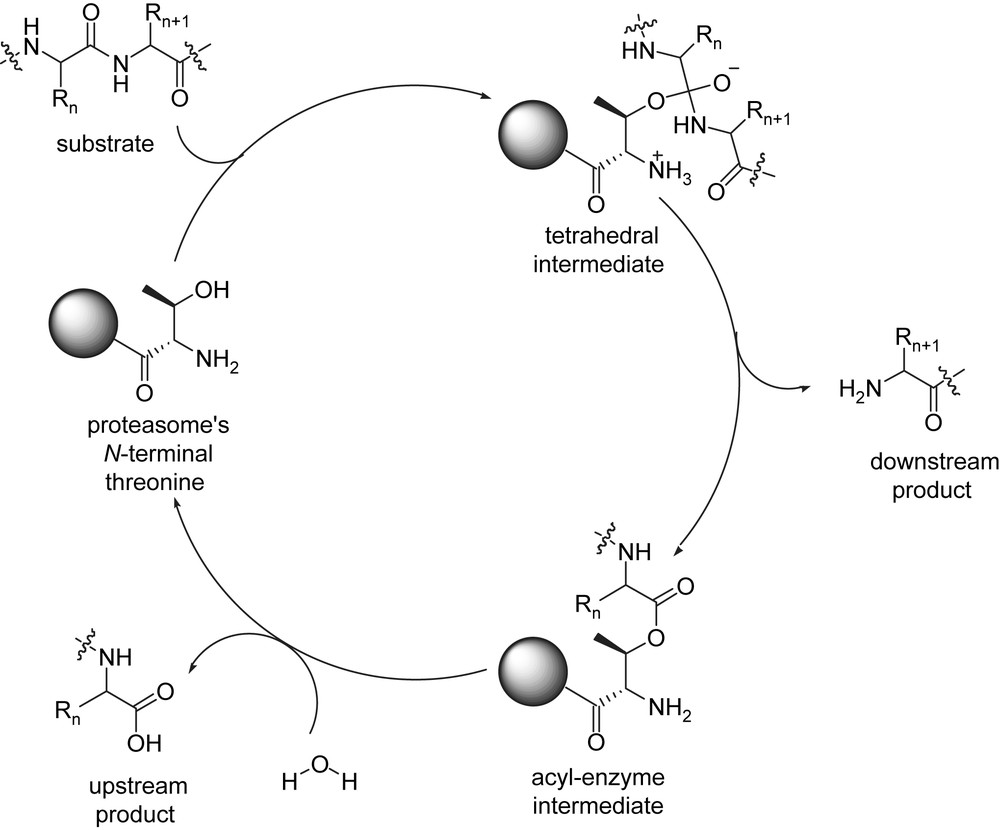
Proteasome catalytic mechanism.
Three proteolytic activities (chymotrypsin-like, trypsin-like and post-glutamyl peptide hydrolyzing) have been characterized in the catalytic domain of the 20S proteasome and, respectively, attributed to active sites located on the β5, β2 and β1 subunits [3].
Recently, great advances have been made in our understanding of the fundamental importance of the ubiquitin–proteasome pathway in diverse biological processes and the proteasome has been shown to play a crucial role in major cellular processes such as T-cell activation, cell-cycle control, cell adhesion and migration, apoptosis or antigen presentation [4].
1.2 Proteasome inhibition in the tumor-targeting approach
Realization of the proteasome's importance in various aspects of cell biology as well as increased crystallographic knowledge concerning the proteasome itself [5] has prompted increased efforts in the field of proteasome inhibition since selective inhibitors offer an amazing therapeutic potential [4,6]. Some of them have now been shown to have impressive potential for treatment of inflammation, autoimmune diseases, and strokes, and they now clearly appear as excellent targets for cancer chemotherapy [7].
Cancer is characterized either by uncontrolled cellular proliferation or by a failure of cells to undergo apoptosis [8]. As the proteasome is an important regulator of both these processes, therapeutic regimes to manipulate proteasomal activity could potentially restore normal cellular homeostasis in cancer patients. Drug resistance and lack of tumor specificity frequently hinder the treatment of neoplastic disease, creating a need for new classes of potent and specific anticancer drugs.
In preclinical cancer models, proteasome inhibitors have been shown to induce potently apoptosis in many types of cancer cells, including tumor cells resistant to conventional chemotherapeutic agents, with reduced cytotoxicity in normal cells. They have also been shown to have in vivo antitumor efficiency, and sensitize malignant cells and tumors to the proapoptotic effects of conventional chemotherapeutics and radiation therapy. Interestingly, transformed cells display greater susceptibility to proteasome inhibition than non-malignant cells. Therefore, proteasome inhibition holds promise as a novel approach to the treatment of cancer, as demonstrated with Bortezomib (Velcade®) (Fig. 3),1 the first such inhibitor to be approved for therapy. All classes of proteasome inhibitors (peptide aldehydes, peptide vinylsulfones, peptide boronates, peptide epoxyketones or β-lactones, Fig. 3) covalently (and most of the time irreversibly) bind to the catalytic threonine and inhibit the proteasome in a more or less selective manner.
To increase the potency and therapeutic utility of proteasome inhibitors, one must improve their specificity. As TMC-95A–D block the active sites of the proteasome non-covalently by a strong array of hydrogen bonds, their structure clearly constitutes a new lead for the design of specific proteasome inhibitors with increased activity in the tumor-targeting approach as demonstrated with the synthesis of analogues with tailored biological activities.
Their especially potent biological profile together with their appealing complex structure immediately generated considerable interest within the synthetic community. After an overview of the different synthetic routes reported to these macrocyclic natural products, the design and synthesis of various analogues will be detailed. First, the structure of these compounds will be briefly described in the following paragraphs.
2 TMC-95A–D: isolation, structure, binding mode and biological activities
TMC-95A (1a) and its diastereoisomers TMC-95B–D (1b–d, Fig. 4), recently isolated as fermentation products of Apiospora montagnei Sacc. TC 1093 (isolated from a soil sample) [9], represent a new class of selective proteasome inhibitors. Their unique structures have been elucidated on the basis of FAB–HRMS, extensive 1D and 2D NMR (DQF–COSY, HMBC, ROESY) analyses, chiral TLC and HPLC as well as degradation analyses.

Structure of TMC-95A–D.
Among their defining structural characteristics are (i) the cyclic polypeptide array containing l-tyrosine, l-asparagine, and highly oxidized l-tryptophan moieties, (ii) a (Z)-1-propenylamide substructure, and (iii) a 3-methyl-2-oxopentanoic acid substructure in the form of an amidic linkage to the tyrosine-like unit of the cyclic peptide [9].
Biological studies showed that TMC-95A inhibited the chymotrypsin-like (CT-L), trypsin-like (TL), and post-glutamyl peptide hydrolytic (PGPH) activities of the proteasome with IC50 values of 5.4, 200, and 60 nM, respectively [10]. TMC-95B inhibited these activities to the same extent as TMC-95A, while TMC-95C and D were 20–150 times weaker (Table 1, ALLN, a known proteasome inhibitor, is given as a reference compound). TMC-95A also showed cytotoxic activities against human cancer cells HCT-116 and HL-60 with IC50 values of 4.4 and 9.8 μM, respectively. Moreover, it is fundamental to note that TMC-95A is specific to the proteasome since it does not inhibit other proteases such as calpain, cathepsin and trypsin.
Inhibitory activities of TMC-95A–D against CT-L, TL and PGPH activities of 20S proteasome (human proteasome)
| Compound | In the presence or absence of 0.02% SDS | IC50 (μM) | ||
| CT-L | TL | PGPH | ||
| TMC-95A | +SDS | 0.0054 | 0.20 | 0.060 |
| −SDS | 0.012 | 1.5 | 6.7 | |
| TMC-95B | +SDS | 0.0087 | 0.49 | 0.060 |
| TMC-95C | +SDS | 0.36 | 14 | 8.7 |
| TMC-95D | +SDS | 0.27 | 9.3 | 3.3 |
| ALLN | +SDS | 6.6 | 6.0 | 21 |
Finally, this reversible proteasome inhibitor was found to possess neuritogenic activity in PC12 rat pheochromocytoma cells since it induces a positive neurite initiation of PC12 cells at concentrations ranging from 1 to 12 mM [11].
The binding mode of these inhibitors to the proteasome has been elucidated by Groll and co-workers in 2001 by X-ray crystallography [12] and provides strong basis for the development of synthetic selective proteasome inhibitors. Unlike other synthetic or natural proteasome inhibitors, TMC-95A does not modify the N-terminal catalytic threonine residue. The backbone of TMC-95A adopts a β-conformation and extends the short β-strand S1 by the generation of an antiparallel β-sheet structure. Additionally, TMC-95A displays a host of hydrogen bonds with the protein giving further stabilization of the compound in its bound status, as shown in an exemplary way for β2 (Fig. 5).
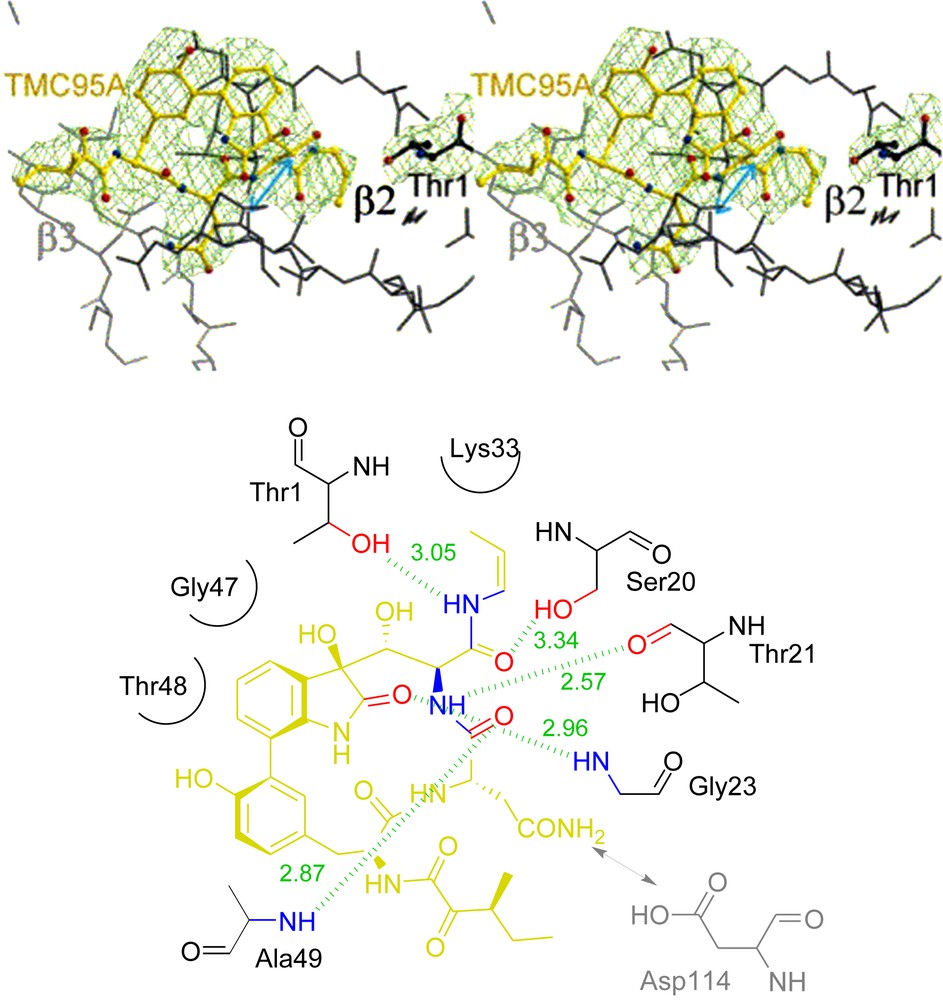
Crystal structure of TMC-95A bound to the proteasomal subunit β2 and schematic overview.
3 Total syntheses of TMC-95A and B
The great interest for proteasome inhibition, the considerable biological activity, and the remarkable structures of the TMC-95 class of natural products provided the motivation to contemplate a total syntheses of these compounds that would be readily adaptable to the synthesis of biologically active analogues. Indeed, immediately after the publication of the structures of these novel cyclic peptide natural products, significant synthetic activity in this field commenced, resulting in total syntheses being reported by the groups of Lin and Danishefsky [13], Hirama et al. [14] and Albrecht and Williams [15].
Examination of these syntheses clearly reveals that the synthetic challenges that must be overcome in designing a synthetic route are (i) formation of the highly oxidized tryptophan core where lies most of the chemical complexity, (ii) installation of the biaryl, (iii) macrocyclization and (iv) installation of the enamide and oxopentanoic side chains. Various strategies have been used to this end and their key steps are highlighted in Fig. 6. Before detailing these syntheses, the key disconnections used for the preparation of the target molecules will be described and the different approaches compared.
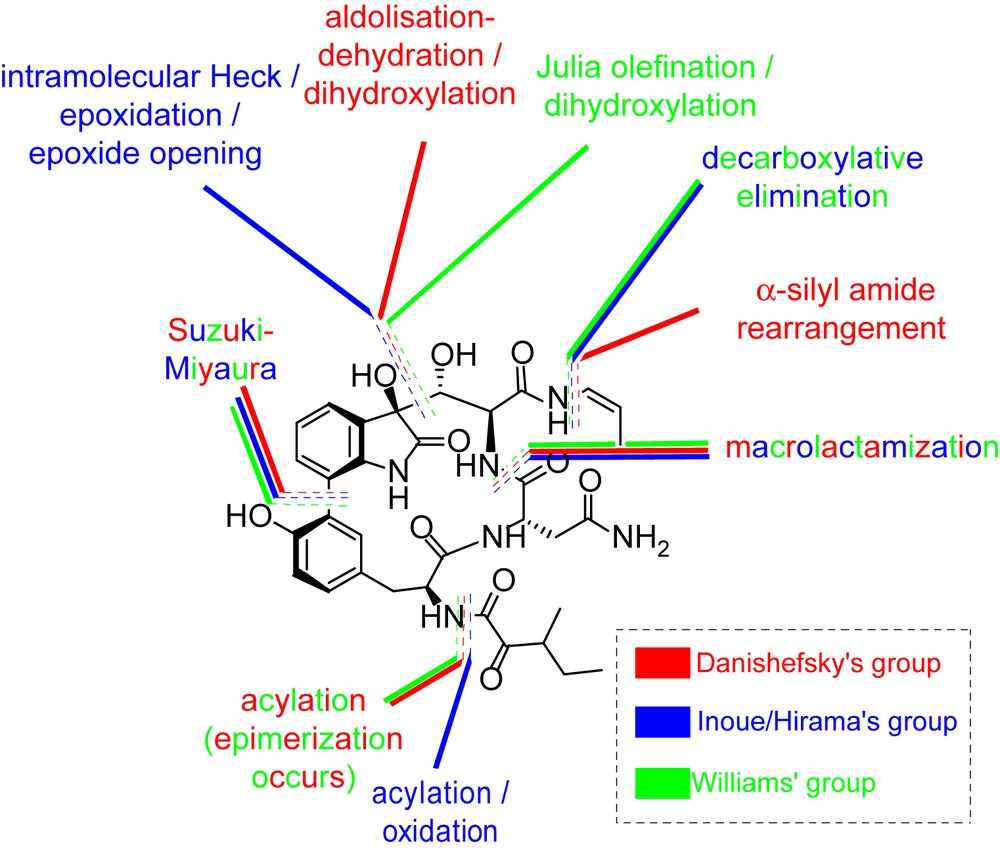
Key bond disconnections used in total syntheses of TMC-95A and B.
A seminal contribution came from the Danishefsky group who investigated a wide number of activating agents to form the macrocycle using a macrolactamization reaction [13]. After extensive investigation, they eventually found that this cyclization was best performed using an EDC/HOAt-mediated coupling, thus forming the macrocycle in 52% yield. In this synthesis, the biaryl bond was formed using a Suzuki–Miyaura coupling, the enamide was installed using an α-silyl amide rearrangement, and the key steps used for the formation of the highly substituted tryptophan core rely on the use of an aldolisation/dehydration/asymmetric dihydroxylation sequence (Fig. 6).
Subsequently, Inoue, Hirama and co-workers reported a synthesis of TMC-95A using an intramolecular Heck reaction/epoxidation/intramolecular epoxide opening sequence to form the tryptophan core while a decarboxylative elimination was used to install the Z-enamide. Interestingly, the use of a two-step installation of the oxopentanoic side chain provided a selective synthesis of TMC-95A while a mixture of TMC95A and B is produced by direct acylation [14].
Finally, Williams reported in 2004 a synthesis where the main difference to the first two syntheses relies on the use of a Julia olefination for the synthesis of the tryptophan precursor (Fig. 6) [15].
3.1 Danishefsky's route to TMC-95A and B
The first total syntheses of TMC-95A and B (obtained as a mixture of isomers and separated by reverse-phase HPLC after completion of the synthesis; see below for details) were reported by Danishefsky and co-workers in 2002 who obtained the macrocyclic natural products using 24 steps and 1% overall yield (longest linear sequence) [13b,c].
The synthesis started with the elaboration of the suitably functionalized oxindole core 7. After unsuccessful attempts at the preparation of 7 using an intramolecular Heck coupling reaction, an alternative route based on crossed-aldol reaction of 7-iodooxindole 5 and Garner aldehyde 6 was designed (Scheme 1). To this end, 2-iodo-aniline 2 was first reacted with chloral hydrate and hydroxylamine in water to give isonitrosoacetanilide 3 which was further cyclized to 7-iodo-isatin 4 under acidic conditions. The latter was finally deoxygenated using a “Wolf-Kishner like” reduction to give 5. Treatment of 5 with 2 equiv. of LDA followed by addition of Garner aldehyde 6 and elimination via mesylation of the intermediate aldol product finally afforded the oxindole core 7 as a mixture of olefin isomers, the minor being converted to the major one through an iodine-mediated isomerization (Scheme 1).
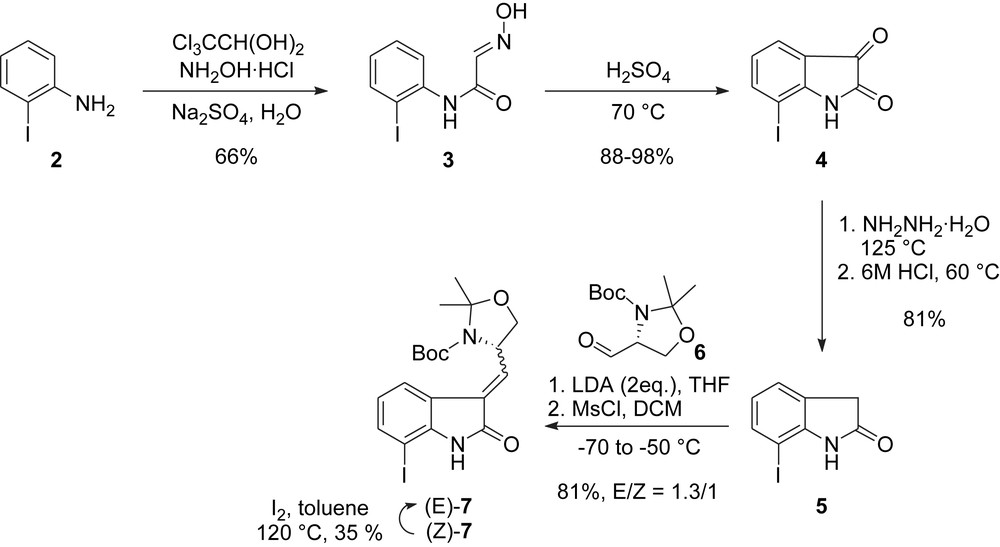
Danishefsky's synthesis: oxindole core.
Next, elaboration of the acyclic skeleton starting from the oxindole core began with a Suzuki-type coupling of 7 with tyrosine-derived boronate 8 which cleanly allowed for the installation of the biaryl domain. The last constitutive aminoacid was introduced after saponification of the methyl ester in 9 and coupling of the resulting free acid with asparagine t-butyl ester. At this stage, a diastereoselective (substrate and reagent controlled) dihydroxylation was used to install the two consecutive hydroxyl groups on the oxindole moiety of 11. It should be mentioned here that the presence of the N,O-acetonide is crucial to ensure useful level of selectivity (Scheme 2). Finally, cleavage of this acetonide and protection of the resulting primary alcohol furnished the fully elaborated acyclic skeleton 12 and set the stage for the macrocyclization step.
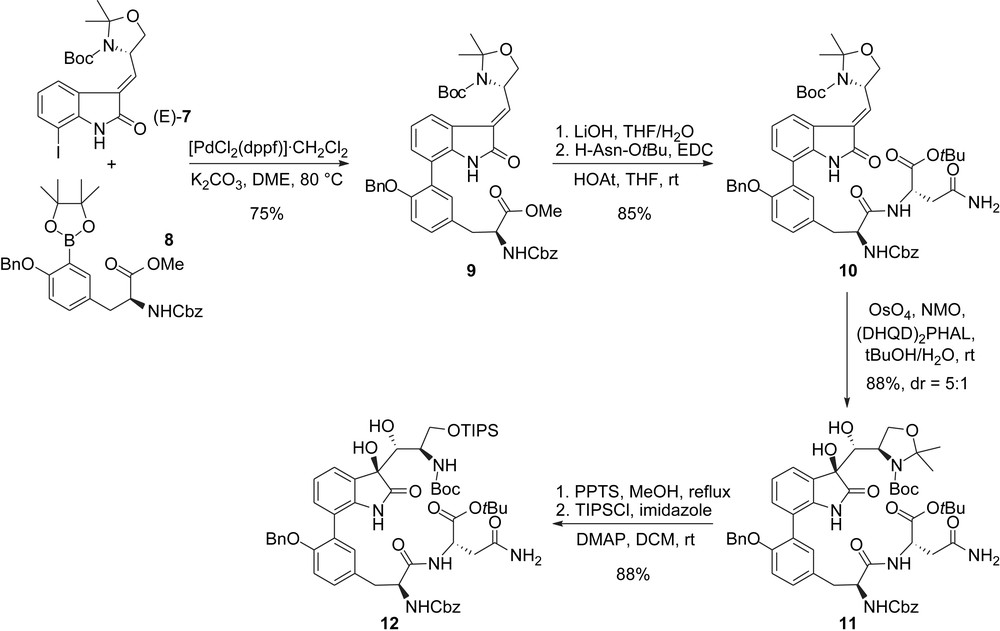
Danishefsky's synthesis: elaboration of the acyclic skeleton.
The macrolactamization reaction was thoroughly investigated and found to be best effected using an EDC/HOAt/DIPEA-mediated coupling in a mixture of DCM and DMF. The TMC-95's macrocyclic core 13 was obtained in 36% yield (Scheme 3).

Danishefsky's synthesis: formation of the macrocyclic core.
The remaining issues to be addressed were the installation of the two side chains. To this end, the carbobenzyloxy group was first deprotected by hydrogenolysis, which also removed the benzyl group. The C36 stereocenter being readily epimerized, the free amine was next acylated with racemic 3-methyl-2-oxopentaoic acid to give 14 as a mixture of two diastereoisomers. At this stage and to install the enamide side chain, the TIPS group was deprotected, all four hydroxyl groups were protected as TES ethers and reaction with Jones reagent selectively oxidized the primary TES ether to the corresponding carboxylic acid. This latter was finally coupled with silylallylamine 15, yielding α-silylallylamide 16 (Scheme 4). Upon heating, this compound underwent a quite spectacular concurrent ene- and silatropic-like bond reorganization leading to the required (Z)-enamide. Final deprotection gave a mixture of TMC-95A 1a and TMC-95B 1b that could be further separated by reverse-phase HPLC and thus completed the first and especially elegant total syntheses of these natural products.
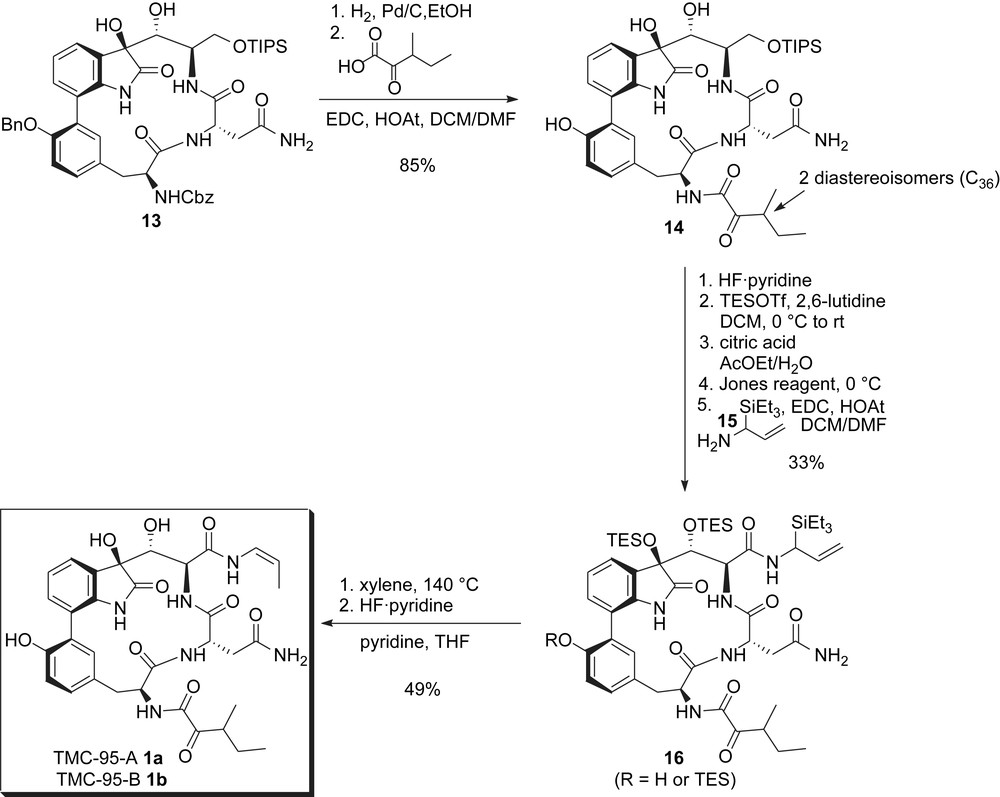
Danishefsky's synthesis: installation of side chains and completion of the synthesis.
3.2 Inoue and Hirama's route to TMC-95A and B
A year later, Hirama and Inoue reported their total syntheses of TMC-95A. A notable feature of their approach is the diastereoselective installation of the oxopentanoic side chain, therefore, avoiding a painful separation of TMC-95A and B at the end of the synthesis. Using their synthetic route, they obtained TMC-95A in 35 steps and 0.1% overall yield (longest linear sequence) [14b].
To reach an optimum level of convergence, they first prepared the oxindole and oxopentanoic-tyrosine fragments. These syntheses will be detailed in the following paragraphs, starting with the oxindole fragment whose synthesis features a stereoselective Mizoroki–Heck reaction and a diastereoslective oxidation/intramolecular epoxide opening reaction sequence [14a].
The synthesis of this northern fragment commences from known ester 17 (obtained from d-serine). A reduction followed by Wittig reaction and subsequent treatment with 2,6-dibromoaniline 18 in the presence of trimethylaluminium gave conjugated dibromo-anilide 19 after Boc protection (Scheme 5). This substrate was then used for the elaboration of the oxindole nucleus using a stereoselective Mizoroki–Heck reaction. After extensive studies, the authors found that this reaction was best carried out employing “ligandless” conditions at room temperature to afford the trisubstituted olefin 20 as a single isomer. This reaction was shown to be especially substrate dependent [16], which explains the little success met by Danishefsky and co-workers with a similar strategy [13c]. Introduction of the two contiguous oxygenated stereocenters of the target molecule was next effected by a diastereoselective epoxidation followed by intramolecular opening of the epoxide by the carbamate, yielding 22 as a single isomer.
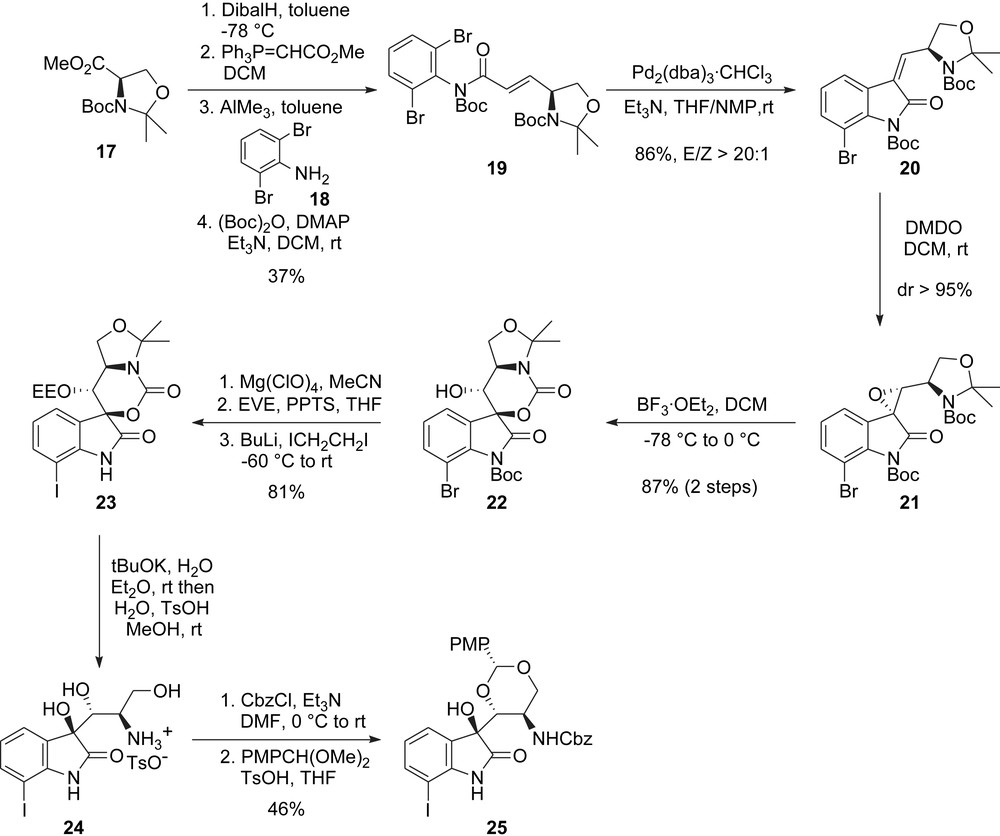
Inoue and Hirama's synthesis: oxindole core.
Next, and in order to install suitable functionalities and protecting groups for the rest of the synthesis, the Boc-group was deprotected, the secondary alcohol protected as an ethoxyethyl ether and the aromatic bromide was converted to the corresponding iodide 23 to ensure optimal biaryl coupling. Treatment of this compound 23 under the “anhydrous hydroxide” conditions reported by Gassman followed by lowering the pH value with p-toluenesulfonic acid then allowed for an interesting simultaneous deprotection of the acetonide, the carbamate and the ethoxyethyl ether to furnish 24 in a most straightforward fashion. Further protection of the amine and selective hydroxyl protection as a PMP acetal finally gave the fully elaborated oxindole fragment 25 (Scheme 5).
The preparation of the second coupling partner was initiated by condensation of iodotyrosine derivative 26 with carboxylic acid 27 (Scheme 6). In contrast with Danishefsky's synthesis, the side chain was installed at an early stage of the synthesis and the use of an α-hydroxy carboxylic acid in place of its α-oxo analogue suppresses epimerization of the side chain methyl group, therefore, allowing for a selective synthesis of TMC-95A. Orthogonal protection of phenol and secondary alcohol followed by conversion of the iodide in 28 to its boronate derivative 29 sets the stage for the crucial fragment coupling.
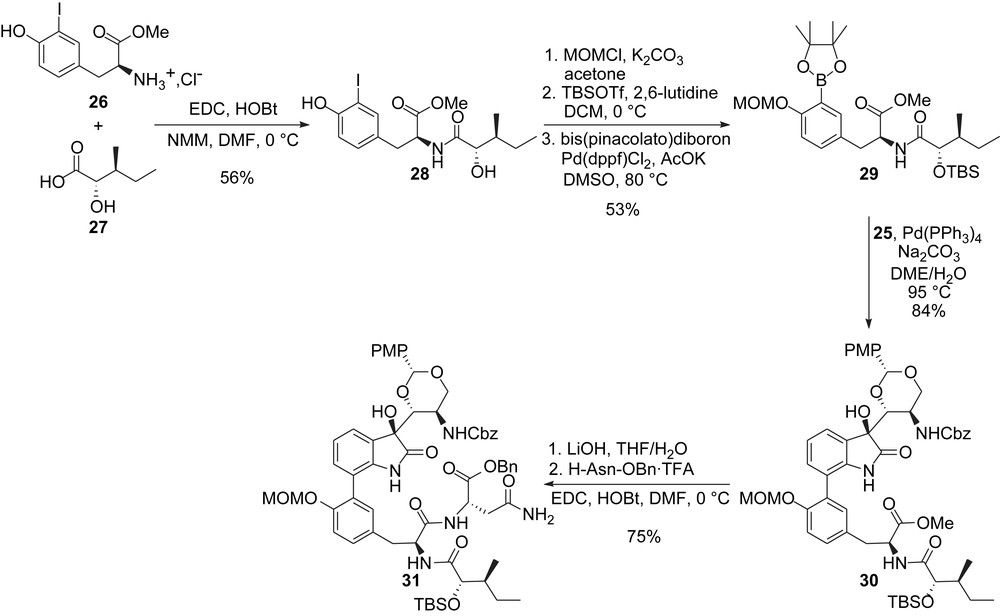
Inoue and Hirama's synthesis: elaboration of the acyclic skeleton.
A Suzuki-type coupling of 29 with iodide 25 then cleanly allowed for the installation of the biaryl domain. The last constitutive aminoacid was next introduced after saponification of the methyl ester in 30 and coupling of the resulting free acid with asparagine benzyl ester (Scheme 6).
Formation of the macrocycle was also envisioned using a macrolactamization strategy at the “tryptophan/asparagine-like” peptide bond. Hydrogenolysis of both carbobenzyloxycarbonyl and benzyl groups gave a seco acid which was cyclized by reaction with EDC and HOAt to give TMC-95A's macrocyclic core 32 in excellent yield (Scheme 7).
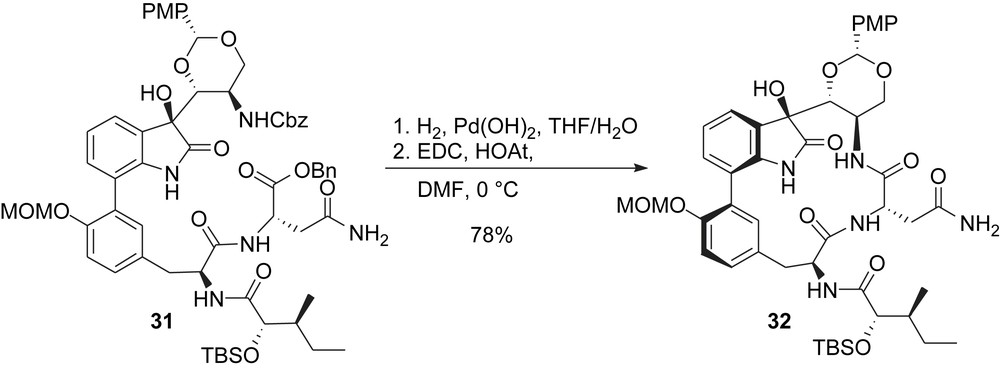
Inoue and Hirama's synthesis: formation of the macrocyclic core.
Few manipulations were required before installing the enamide side chain: first, the TBS group was replaced by a more labile TES for easier deprotection at the end of the synthesis (late stage deprotection of this TBS failed). The PMP acetal was then cleaved with buffered zinc triflate and ethanethiol to give a triol and selective oxidation of the primary alcohol under Parikh–Doering conditions followed by treatment with sodium chlorite gave the corresponding carboxylic acid that was finally coupled with allothreonine benzyl ester to give 33 (Scheme 8). Deprotection of the benzyl group by hydrogenolysis followed by subsequent treatment of the β-hydroxy carboxylic acid with DEAD and triphenylphosphine induced dehydrative decarboxylation (anti-elimination of the activated alcohol) and allowed for an especially efficient installation of the enamide in 34, obtained as a single diastereoisomer [17].
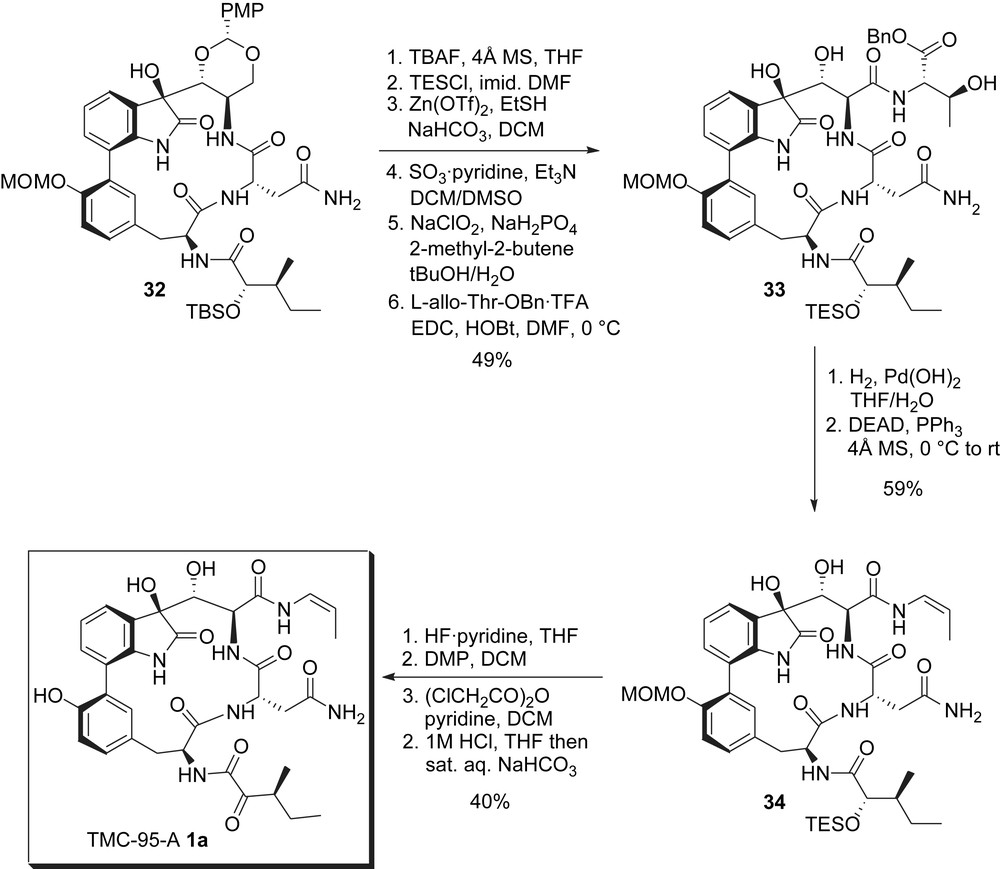
Inoue and Hirama's synthesis: installation of the enamide and completion of the synthesis.
To complete the synthesis, the TES group was removed and the resulting alcohol was selectively oxidized with Dess–Martin periodinane, therefore, adjusting the oxidation level of the side chain to that of the natural product. Deprotection of the MOM group required temporary protection of the secondary alcohol at C7 as its chloroacetyl ester to avoid its reaction with the enamide under acidic conditions. Successive and one-pot exposure of this ester to acidic and basic conditions then afforded synthetic TMC-95A 1a (Scheme 8).
3.3 William's route to TMC-95A and B
In 2003 as well, Albrecht and Williams reported a concise formal synthesis of these proteasome inhibitors [15a]. This preliminary communication was followed in 2004 by a full account detailing their total syntheses of TMC-95A and B [15b]. The strategy also uses a Suzuki coupling to form the biaryl subunit and a macrolactamization strategy to form the macrocyclic core. The main difference relies on the synthesis of the oxindole core 37 which features a stereoselective modified Julia olefination between sulfone 36, prepared in four steps from Cbz-serine methyl ester 35 and iodo-isatin 4 (Scheme 9).
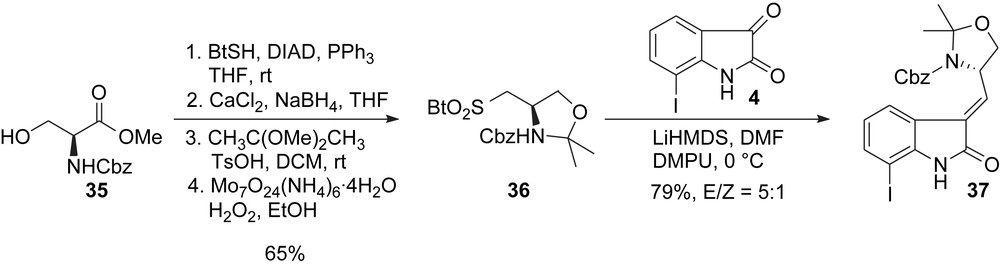
Williams synthesis: oxindole core.
After unsuccessful Stille coupling reaction between iodide 37 and tyrosine-derived boronate 38, the biaryl moiety was finally formed using a Suzuki coupling reaction (Scheme 10). Saponification of the methyl ester in 39 followed by coupling with asparagine benzyl ester then gave 40 whose double bond was used to introduce the 1,2-diol group of the natural product. Interestingly, while the use of a mixture of OsO4, NMO, and (DHQD)2PHAL by Danishefsky on a similar substrate bearing a Boc-group in place of the Cbz gave a 5:1 ratio of diastereoisomers, the use of OsO4 alone here provided 41 as a single stereoisomer. The oxygenated side chain was introduced at this stage after Boc-deprotection and coupling of the resulting free amine with 3-methyl-2-oxo-pentanoic acid sodium salt and gave the acyclic skeleton 42 (Scheme 10). It ought to be mentioned that the MOM group was also cleaved during the last deprotection step, therefore, avoiding its painful deprotection at the end of the synthesis (see Scheme 8).
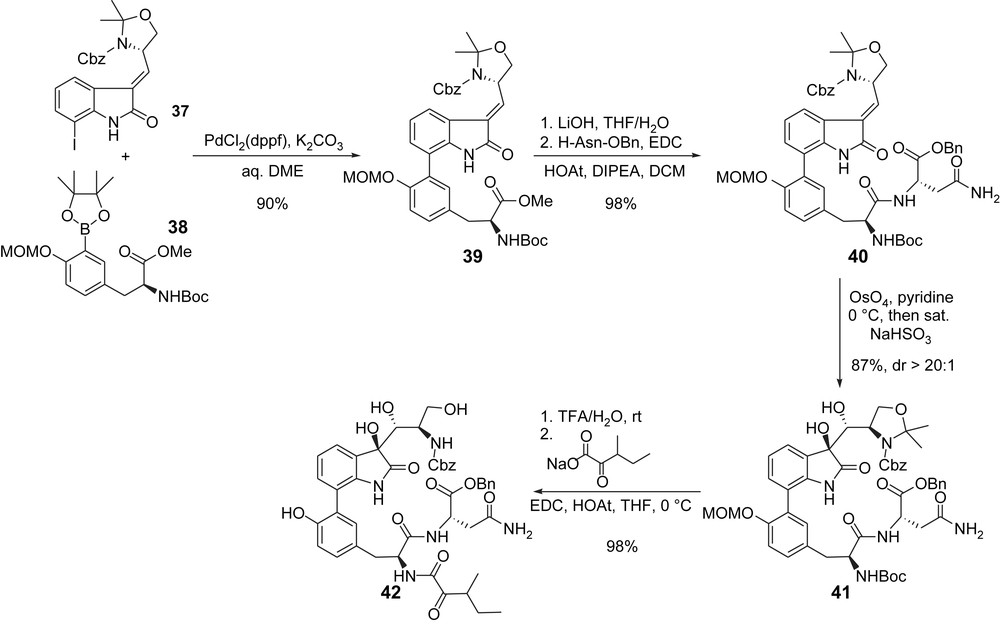
Williams synthesis: elaboration of the acyclic skeleton.
Similarly to the two previous syntheses, formation of the macrocyclic skeleton was envisioned through macrolactamization of the seco acid obtained after deprotection of 42 and gave macrocyclic product 43 in 49% yield (Scheme 11).
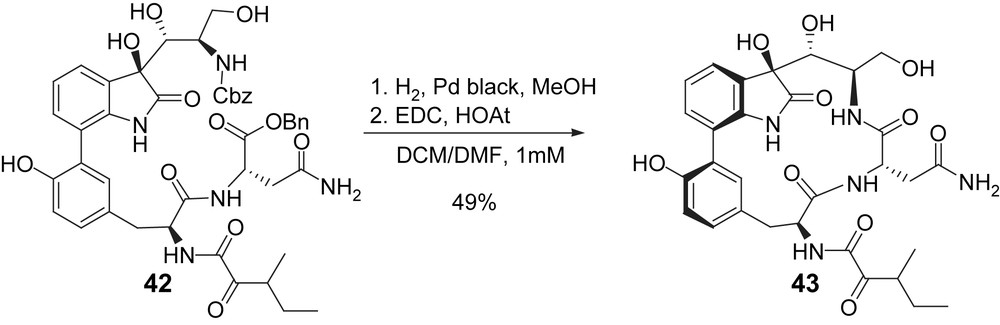
Williams synthesis: formation of the macrocyclic core.
After unsuccessful attempts at the formation of the enamide side chain based on Fürstner's enamide synthesis that uses hydroxyalkyl silanes for the preparation of enamides via a Peterson olefination, an endgame strategy similar to that of Inoue and Hirama was used (Scheme 12). Here again, selective oxidation of the primary alcohol in 43, coupling with allothreonine benzyl ester followed by debenzylation and decarboxylative dehydration allowed for the installation of the (Z)-enamide and gave a mixture of TMC-95A 1a and TMC-95B 1b.
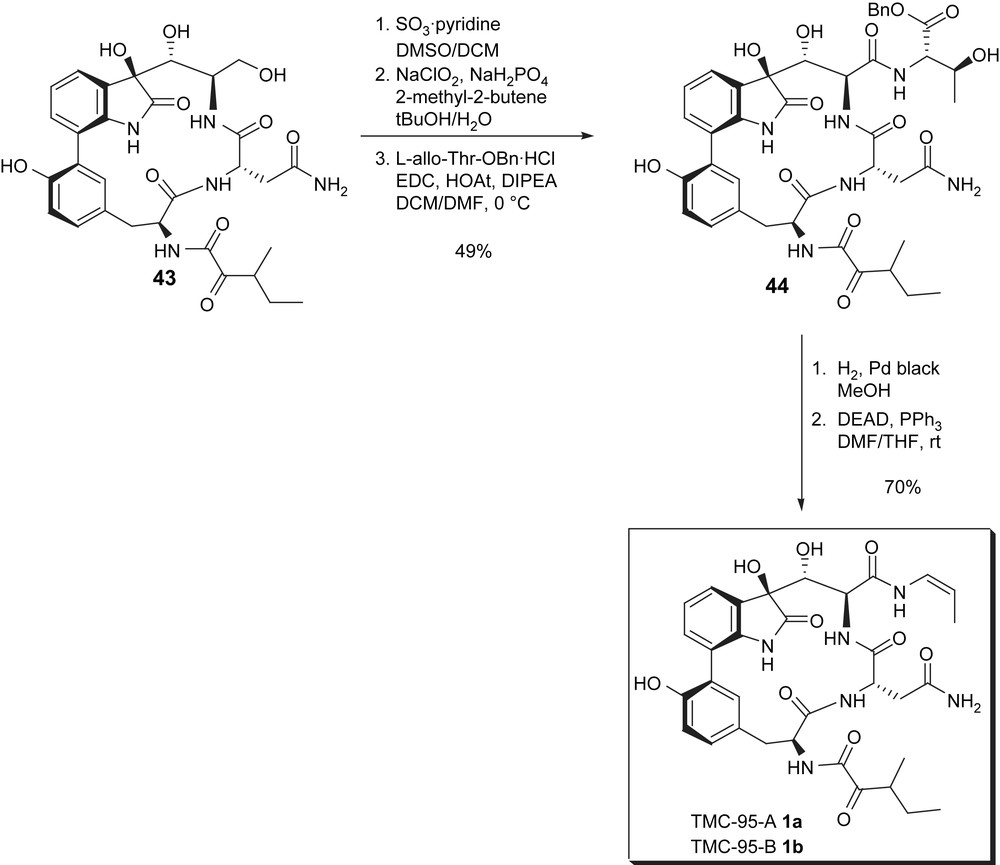
Williams synthesis: installation of the enamide and completion of the synthesis.
A striking feature of this approach is the minimal protecting group manipulation which allowed for the preparation of the natural products in 18 steps and 4% overall yield (longest linear sequence).
The three total syntheses recorded here along with the synthetic approach to these natural products by the group of Ma [18] constitute excellent basis for the preparation of analogues. Moreover, due to the exceptional activity of these proteasome inhibitors and their unique binding mode, their core structure clearly appears as an excellent platform for drug development and has led to intense interest in the development of related compounds as potential medicinal agents or biological probes. Advances in this field will be overviewed in the next section.
4 TMC-95 analogues: chemistry and biology
The most significant contributions aiming to design and synthesize simplified analogues of TMC-95A and B retaining the biological activity of the promising lead structure of the natural product will be presented in this section. To date, three research groups have investigated this matter and their results will be overviewed independently.
4.1 Moroder's TMC-95A analogues
A seminal contribution in this field came from the Moroder group [19], shortly after the publication of Danishefsky's total syntheses (see Section 3.1). Based on molecular modelling starting from the X-ray structure of TMC95-A bound to the proteasomal subunit β2 (Fig. 5), the minimum structural elements of TMC-95A required for binding to the active site were determined (compound 45, Fig. 7), and analogue 46 was identified as the first synthetic target and for validation of the design concept. In this latter compound, the propyl side chain (R1) replaces the propenylamide in the parent compound and is expected to interact with the S1 pocket, while the asparagine side chain (R3), is expected to occupy the S3 pocket.
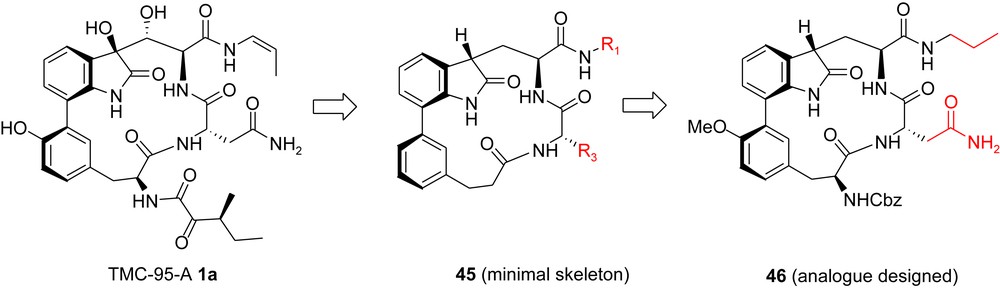
Moroder's analogue: minimal skeleton for binding.
Construction of this strained ring structure was based on a macrolactamisation strategy from a suitably functionalized acyclic precursor. The first step of this synthesis involved a Suzuki coupling of N-Boc protected propylamide derivative 47 with aryl boronate 48 and gave key intermediate 49. C-terminal elongation with H-Asn-OtBu then gave an intermediate peptide that was finally oxidized with DMSO in acidic conditions to give 50. Macrolactamization of this compound proceeded in fair yield to give the desired analogue 46 as a single macrocyclic diastereoisomer in which the 3S absolute configuration at the oxindole moiety was determined on the basis of NMR data (Scheme 13). This quite surprising stereoselectivity was rationalized by the steric requirements for macrolactamization (only the 3S diastereoisomer can undergo cyclization to the constrained macrolactam).
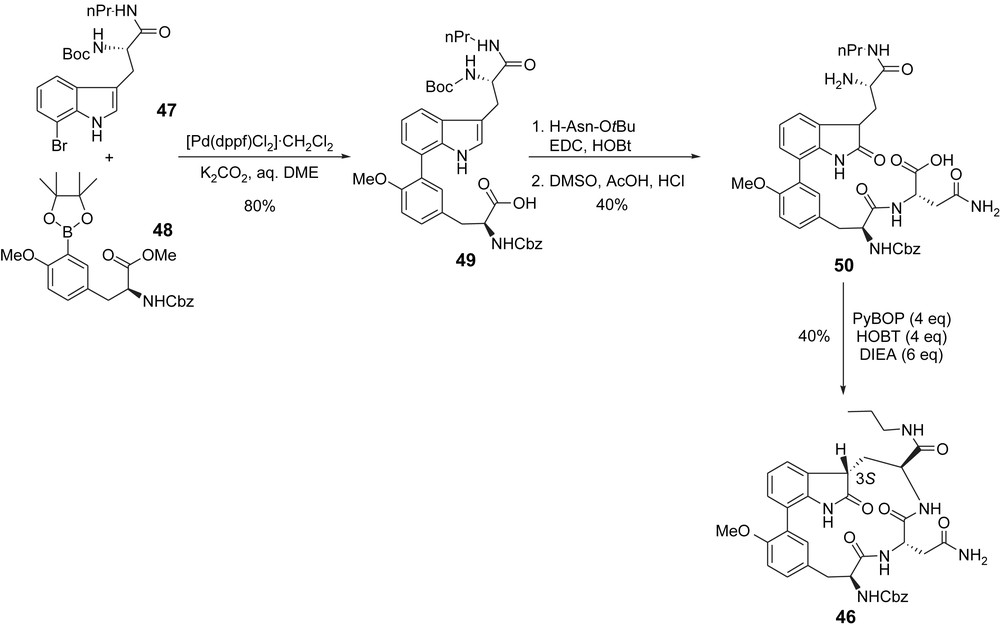
Moroder's synthesis of simplified TMC95-A analogue.
Inhibitory activity of this first analogue 46 for the CT-L (chymotripsin-like), TL (trypsin-like) and PGPH (peptidyl-glutamil-peptide-like) hydrolase activities of yeast proteasome was next evaluated and compared with those of TMC95-A (Table 2, entry 9). While compound 46 retains almost full inhibition of TL and PGPH activities, it is significantly less potent against the CT-L activity, a difference that was attributed to the exchange of the (Z)-1-propenylamide with the more flexible propylamide for occupancy of the S1 subsite of the enzyme. However, full retainment of affinity for the two other active sites of the proteasome confirms that the N-terminal acyl residue as well as the tyrosine hydroxy group is not critically involved in the interaction with the protein counterpart, while the biaryl moiety restricts the peptide backbone into the extended β-strand conformation for optimal hydrogen bonding to the active site clefts. The data also confirm that the degree of oxidation of the tryptophan residue can be reduced and that the oxindole group is sufficient for the additional hydrogen bond to the protein backbone to be established. Overall, these results nicely demonstrate that the sophisticated structure of TMC-95A can be considerably reduced.
Inhibitory activities of TMC-95A–D and analogues against CT-L, TL and PGPH activities of 20S proteasome
| Compound | ±0.02% SDS | IC50 (μM) | Ki (μM) | Ref. | |||||
| CT-L | TL | PGPH | CT-L | TL | PGPH | ||||
| 1 | +SDS | 0.0054c | 0.20c | 0.060c | – | – | – | [10] | |
| 2 | −SDS | 0.012c | 1.5c | 6.7c | – | – | – | [10] | |
| 3 | +SDSg | – | – | – | 0.0011a | 0.043a | 0.65a | [20] | |
| 4 | +SDSg | – | – | – | 0.0011b | 0.81b | 0.029b | [13c,26] | |
| 5 | +SDS | 0.0087c | 0.49c | 0.060c | – | – | – | [10] | |
| 6 | +SDSg | – | – | – | 0.0017b | 1.1b | 0.023b | [13c,26] | |
| 7 | +SDS | 0.36c | 14c | 8.7c | – | – | – | [10] | |
| 8 | +SDS | 0.27c | 9.3c | 3.3c | – | – | – | [10] | |
| 9 | −SDS | 8.0a | 10.6a | 7.4a | – | – | – | [19a] | |
| −SDS | 1.9c | 3.7c | 1.8c | – | – | – | [19a] | ||
| +SDSg | – | – | – | 2.4a | 55a | >2000a | [21,22] | ||
| +SDSg | – | – | – | 55a | 0.15a | 90a | [20] | ||
| 10 | +SDSg | – | – | – | 1.2 (Kit), 2.4a,d | 5.3 (Kit), 22a,d | >1000a | [20] | |
| 11 | +SDSg | – | – | – | 9.1a | 60a | >2000a | [21] | |
| 12 | +SDSg | – | – | – | 5.5a | 74a | >2000a | [22] | |
| 13 | +SDSg | – | – | – | 65a | >2000a | >2000a | [22] | |
| 14 | +SDSg | – | – | – | nie,f | x = 1.4e | >100e | [25] | |
| 15 | +SDSg | – | – | – | nie,f | x = 1.8e | >100e | [25] | |
| 16 | +SDSg | – | – | – | nie,f | nie,f | 37.1e | [25] | |
| 17 | +SDSg | – | – | – | nie,f | x = 1.4e | nie,f | [25] | |
| 18 | +SDSg | – | – | – | nie,f | 69e | nie,f | [25] | |
| 19 | +SDSg | – | – | – | nie,f | 21.5e,f | >100e | [25] | |
| 20 | +SDSg | – | – | – | nie,f | x = 2e | nie,f | [25] | |
| 21 | +SDSg | – | – | – | nie,f | x = 1.9e | >100e | [25] | |
| 22 | +SDSg | – | – | – | nie,f | nie,f | 96.4e | [25] | |
| 23 | +SDSg | – | – | – | 59.3e | nie,f | 74.0e | [25] | |
| 24 | +SDSg | – | – | – | 14.1e | nie,f | 55.4e | [25] | |
| 25 | +SDSg | – | – | – | nie,f | nie,f | nie,f | [25] | |
| 26 | +SDSg | – | – | – | nie,f | nie,f | >100e | [25] | |
| 27 | +SDSg | – | – | – | 1.6e | 3.0e | 2.8e | [25] | |
| 28 | +SDSg | – | – | – | 59.3e | nie,f | 30.2e | [25] | |
| 29 | +SDSg | – | – | – | 44.8e | x = 9.8e | >100e | [25] | |
| 30 | +SDSg | – | – | – | 9.2e | x = 6e | 8.7e | [25] | |
| 31 | +SDSg | – | – | – | nie,f | x = 1.8e | nie,f | [25] | |
| 32 | +SDSg | – | – | – | nie,f | x = 1.5e | nie,f | [25] | |
| 33 | +SDSg | – | – | – | nie,f | x = 6.8e | nie,f | [25] | |
| 34 | +SDSg | – | – | – | nie,f | x = 1.7e | 31.5e | [25] | |
| 35 | +SDSg | – | – | – | nie,f | x = 1.7e | 55.4e | [25] | |
| 36 | +SDSg | – | – | – | 59.3e | x = 2.2e | 84.2e | [25] | |
| 37 | +SDSg | – | – | – | x = 1.3e | x = 1.9e | nie,f | [25] | |
| 38 | +SDSg | – | – | – | x = 1.3e | x = 1.8e | nie,f | [25] | |
| 39 | +SDSg | – | – | – | nie,f | x = 1.9e | nie,f | [25] | |
| 40 | +SDSg | – | – | – | 1.2e | 3.9e | 0.6e | [25] | |
| 41 | +SDSg | – | – | – | x = 1.4e | x = 1.6e | nie,f | [25] | |
| 42 | +SDSg | – | – | – | x = 1.5e | x = 2.5e | nie,f | [25] | |
| 43 | +SDSg | – | – | – | nie,f | x = 1.9e | nie,f | [25] | |
| 44 | +SDSg | – | – | – | nie,f | x = 1.6e | nie,f | [25] | |
| 45 | +SDSg | – | – | – | 65.7e | nie,f | 1e | [25] | |
| 46 | +SDSg | – | – | – | >100e | nie,f | 23.4e | [25] | |
| 47 | +SDSg | – | – | – | nie,f | x = 1.6e | 96.4e | [25] | |
| 48 | +SDSg | – | – | – | 65.7e | >100e | nie,f | [25] | |
| 49 | +SDSg | – | – | – | nie,f | nie,f | nie,f | [25] | |
| 50 | +SDSg | – | – | – | 2.1e | 4.3e | 0.7e | [25] | |
| 51 | +SDSg | – | – | – | x = 1.5e | x = 3.2e | nie,f | [25] | |
| 52 | +SDSg | – | – | – | x = 1.6e | x = 2.2e | nie,f | [25] | |
| 53 | +SDSg | – | – | – | 81.8e | x = 2.3e | 31.5e | [25] | |
| 54 | +SDSg | – | – | – | x = 1.6e | x = 1.9e | nie,f | [25] | |
| 55 | +SDSg | – | – | – | 1.07e | 0.32e | nie,f | [25] | |
| 56 | +SDSg | – | – | – | 2.22e | 1.12e | nie,f | [25] | |
| 57 | +SDSg | – | – | – | 0.85e | 0.98e | nie,f | [25] | |
| 58 | +SDSg | – | – | – | 3.69e | 0.33e | 22.5e | [25] | |
| 59 | +SDSg | – | – | – | 0.48e | >100e | >100e | [25] | |
| 60 | +SDSg | – | – | – | 0.0019b | 1.2b | 0.023b | [13c,26] | |
| 61 | +SDSg | – | – | – | 0.024b | 13b | 0.11b | [13c,26] | |
| 62 | +SDSg | – | – | – | 0.0014b | 0.93b | 0.011b | [26] | |
| 63 | +SDSg | – | – | – | 0.0057b | 9.7b | 0.15b | [26] | |
| 64 | +SDSg | – | – | – | 0.0062b | 9.7b | 0.13b | [26] | |
| 65 | +SDSg | – | – | – | 0.0049b | 2.6b | 0.063b | [26] | |
| 66 | +SDSg | – | – | – | 0.0053b | 5.1b | 0.087b | [26] | |
| 67 | +SDSg | – | – | – | 0.0070b | 49b | 5.3b | [26] | |
| 68 | +SDSg | – | – | – | >100b | >100b | >100b | [26] | |
| 69 | +SDSg | – | – | – | 22b | >100b | 65b | [26] |
a Yeast 20S proteasome.
b Bovine 20S proteasome.
c Human 20S proteasome.
d Two phases with temporary Kit values explained by hydrolysis of the C-terminal amide.
e Rabbit 20S proteasome.
f ni: No inhibition at 100 μM.
g SDS omitted in assays for TL activity.
In a second analogue, 51 (Fig. 8), the (Z)-prop-1-enyl group of TMC-95A was replaced by a norleucine side chain and the asparagine with a leucine, with the aim of increasing the specificity for the β5 subsite, which is responsible for the chymotrypsin-like activity. In the case of calpain inhibitor I (Ac–Leu–Leu–Nle–H) and peptide vinylsulfones, similar P1 and P3 residues were found to be well suited for this purpose. The superimposition of the X-ray structures of these inhibitors with that of TMC-95A in complexes with yeast CP showed an almost identical peptide backbone display, thus suggesting similar orientations of the Leu and Nle side chains when incorporated into TMC-95A to act as P1 and P3 residues, respectively. This, however, proved to be an unfavorable approach since most inhibitory activities were considerably reduced (Table 2, entry 10) [20].
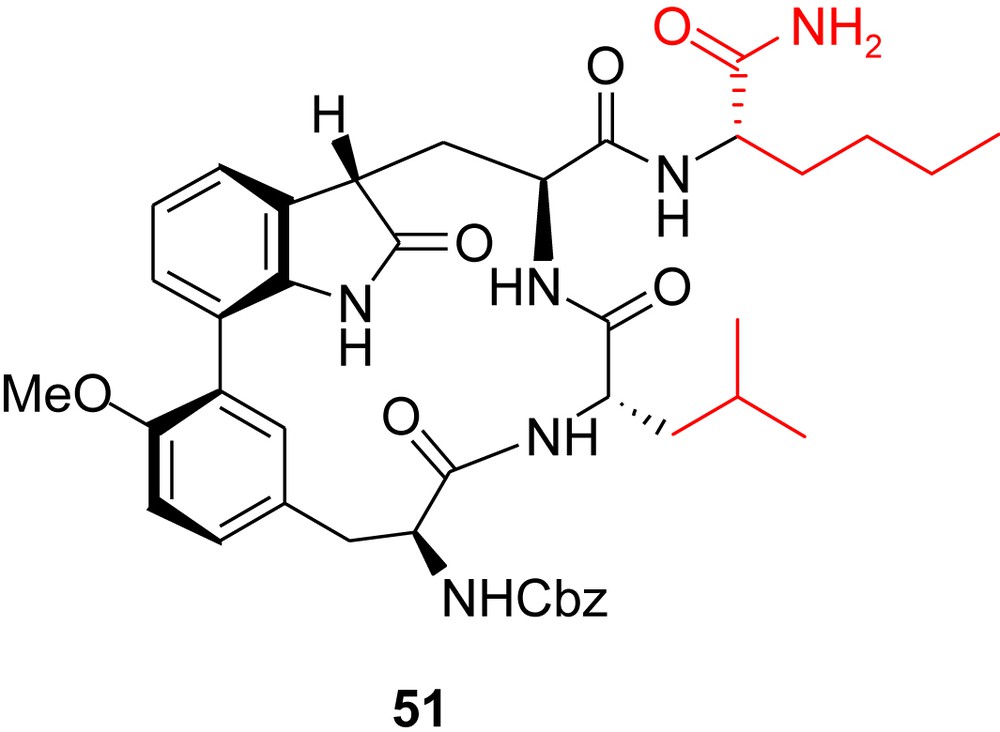
Moroder's second analogue: targeting specificity for the β5 subsite.
Another contribution from this group came one year later [21]. On the basis of the simplified skeleton mentioned above, they designed an analogue 53 in which the long R1 side chain was expected to interact with the S′-subsite via a nonscissible ketomethylene group. A different route was designed for the preparation of this compound and featured an intramolecular Suzuki cross-coupling performed on linear peptide 52. This macrocyclization was found to be especially efficient and gave the expected analogue as a single diastereoisomer. Here again, only the 3S diastereoisomer at the 2-oxoindole moiety undergoes ring-closure, and though the reaction was performed starting with an epimeric mixture at this stereocenter, a base catalyzed epimerization accounts for the good yield (57%) of purified product. However, the inhibitory potency of this analogue (Table 2, entry 11) was not significantly enhanced compared to 46 (Scheme 14).
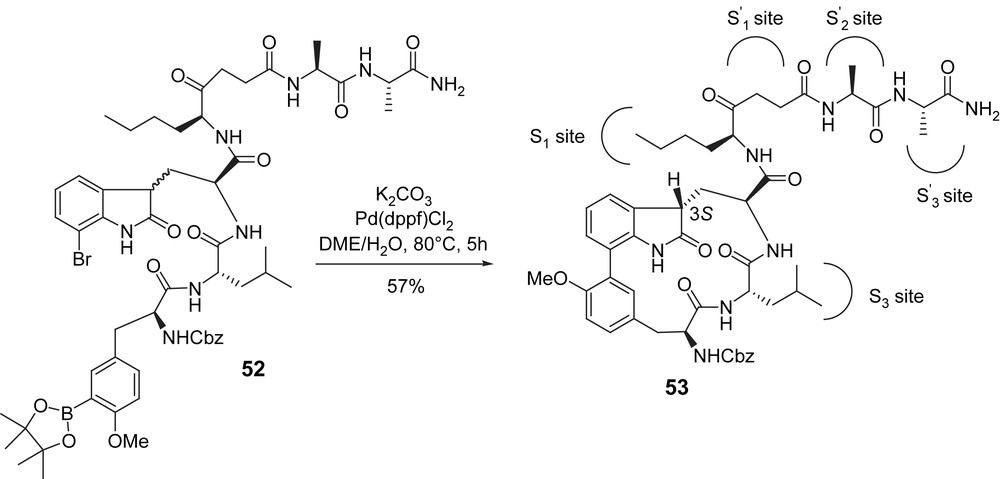
Moroder's second-generation analogue: trying to reach the S′ subsite.
Another key structural modification was disclosed by the Moroder group in 2004 [22]. Considering that the role of the phenol–oxindole biaryl system in TMC-95 is to induce and stabilize an extended β-type peptide backbone conformation, which is mandatory for optimal interaction with the proteasome, they planned to replace this complex framework by an endocyclic biaryl ether. Since this less demanding structure should be able to induce the same conformation, they targeted compounds 55 and 57. Key step for the macrocyclization leading to these analogues now rely on an intramolecular aromatic nucleophilic substitution involving acyclic substrate 54 or Rink-amide linked precursor 56. If the macrocyclization of 54 and 55 proceeded with a modest yield in solution (13%), the overall yield (stepwise peptide couplings and macrocyclization) reached as much as 9% when the synthesis was done on solid phase. Because of the facile synthetic access of tripeptides containing in i, i + 2 position residues of the isodityrosine type, this considerably simplified access to TMC-95 analogues and the strategy appeared to be quite successful since compound 55 was found to retain the activity of the more complex analogue 46, while 57 was found to inhibit only the CL activity, with a somehow lower potency compared to 46 (Scheme 15 and Table 2, entries 12 and 13). Later on, these analogues were successfully crystallized into the yeast 20S proteasome complex, highlighting the plasticity of the proteasomal tryptic-like specificity pocket [23].
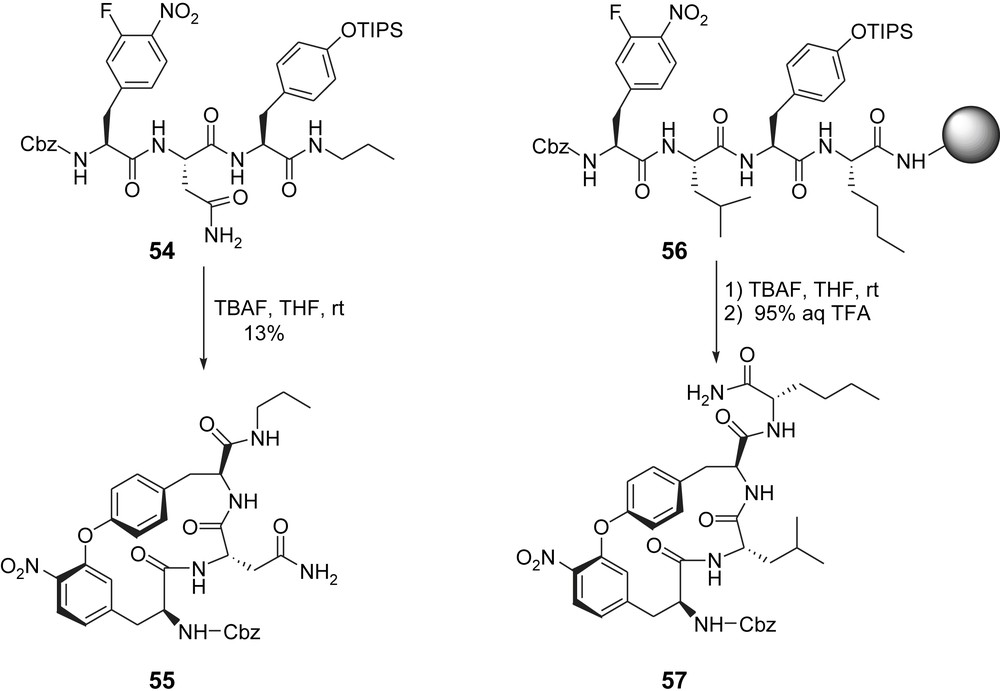
Moroder's biaryl ether analogues of TMC95-A.
4.2 Vidal's TMC-95A analogues
In 2003, soon after the first total syntheses of TMC95-A, Vidal and co-workers [24] disclosed their efforts aiming at the synthesis of simplified analogues of this molecule. They based their design on retaining the macrocyclic peptidic structure but replacing the complex oxindole by a simple indole. Biaryl compounds 59 were, therefore, identified as synthetic targets and the synthetic plan was based on an intramolecular Ullmann-type reaction to form the macrocycle starting from suitably functionalized tripeptides 58. This strategy, however, led to rather disappointing results since the desired macrocycles could only be obtained in low yields using nickel(0)-promoted intramolecular cross-coupling (Scheme 16). Interestingly, these results corroborate the work of Moroder (see Scheme 14) who demonstrated the need of an sp3 hybridization of C3 indole carbon atom for the cyclization to proceed. It should be noted that the alternative strategy based on the macrolactamization (through intramolecular peptide coupling) failed to give the expected macrocycles.
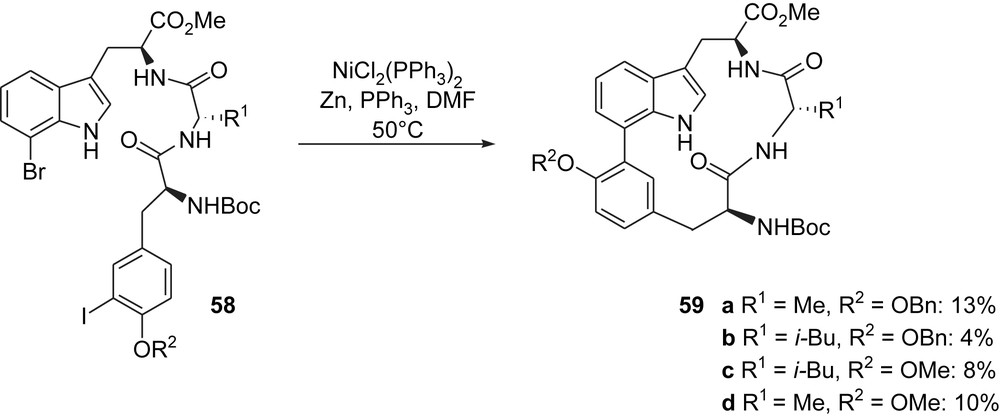
Vidal's TMC95-A analogues.
Biological activities of compounds 59 (Table 2, entries 14–16) as well as those of 45 acyclic precursors based on the tripeptide sequence of TMC-95A were recently reported (Table 2, entries 17–59): while compounds 59 were poor inhibitors, some acyclic precursors displayed interesting activities. Inhibition constants were submicromolar despite the absence of the entropically favorable constrained conformation that is characteristic of TMC-95A and its cyclic analogues. These linear compounds were readily prepared and reasonably stable in culture medium and could be optimized to block one, two, or all three proteasome catalytic sites. Furthermore, cytotoxicity assays performed on a series of human tumor cell lines identified the most potent inhibitors in cells [25].
4.3 Danishefsky's TMC-95A analogues
The first total syntheses of TMC95-A allowed to prepare some simplified analogues with minor alterations of the skeleton of the natural product. In an effort to get rid of the structural features that could render the synthesis impractical for producing amounts of materials appropriate for clinical follow-up, a set of analogues 62–71 (Fig. 9) was prepared by Danishefsky and co-workers according to the synthetic route shown for the preparation of 62 (Scheme 17) [26].
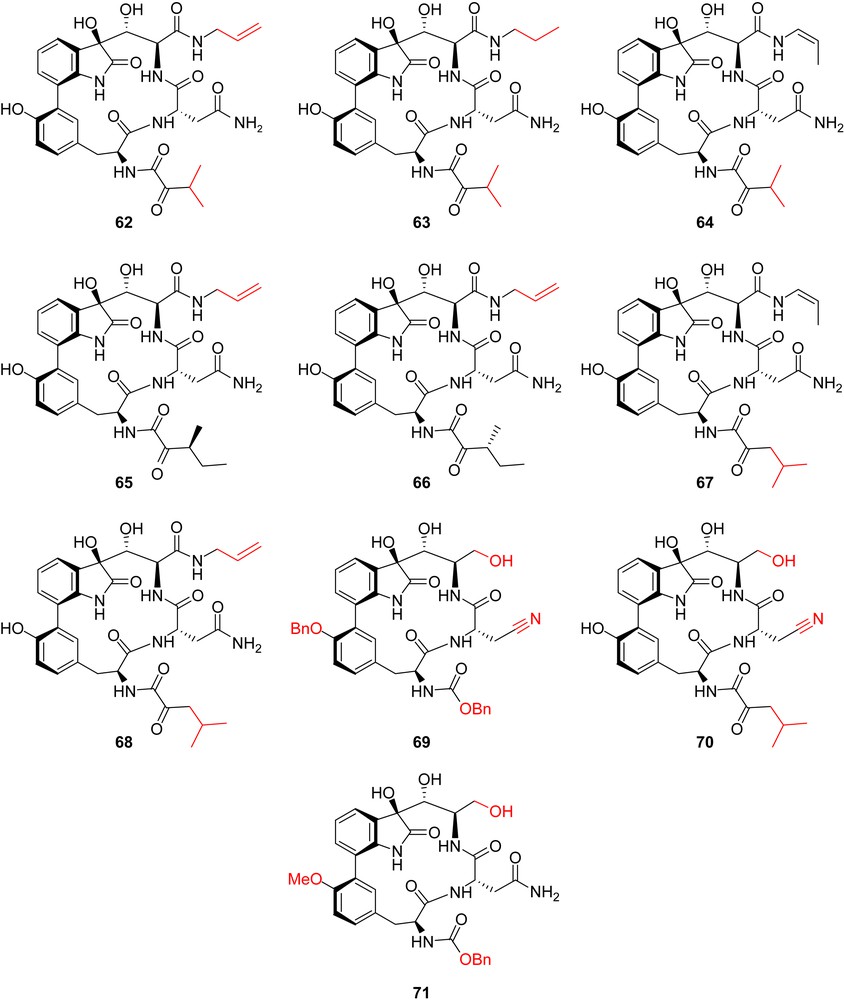
Danishefsky's TMC-95 analogues.
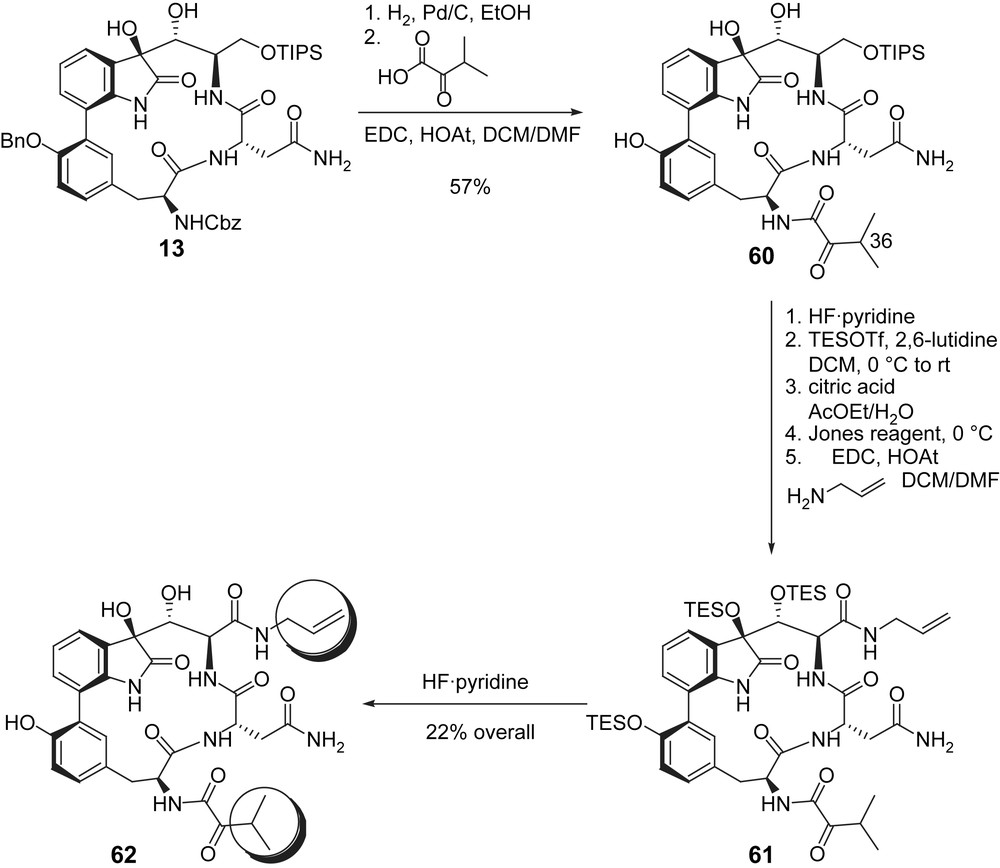
Danishefsky's route for the preparation of TMC-95 analogues.
Important preliminary findings as to SAR could be obtained after biological evaluation of these synthetic analogues (Table 2, entries 60–69). First, it was demonstrated that the cis-enamide, which interacts with the S3 pocket, and for which a new synthetic methodology had to be devised, could be replaced by an allyl amide without alteration of the activity. However, a saturated propyl side chain induced a considerable loss of activity which shows that this side chain requires a certain degree of rigidity for optimal interaction with the S3 pocket. Reduction of the enamide to a simple alcohol, which involves loss of a favorable hydrogen bond and hydrophobic interaction, was shown to be responsible for the loss of activity.
Importantly, the difficult installation of the side chain's C36 stereocenter (S in TMC-95A and C, R configuration in TMC-95B and D) and the tedious separation of diastereoisomers at the end of the synthesis could be circumvented by replacing the sec-butyl carbon chain by an isopropy group.
These results clearly demonstrate the power of chemical synthesis to identify simpler effective proteasome inhibitors derived from natural products and should pave the way for the preparation of analogues that would now possess a greater activity and specificity.
4.4 Inhibition of 20S proteasome with TMC-95 analogues
For clarity, we chose to report all values concerning the proteasome inhibition in a single section at the end of this manuscript. The values are reported in Table 2 and it is important to notice that the origin of the 20S proteasome varies from one study to another (yeast, bovine, rabbit or human proteasome were used) as well as the protocol used for the determination of IC50 or Ki values. Care should then be taken when comparing values from different studies.
5 Conclusion
Since their isolation in 2000, TMC95-A and B have motivated intensive research: these complex natural cyclopeptides with a unique reversible mode of action, interacting with a biological target of growing interest, have stimulated new knowledge in chemistry, biology and pharmacology. Yet, simplified analogues of these fascinating molecules have not fulfilled the purpose in view, that is to inhibit at a subnanomolar level in a reversible way the catalytic activity of the proteasome. Considering the potential applications in biology, and especially for cancer treatments, the pioneering researches described in this review will certainly continue, and the first SAR summarized here will help future prospects for the search of potent inhibitors based on this skeleton, for which efficient synthetic routes will have to be devised.
Acknowledgements
The authors thank Dr. Karen Wright, Dr. Bruno Drouillat, Dr. Jean-Paul Mazaleyrat and Dr. Michel Wakselman (Institut Lavoisier, Versailles) as well as Dr. Sarah Bregant (Commissariat à l'Energie Atomique, Saclay) for helpful and stimulating discussions. A.C. thanks the Institut du Cancer for a fellowship.
1 See: http://www.bortezomib.com or http://www.velcade.com.