

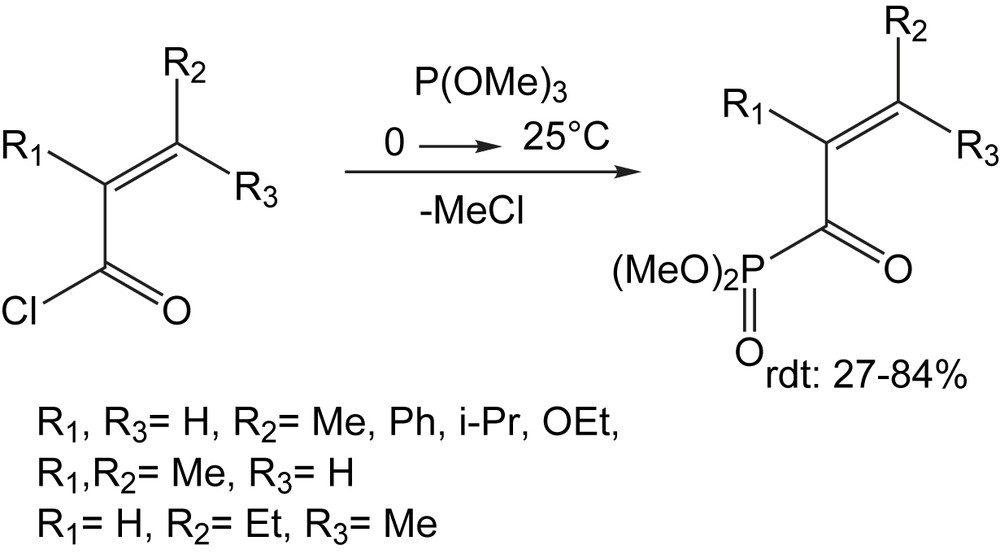
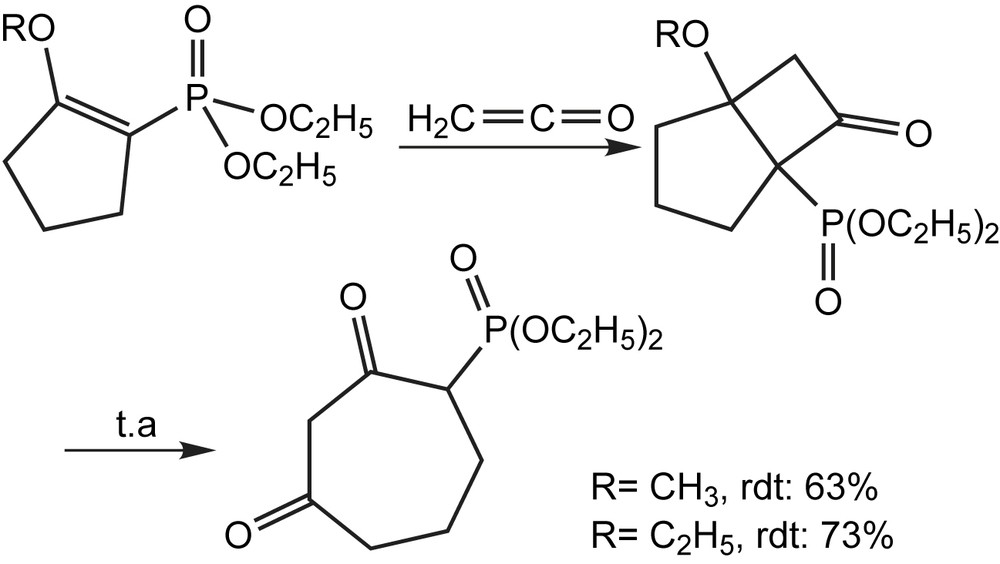
1 Introduction
Parmi les dérivés phosphorés utilisés en synthèse organique, les oxoalkylphosphonates est l'une des classes les plus intéressantes. L'acide bis-naphtyl β-cétophosphonate, par exemple, se présente comme un nouvel inhibiteur non peptidique de la cathepsine neutrophile G (Ki = 38 μM), et chymase (Ki = 2,3 μM) [1,2]. Le glutamyl-β-cétophosphonate-adénosine (Glu-KPA) est un inhibiteur compétitif de la synthétase de glutamyle-tRNA d'Escherichia coli (GluRS) avec le Ki 18 μm [3a] tandis que les α,α-difluoro-β-cétophosphonates sert d'inhibiteurs efficaces des protéines tyrosine phosphatase 1B [3b]. Concernant les α-cétophosphonates analogues de l'α-glutarate, ils sont connus pour inhiber l'activité du complexe α-cétoglutarate déshydrogénase isolé à partir du cerveau et les cellules cultivées [4].
Sur le plan synthétique, ils donnent lieu à une gamme diverse de réactions et permettent l'incorporation de nouvelles fonctionnalités. Après la revue de Kolodiazhnyi sur les hydroxyphosphonates [5] et de Palacios sur les β-aminophosphonates et –phosphinates [6], ainsi que le livre de Savignac consacré à la chimie des phosphonates [7], cette revue sur les oxophosphonates va couvrir la période des années 2000 à ce jour. Nous développerons dans une première partie, les différentes méthodes de synthèse des α- et β-cétophosphonates, ainsi que quelques exemples de γ- et δ-oxophosphonates. Une deuxième partie traitera des réactions de ces oxophosphonates (réductions, amination, substitution, silylation, cycloaddition,…).
2 Synthèse des oxophosphonates
2.1 Les β-cétophosphonates
Les phosphonates portant une fonction cétone en position β (β-cétophosphonates) sont d'un grand intérêt pour la synthèse organique. Leur utilisation dans l'oléfination de Horner–Wadsworth–Emmons (HWE), est d'une importance particulière. Les β-cétophosphonates sont également des intermédiaires utiles dans la synthèse des noyaux hétérocycliques tels que des quinoléines, des pyrroles, des pyrazoles, et des naphthydrines. Leur synthèse est généralement réalisée par l'intermédiaire de la réaction d'Arbuzov, la réaction de Michaelis–Becker, ou par acylation d'anion d'alkylphosphonate. Ils peuvent également être formés à partir des phosphonates de vinyle via la migration 1,3-phosphore; cependant, cette stratégie semble être limitée à la formation des β-cétophosphonates cycliques.
2.1.1 Réaction d'Arbuzov
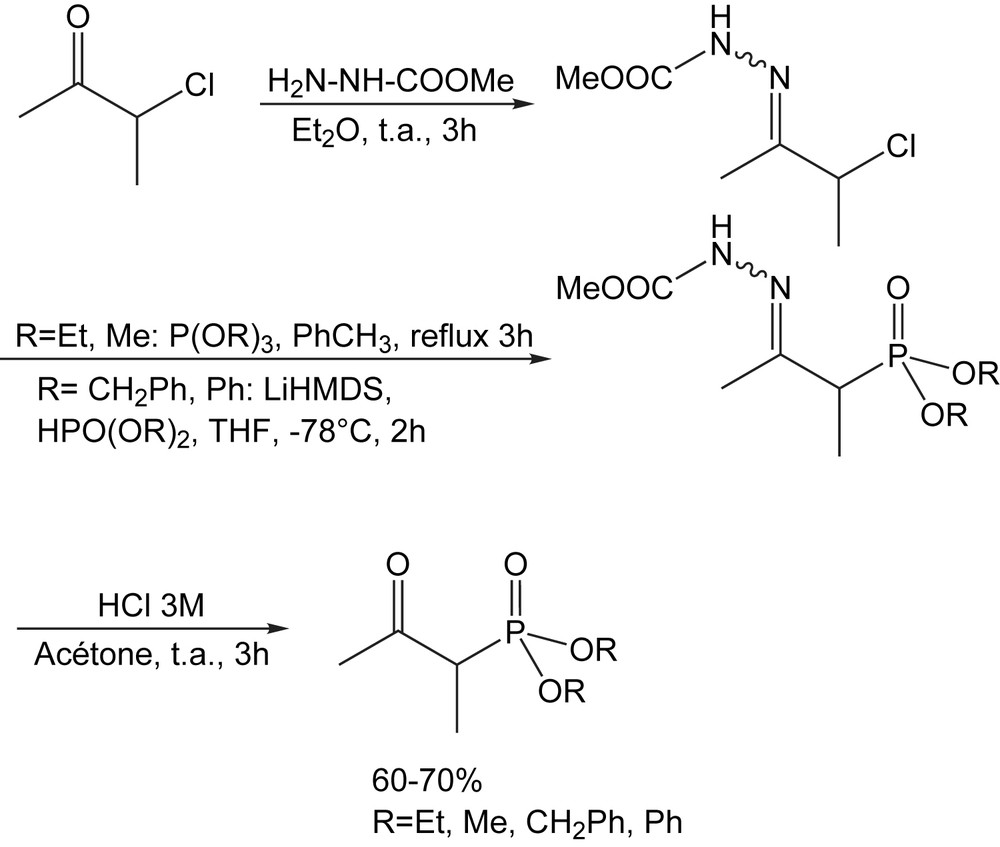
Les β-cétophosphonates non commerciaux substitués par un groupement benzyle ou phényle sont synthétisés par un procédé en trois étapes. Pour les dialkyles phosphonates (R = Me) et (R = Et), la synthèse débute, tout d'abord, par la protection du groupe cétonique pour conduire à l'hydrazone comme intermédiaire, suivi d'une réaction d'Arbuzov pour finir par une déprotection. Dans ce processus, les rendements sont autour de 60–70%. Pour le dibenzylphosphonate, la réaction d'Arbuzov a été réalisée sous vide afin d'éliminer le chlorure de benzyle formé dans la réaction. Une stratégie alternative, consiste à utiliser une substitution nucléophile directe de l'atome de chlore par l'action de l'anion du dibenzylphosphite. Quand la réaction s'effectue à température ambiante en présence de Cs2CO3 comme base, le produit de β-élimination est l'unique produit isolé. Cependant, l'utilisation de LiHMDS à basse température (−78 °C) mène, par une séquence en trois étapes et après hydrolyse, au composé désiré avec un bon rendement (63%). Le même procédé, toujours en trois étapes, a été utilisé avec succès pour le diphénylphosphonate, mais avec un rendement modeste de 35% (Schéma 1) [8].
2.1.2 Acylation des alkylphosphonates avec les chlorures d'acyle, les esters et les amides
La préparation des dialkyl β-céto phosphonates par acylation directe des α-lithioalkylphosphonates présente beaucoup d'avantages. L'utilisation des esters, par exemple, permet d'éviter les problèmes liés à la réaction d'Arbuzov. Comme application dans le domaine des sucres, la condensation d'un ester méthylique d'acide aldonique avec l'anion du diméthyl méthylphosphonate se fait selon la méthodologie standard (Schéma 2). Les sucres insaturés dérivés de phosphonates sont obtenus avec de bons rendements [9].
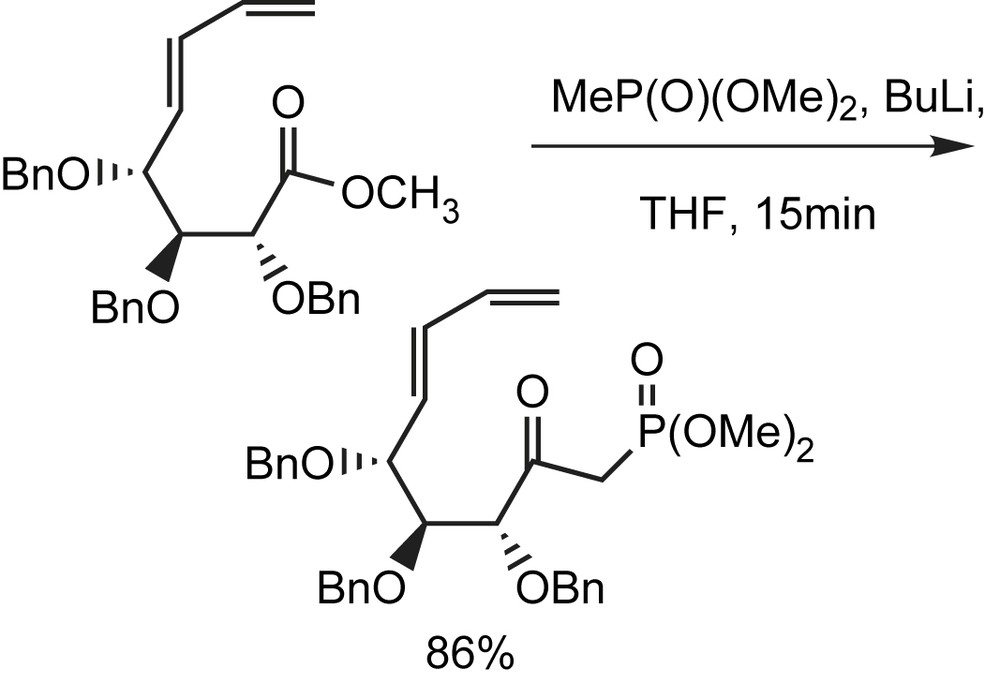
Ces phosphonates sont habituellement plus réactifs vis-à-vis des aldéhydes de sucres et peuvent être préparés avec des rendements beaucoup plus grands (86%) que les phosphoranes correspondants.
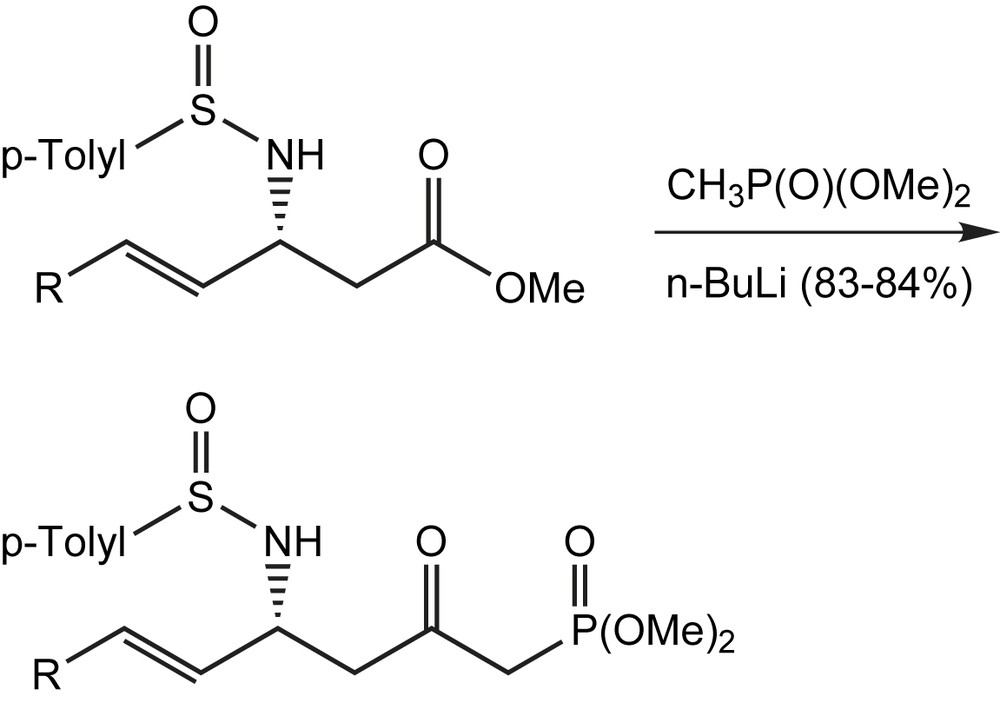
Le traitement des (SS,R)-(+)-N-sulfinyl β-amino esters avec le diméthyl lithiométhylphosphonate conduit aux (SS,R)-(+)-N-sulfinyl δ-amino-β-cétophosphonates [10] avec des rendements excellents (Schéma 3) et constitue un autre exemple d'acylation par les esters.
Dans ce même contexte, l'acylation des héréocycles pentavalents de configuration cis, par l'acétate d'éthyle, est utilisée pour la préparation des β-cétophosphonamidates [11] avec des rendements modérés (38–52%). Le produit de départ qui n'a pas réagi, a été facilement séparé par chromatographie sur colonne. L'acylation du phosphonamidate benzylique (R1 = Ph) conduit uniquement à la forme énolique (Schéma 4).

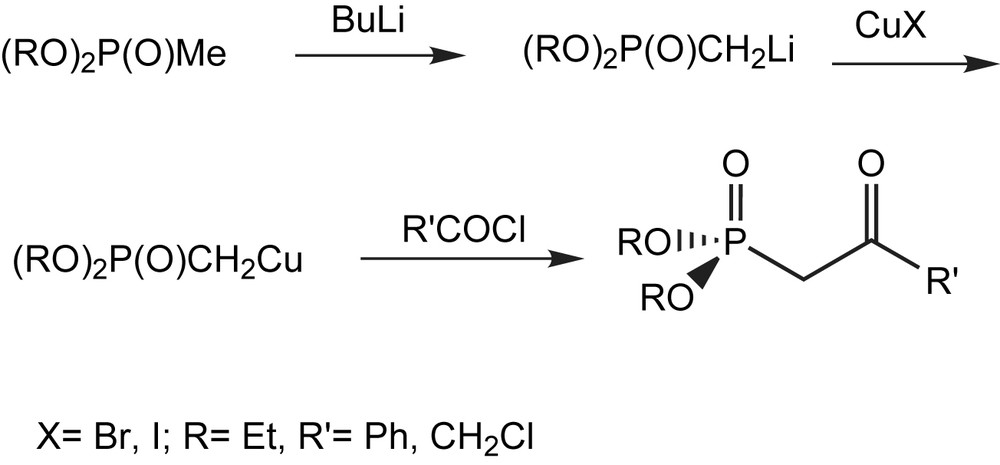
Afin d'atténuer la réactivité des dialkyl α-lithioalkylphosphonates, ils sont convertis quantitativement par transmétallation en dialkyl α-cupro[I] alkylphosphonates par traitement avec CuI [12] (Schéma 5).
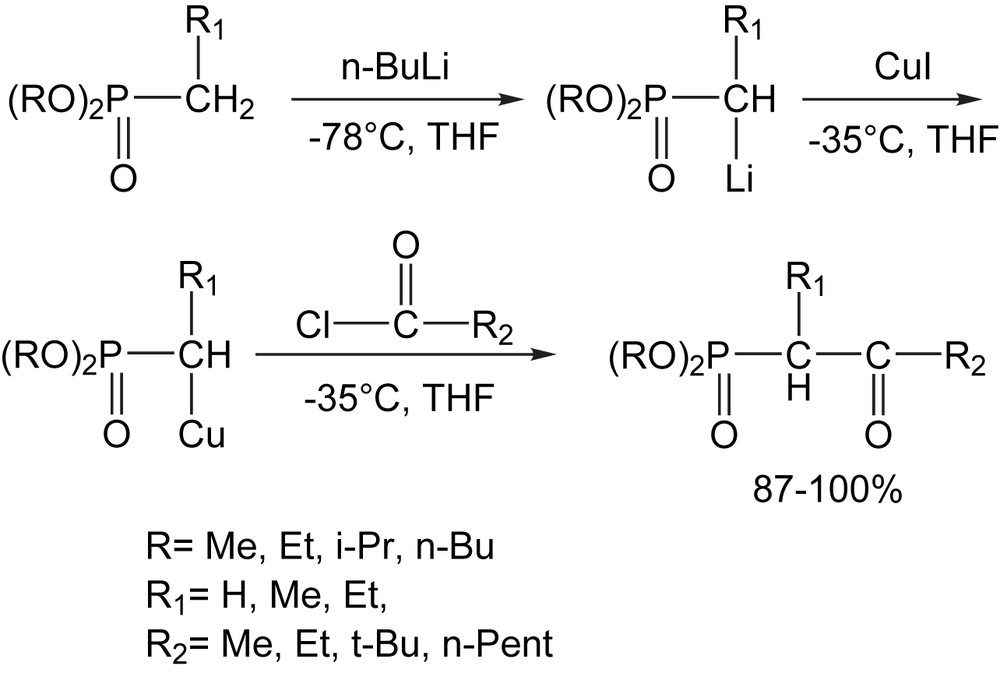
Le bromure cuivreux (CuBr), a été aussi utilisé pour la préparation du diéthyl 3-chloro-2-oxopropylphosphonate [13–15], un précurseur pour la synthèse de (R)-phosphocarnitine (Schéma 6).
Un autre exemple concernant l'utilisation de ce type de sels (CuI), concerne la synthèse de la méthylenomycine A. L'ester méthylique du depoxy-4,5-dihydro-méthylenomycine présente une activité inhibitrice contre les bactéries gram-positives et gram-négatives. Balczewski et coll. décrivent une nouvelle synthèse totale des composés ci-dessus en passant par le β-céto-phosphonate diéthylique [16]. Une séquence en plusieurs étapes pour aboutir à la molécule cible est représentée dans le Schéma 7.
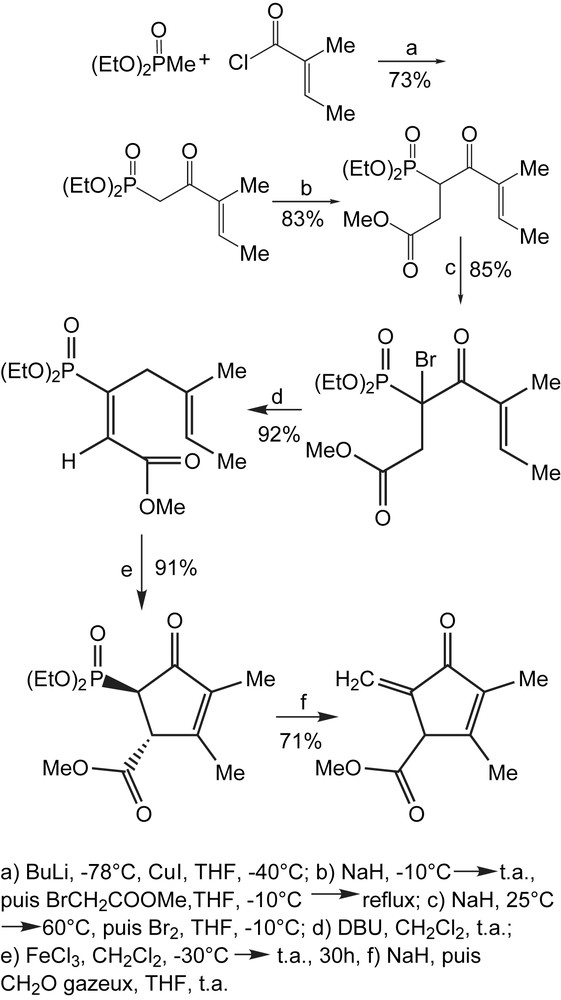
Le β-cétophosphonate obtenu dans la première étape se présente comme un mélange d'isomères E et Z dans le rapport 22/1. L'isomère E du sel de sodium est traité par le bromoacétate de méthyle pour conduire au β-cétophosphonate α-substitué. Une simple bromation du carbanion issu de ce dernier, suivi d'une déhydrobromation avec le DBU conduit à la diénone, un intermédiaire clé car dans cette stratégie la construction de la cyclopentènone peut être accomplie via la réaction de Nazarov.
La synthèse totale de (+)-Amphidinolide W, un nouveau macrolide à 12 chaînons [17] (le premier et l'unique dans son genre qui ne possède pas d'unité exométhylénique et qui fait partie d'une famille connue pour leurs propriétés antitumorales significatives), nécessite la connection de deux fragments plus simples, à savoir le fragment C1–C9 et le fragment C10–C20. La construction du fragment C1–C9 fait intervenir un β-cétophosphonate, et illustre un exemple d'acylation par les amides (Schéma 8).

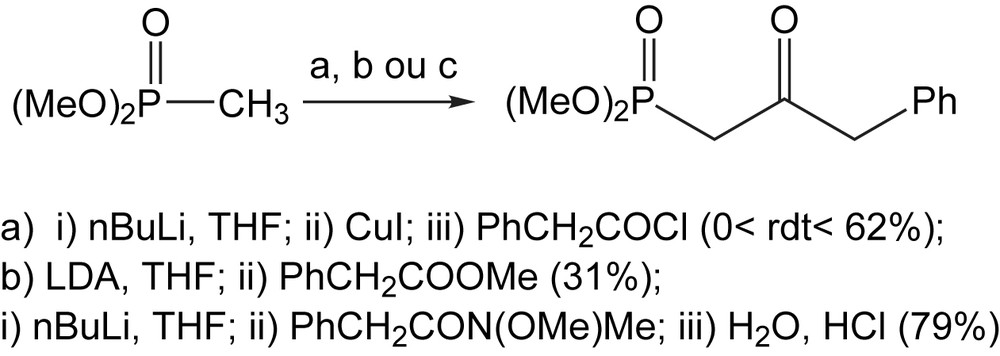
La synthèse du diméthyl (2-oxo-3-phényl) propane-phosphonate requis pour la synthèse des C-glucosinolates (famille de thiosucres naturels), représente un cas d'acylation réalisée par les trois méthodes. La très faible reproductibilité de cette réaction avec le chlorure de phénylacétyle (62°%), conduit à essayer le procédé impliquant l'utilisation du phényl acétate de méth-yle, mais cette dernière réaction a également été décevante (31%). Le remplacement de l'ester par l'amide de Wienrb correspondant permet enfin d'obtenir des β-céto phosphonates avec de bons rendements [18] (Schéma 9).
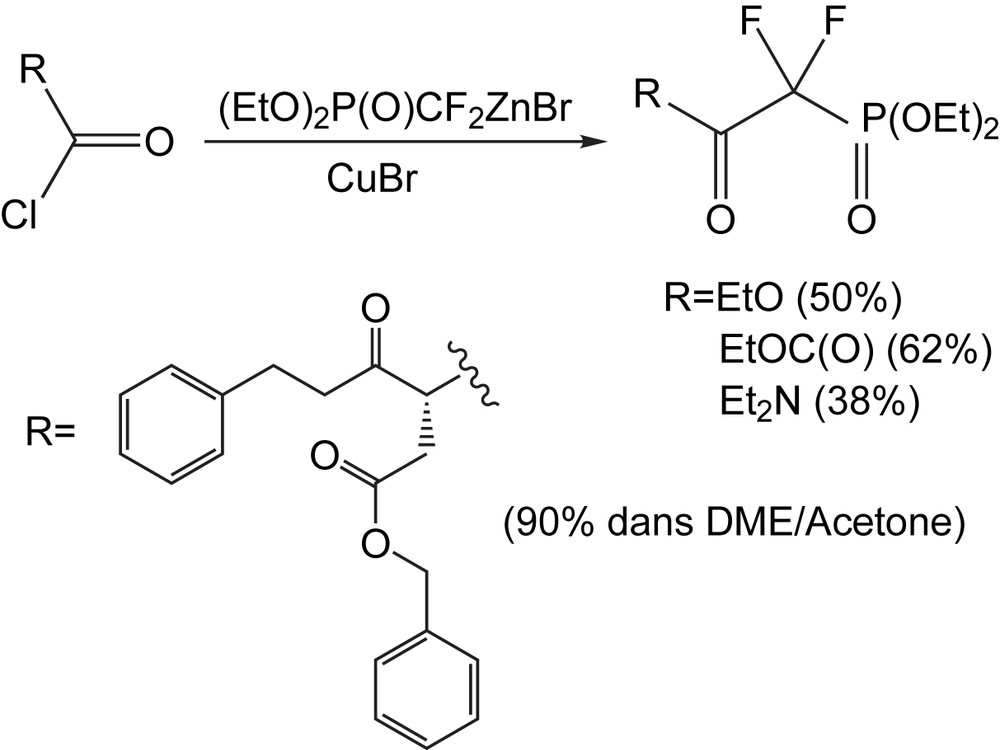
L'acylation par la combinaison d'un organozincique avec des chlorures d'acyle, est un travail pionnier réalisé par Burton et coll. qui démontre qu'un éventail d'α,α-difluoro-β-cétophosphonates peut être synthétisé. Cependant, le chloroformate d'éthyle et le diéthylcarbamoyle ne réagissent pas avec ce réactif. Néanmoins, il a été constaté par la suite que la réactivité de l'organozincique peut être augmentée par catalyse au bromure cuivreux et qu'une telle catalyse permet de donner des réactions efficaces même avec les halogénures d'acyle non réactifs. L'aspect remarquable de cette réaction est sa tolérance élevée vis-à-vis du groupe fonctionnel, comme le schématise la synthèse d'un analogue difluorométhylénique du β-aspartyl phosphate (Schéma 10) [19].
2.1.3 Oxydation
L'azaspiracide est une famille de composés identifiés pour la première fois en 1995 en Hollande. Ce sont des agents responsables d'intoxications humaines liées à la consommation des mollusques et crustacés. Le β-cétophosphonate requis pour la synthèse de cette molécule, est obtenu par un procédé en plusieurs étapes dont l'étape cruciale consiste à utiliser une oxydation dans les conditions de Dess–Martin pour isoler le produit avec un rendement de 81% (Schéma 11) [20].
2.1.4 Substitution par les hétéroatomes, alkylation et cyclisation
Concernant les β-cétophosphonates substitués en position α par un hétéroatome, Rodrigues et coll. [21] décrivent une méthode simple pour la synthèse du diéthyl 1-méthylthio-2-oxoaryléthyl-phosphonate à partir du diéthyl méthylthiométhyl-phosphonate, avec des rendements modérés, comme le montre le Schéma 12.

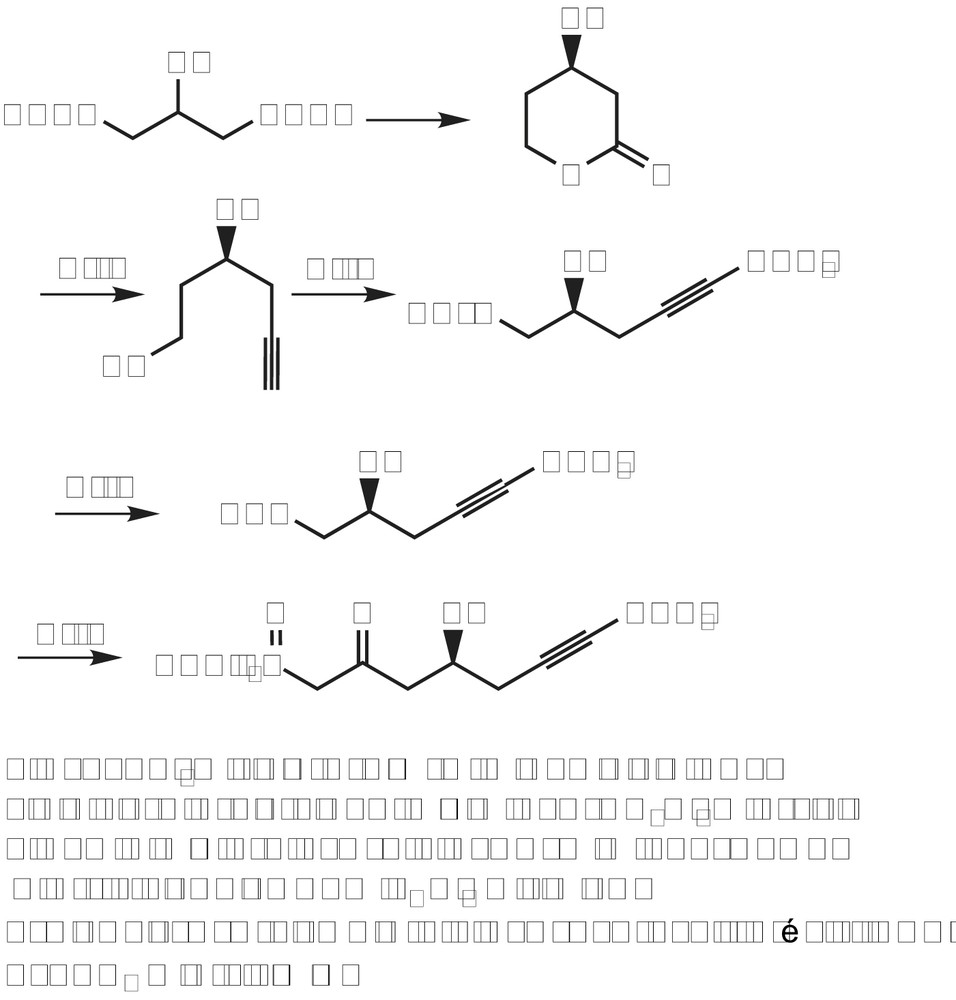
Les β-cétophosphonates cycliques ne sont pas facilement accessibles par les méthodes classiques; pourtant ils peuvent servir comme intermédiaires clés pour la synthèse de nombreux produits d'intérêt. Dans ce contexte, la Sarkomycine est connue pour son effet inhibiteur puissant sur des tumeurs d'ascite d'Ehrlich chez les souris. En raison des activités biologiques liées à la Sarkomycine et à quelques analogues, beaucoup de synthèses de ces cibles ont été rapportées en tant que synthèses multi-étapes avec des rendements globaux faibles. A cet effet, Samarat et coll. décrivent une synthèse courte à grande échelle, des esters et dérivés de (±)-Sarkomycine comprenant un β-cétophosphonate cyclique [22] précurseur de l'homosarkomycine (Schéma 13).
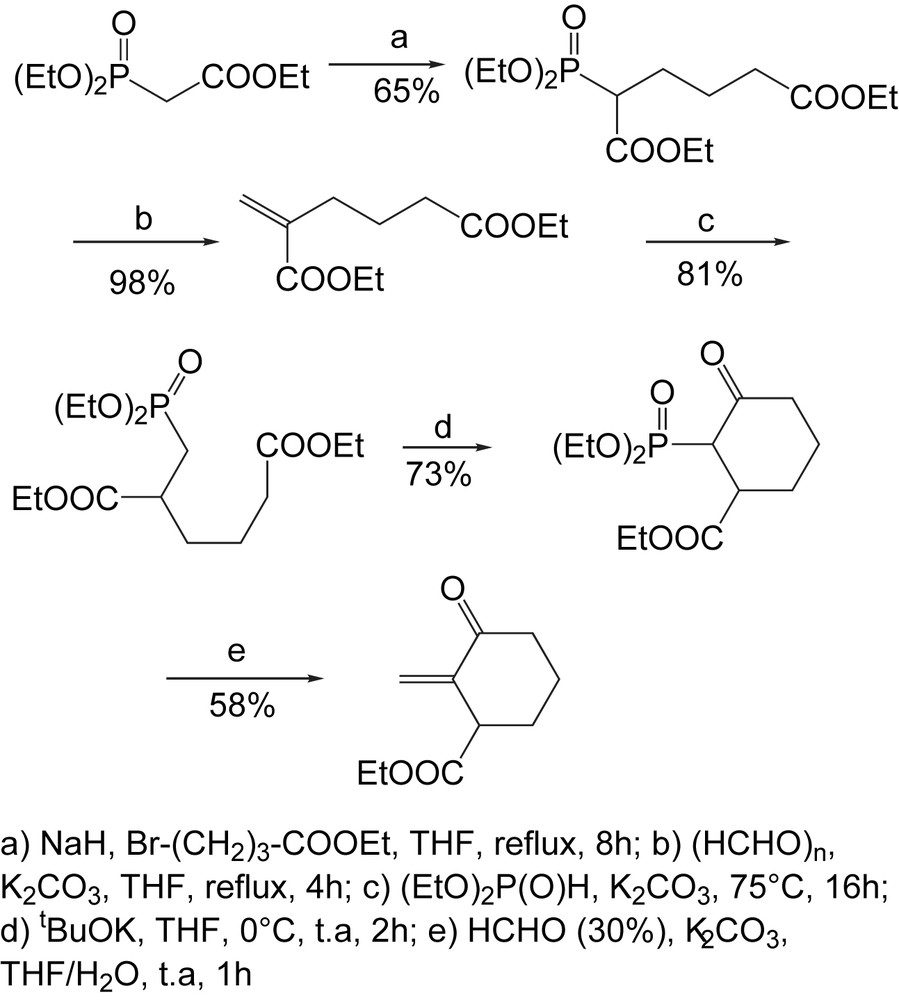
Cette méthodologie a été appliquée à la synthèse de l'ester éthylique analogue de (±)-bis-homo-sarkomycine, cycle à sept chaînons [23]. La cyclisation de type-Dieckman du phosphonate utilise l'hydrure de sodium comme base dans le DME anhydre pour conduire à un mélange de deux diastéréoisomeres du β-cétophosphonate avec 78% de rendement (Schéma 14). L'introduction de la partie exocyclique a été alors effectuée par l'intermédiaire de la réaction de Horner–Wadsworth–Emmons (H-W-E) dans un milieu hétérogène en présence du formaldéhyde aqueux (30%) et d'une solution concentrée de carbonate de potassium (10 M) dans le THF, pour donner l'ester éthylique (±)-bis-homosarkomycine avec un rendement modéré (51%).
2.2 Les α-cétophosphonates
2.2.1 Synthèse des α-acylphosphonates insaturés
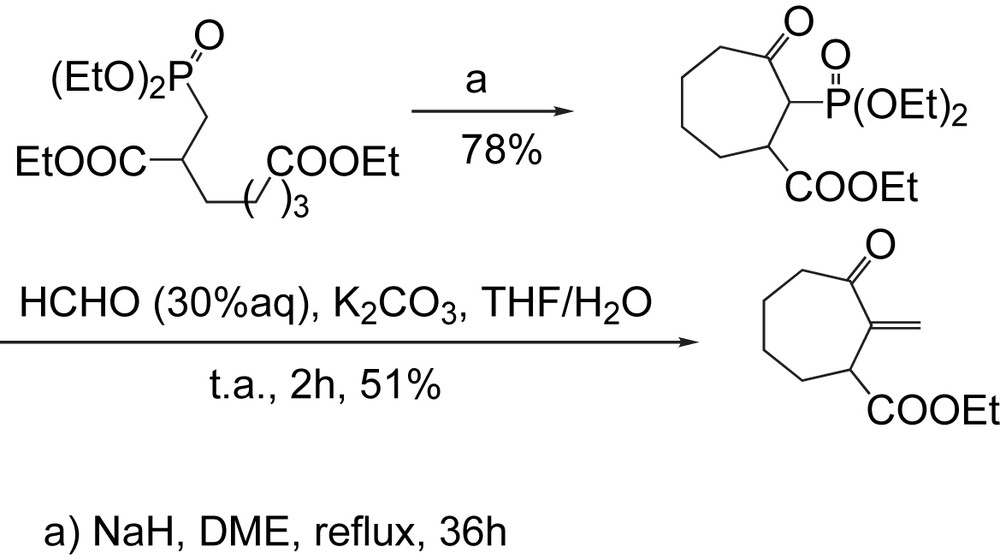
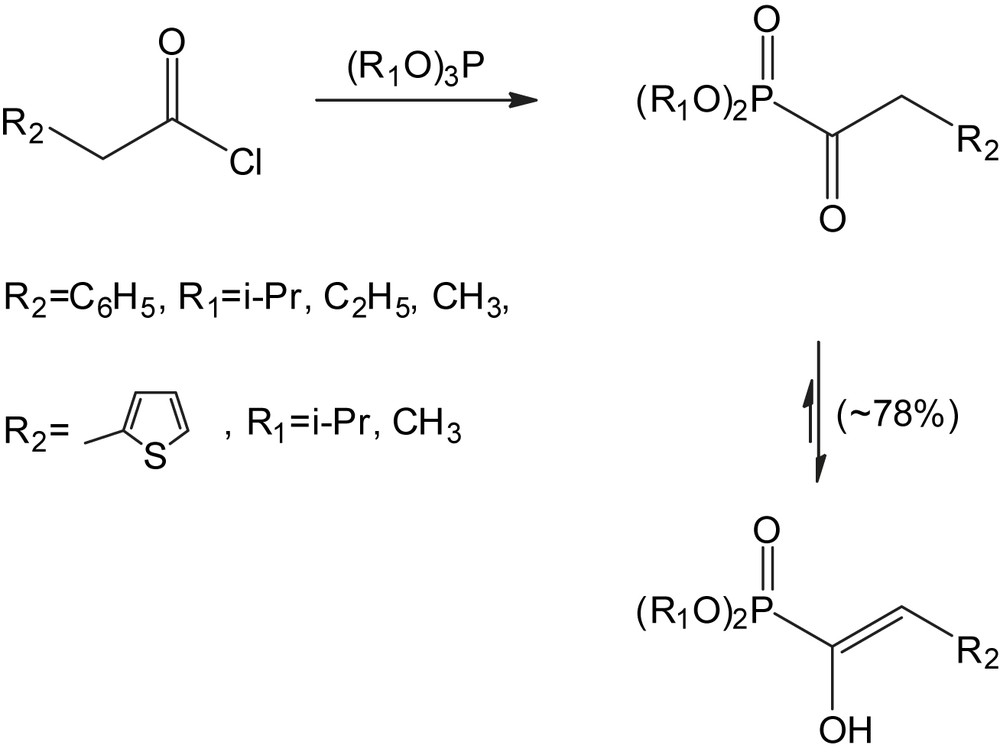
La réaction de Michaelis–Arbuzov est la méthode classique la plus utilisée pour la préparation des α-acylphosphonates. Cette réaction met en jeu un trialkylphosphite et un chlorure d'acide. Elle donne de bons résultats avec les chlorures d'alcanoyle et aroyle.
Les différentes versions de cette réaction, qui se distinguent par la nature de l'agent d'acylation et de la structure du phosphite, ont été couvertes récemment par la revue de McKenna et de Kashemirov en 2002 [24]. Plus récemment, cette réaction a été appliquée avec succès aux chlorures d'acyle insaturés [25,26] (Schéma 15).
Le (E)-diméthyl-1-oxo-2-butènoylphosphonate peut être préparé avec un rendement de 53% par l'addition lente du triméthylphosphite à un excès de chlorure-(E)-2-butènoyle.
Par contre, avec des quantités équimolaires de triméthylphosphite et de chlorure–(E)-2-butènoyle, le produit principal est l'ester trans-2-butènoyl diphosphonylé.
Le traitement d'un certain nombre de chlorures d'acides α,β-insaturés avec le triméthylphosphite fournit les acylphosphonates avec des rendements variables (Schéma 16) après distillation sous pression réduite [27].
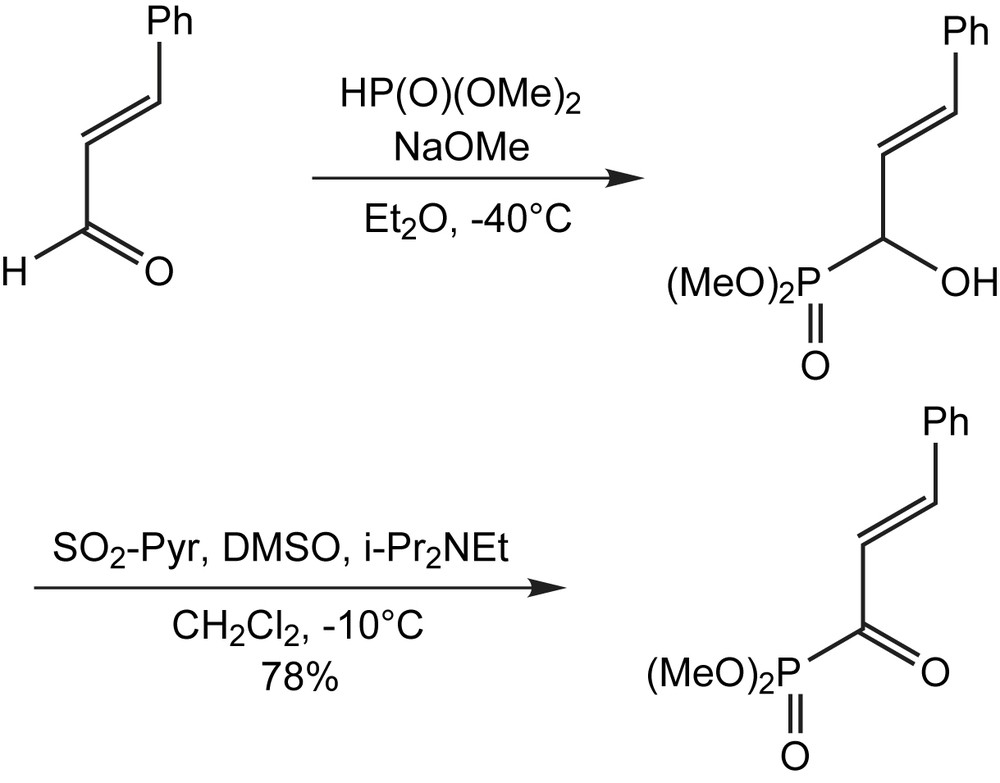
Les tentatives d'utiliser la réaction d'Arbuzov pour synthétiser les acylphosphonates dérivés d'acides cinnamiques se sont révélées sans succès ; par contre, le recours à un procédé en deux étapes a été efficace. La première consiste à ajouter le diméthylphosphite au cinnamaldéhyde en présence d'une base. L'alcool allylique résultant est oxydé suivant les conditions de Parikh–Doering (Schéma 17) [27].
2.2.2 α-Acylphosphonates saturés
2.2.2.1 Réaction type Arbuzov
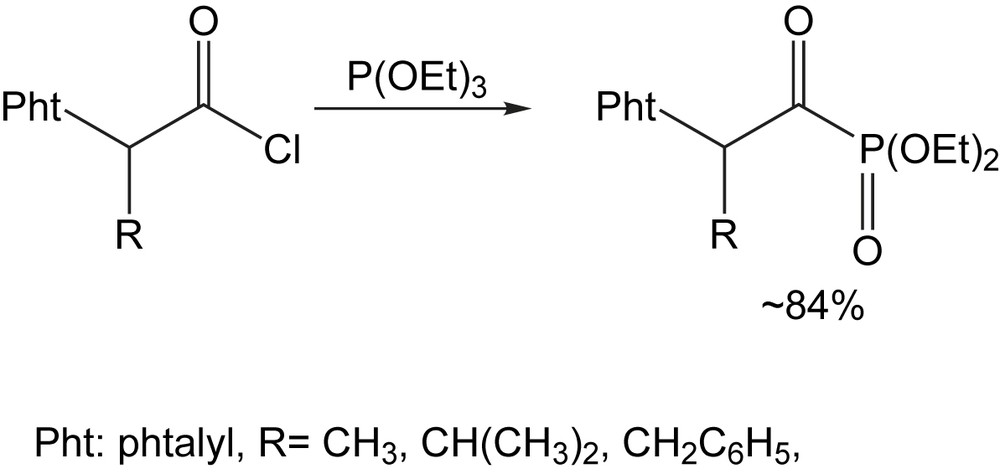
Les réactions du triéthyl phosphite (disponible dans le commerce) avec les chlorures du phtalylglycine, N-phtalyl-l-alanine, N-phtalyl-l-valine, N-phtalyl-l-leucine et N-phtalyl-l-phénylalanine conduisent à des mélanges complexes de plusieurs produits. Ceci peut être évité par distillation du triéthyl phosphite sur sodium, avec comme conséquence la formation des diéthyl 1-oxo-2-(N-phtalylamino) alcane-phosphonates [28] correspondants avec des rendements et puretés satisfaisants (Schéma 18).
Dans l'éther, le tétrahydrofurane ou le benzène, les chlorures d'arylacétyle [29] réagissent à 0 °C pour conduire aux α-cétophosphonates. L'analyse spectroscopique IR et RMN (1H, 13C, 31P) de ces composés montre l'existence d'un mélange de deux tautomères cétonique et énolique (Schéma 19). L'encombrement stérique du motif phosphoré influe sur la proportion relative de la forme énolique. Cette proportion décroit en allant du groupe méthyle (96%) à l'éthyle (88%) et à l'isopropyle (80%).
La phosphorylation des chlorures de l'acide 1-méthyl-2,2-dichloro-cyclopropanecarboxylique avec des dérivés du phosphore tri- et tétravalent, permet la formation d'un nouveau type de phosphonates contenant des cyclopropanes [30] (Schéma 20). Cette nouvelle classe de produits s'avère augmenter la germination en laboratoire des céréales, légumes et graines végétales.

2.2.2.2 Oxydation par les chromates
Contrairement au grand nombre de méthodes existantes pour la conversion des alcools en composés carbonylés, peu de méthodes sont connues pour la préparation des α-céto-phosphonates diéthyliques à partir des α-hydroxy-phosphonates. L'oxydation constitue une méthode alternative à celle habituellement utilisée (Réaction ole Michaelis–Arbuzov) pour la synthèse des acylphosphonates, puisqu'elle débute à partir des α-hydroxyphosphonates facilement accessibles et stables. L'oxydation par les réactifs CrO3/Al2O˙3 [31,32] exige des temps de réaction longs, un rapport molaire élevé pour l'oxydant/substrat, ou un traitement spécial pour l'activation des réactifs. De nouvelles méthodes pour la préparation des phosphonates α-fonctionels à partir des diéthyl α-hydroxyphosphonates ont été publiées ces dernières années. L'utilisation de plusieurs oxydants tels que le chromate de pyridinium (PCC), le dichromate de nicotinium (NDC), le dichromate de pyridinium (PDC) et le dichromate d'isonicotinium (IDNC) ont permis un accès facile aux α-cétophosphonates, à partir des, 1-hydroxyalkyl-, benzyl-, furylméthyl-, naphtylméthyl-, crotyl-, et cinnamylphosphonates (Schéma 21) [33–38]. L'absence de sous-produits dû au clivage de la liaison C(O)–P, le traitement facile de la manipulation ainsi que les temps de réaction relativement courts sont les grands intérêts de cette méthode.
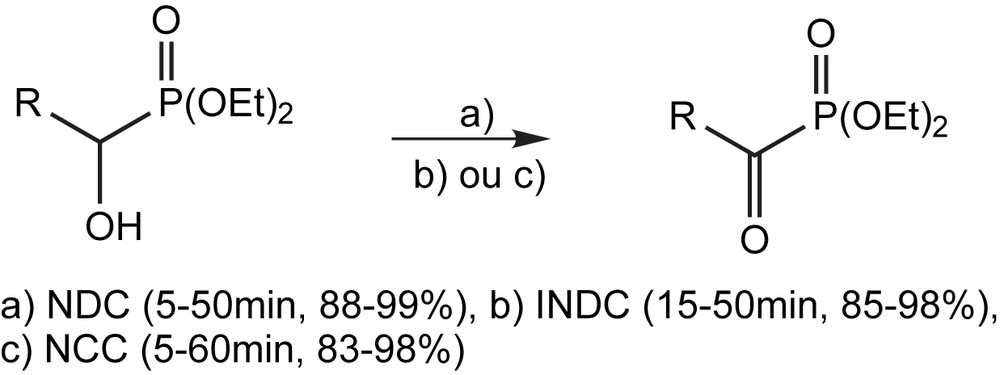
Le système oxydant constitué de bichromate de pyridinium et de triméthylchlorosilane est utilisé pour préparer les α-cétophosphonates par un procédé « one pot » en deux étapes [39,40]. Cette procédure consiste à préparer les α-hydroxyphosphonates par l'action des dialkyl phosphites sur les aldéhydes suivi de leur conversion en cétophosphonates correspondants (Schéma 22). La pureté chimique ainsi que le rendement des cétophosphonates obtenus sont considérablement élevés (90–100%) de sorte qu'ils sont directement engagés dans d'autres transformations sans aucune purification.
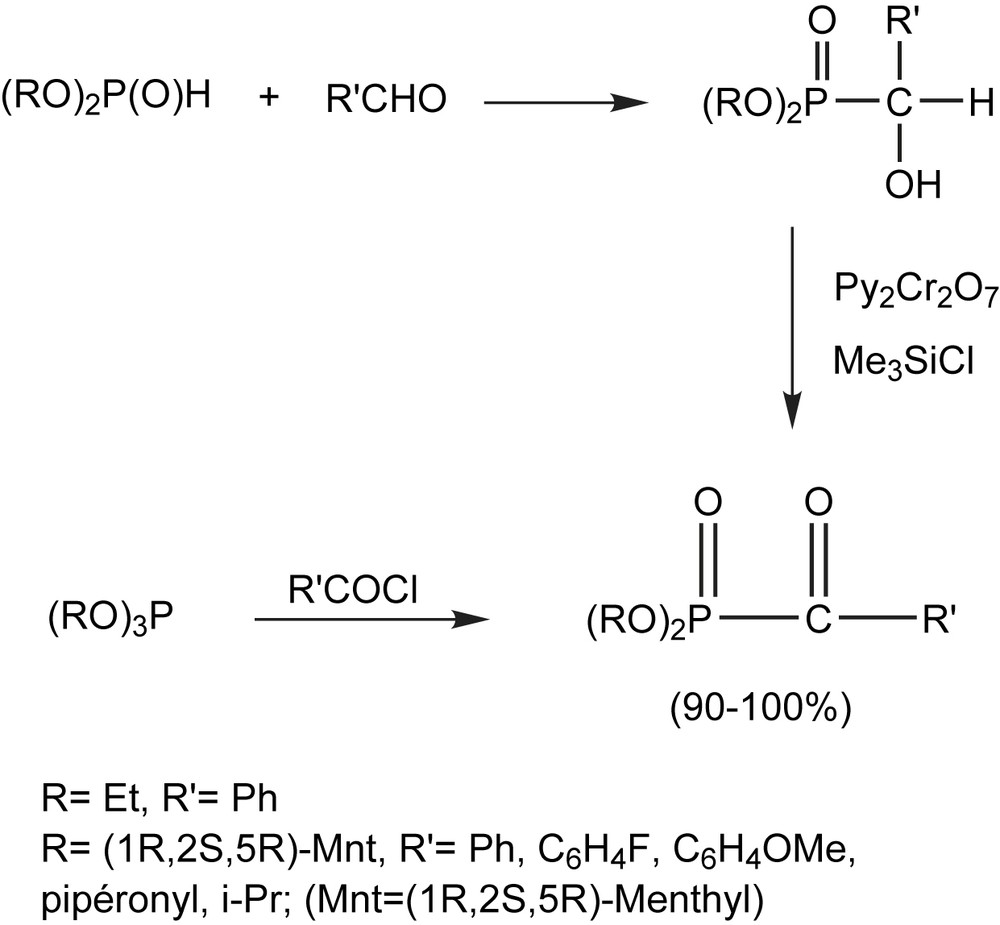
Par la méthode classique, l'action du chlorure de 4-fluorobenzoyle avec le trimenthyl phosphite (Mnt) dans le toluène procède lentement à température ambiante pour donner le cétophosphonate avec un rendement de 80%.
L'utilisation de matériaux en phase solide en tant que catalyseurs basiques a été développée avec succès ces dernières années. Des systèmes font intervenir l'addition de KF ou de CsF sur le mélange du dérivé carbonylé et du dialkyl phosphite (Schéma 23). Un tel procédé est très flexible et s'adapte à une grande variété de composés carbonylés pour produire des α-hydroxy-alkylphosphonates dialkylles. L'oxydation de l'hydroxy-phosphonate dans les conditions de Dess–Martin, conduit à l'α-oxophosphonate [41], qui sera engagé directement pour la fluoration en α,α-difluorophosphonate.

2.2.2.3 Oxydation par des complexes chiraux
Récemment, une nouvelle stratégie sur l'oxydation aérobie du dibenzyl phénylhydroxyméthylphosphonate a été développée (Schéma 24). Elle utilise des complexes chiraux de méthylate de vanadyle (V) porteur de N-salicylidène-α-aminocarboxylates, comme catalyseurs modèles. Les conditions optimales sont obtenues avec le 3,5-dibromo analogue du méthylate de Vanadyle(V). Ce protocole fonctionne bien pour une série d'α-aryl et α-hétéroaryl-α-hydroxyphosphonates et s'annonce prometteur pour des applications potentielles en chimie biomédicale [42].

2.3 Autres cétophosphonates
2.3.1 Extension de la chaîne
Les plus récentes approches synthétiques des γ-cétophosphonates consistent à l'extension de chaîne des β-cétophosphonates, par l'intermédiaire d'une réaction avec l'éthyl-(iodométhyl)-zinc, en une seule étape [43]. Une étude comparée avec les β-cétoesters montre que la transformation de ces derniers nécessite quelques minutes seulement. Par contre, la transformation des β-cétophosphonates n'est complète qu'après 2 h de réaction, avec 3 équiv. d'éthyl-(iodométhyl)-zinc et une température de 0 °C (Schéma 25).

La conversion des β-cétophosphonates est optimisée avec 6 équiv. aussi bien pour le diéthyl zinc que l'iodure de méthyle, à température ambiante. Le produit de réaction est unique et remarquablement propre.
Une variété de β-cétophosphonates peut être convertie en γ-cétophosphonates selon cette méthode. La présence des substituants alkyle en α des groupements fonctionnels, tels qu'une (base de Lewis) ainsi que l'acidité modérée des protons NH sont bien tolérées. Pour conclure, la simplicité, l'étendue, et l'efficacité de cette méthode, font d'elle une alternative attrayante par rapport aux méthodes établies pour la formation des γ-cétophosphonates.
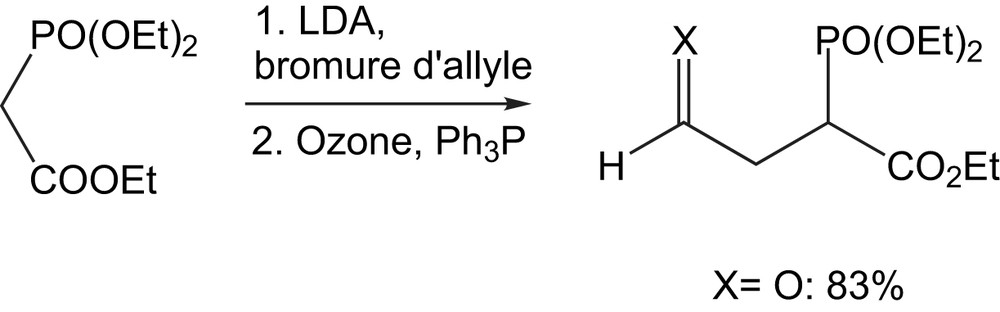
2.3.2 Ozonolyse des γ-alcénylphosphonates
Dans le même contexte et pour étudier le comportement de la réaction des énolates d'γ-cétophosphonates [44], Kraus et coll. ont eu recours, à l'ozonolyse du γ-alcénylphosphonate pour isoler le produit avec un rendement de 83% (Schéma 26).
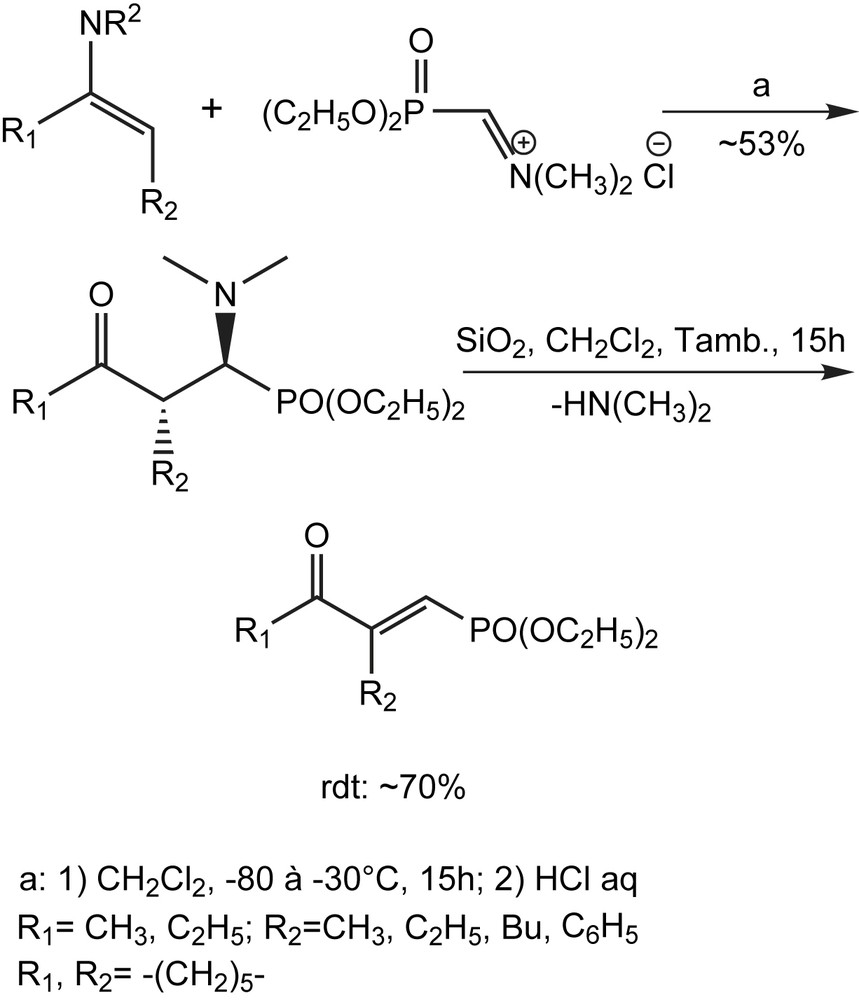
2.3.3 Utilisation d'une réaction type-Mannich
Les γ-cétophosphonates vinyliques peuvent être obtenus à partir des bases de Mannich phosphonatées très peu décrites dans la littérature. Les procédures utilisées pour préparer ces derniers, manquent malheureusement de généralités. Risch et coll. décrivent une méthode générale hautement diastéréosélective de synthèse des α-aminophosphonates diversement substitués [45]. Elle consiste en l'aminoalkylation d'énamines dans des conditions douces. Les bases de Mannich obtenues éliminent facilement le groupe N,N-diméthylamino pour donner des phosphonates vinyliques de configuration E (Schéma 27).
2.3.4 Utilisation des 1,3-dicétones trifluorométhylées
Pour accéder aux γ-cétophosphonates fluorés [46], on a recours aux 1,3-dicétones trifluorométhylées (Schéma 28) par une réaction avec le chlorodiéthylphosphite en présence d'une base.
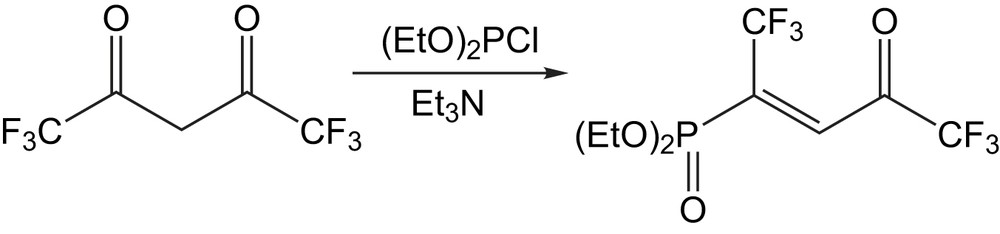
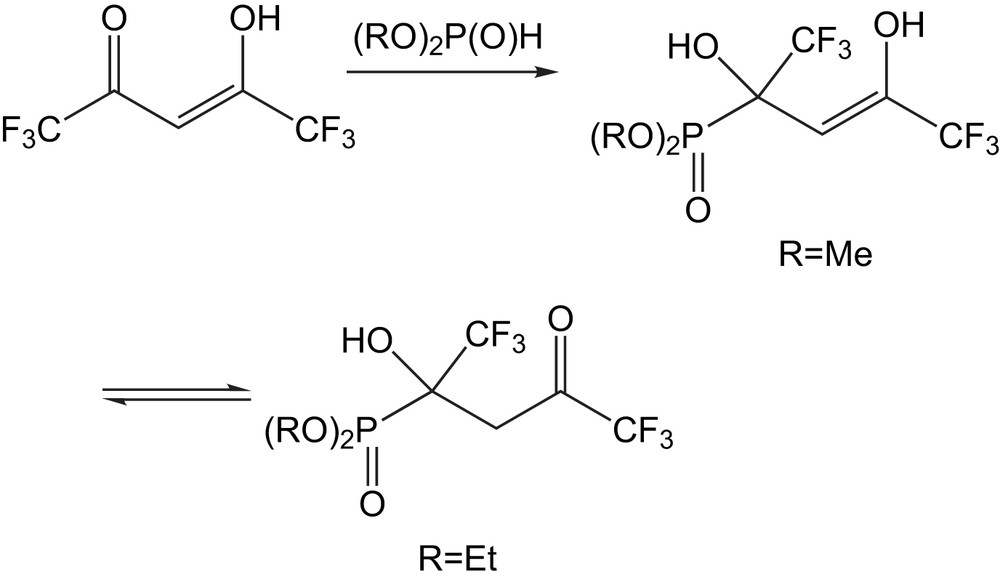
Cette dicétone sous sa forme tautomère réagit avec le dialkylphosphite (R = Me, Et,) pour donner le (Z)-énolphosphonate (R = Me) ou les cétophosphonates respectifs (R = Et) respectivement (Schéma 29).
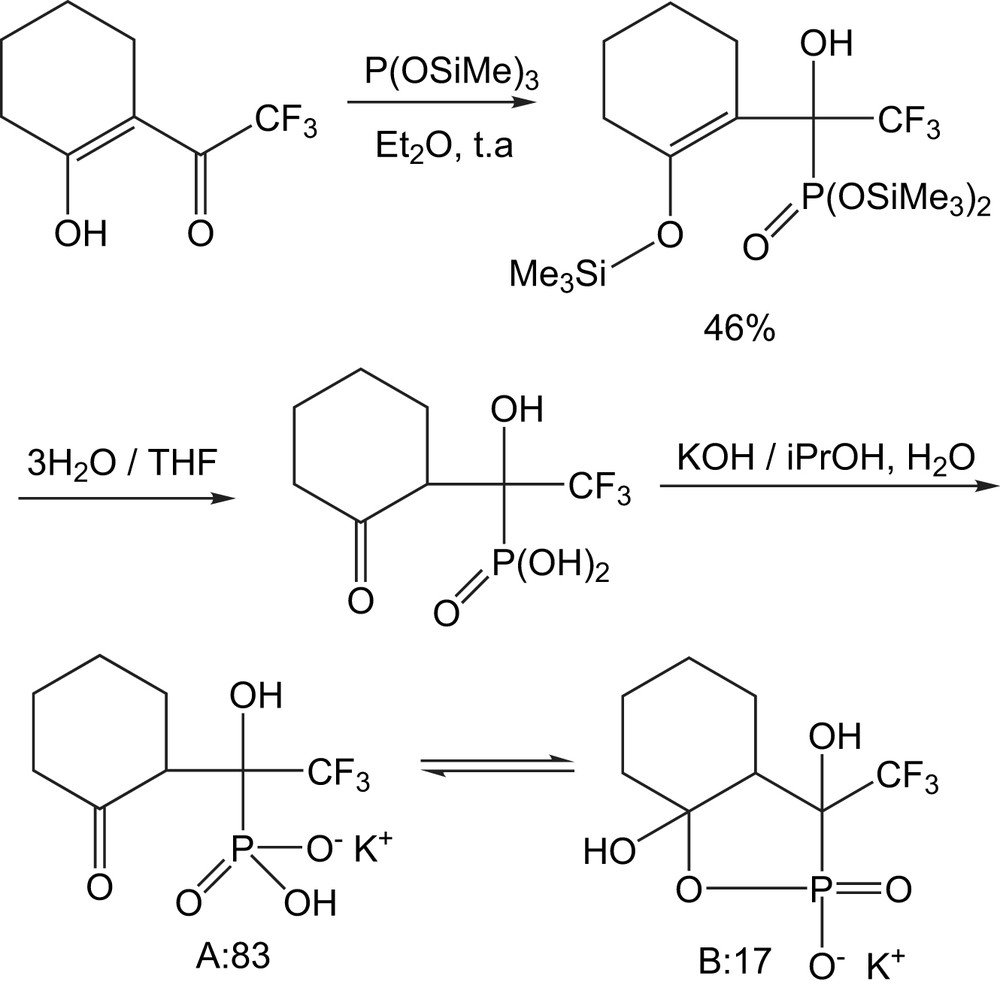
Le 2-trifluoroacétyl cyclohexanone et le phosphite de tris(triméthylsilyle) réagissent à température ambiante pour donner un solide incolore avec 46% de rendement (Schéma 30). Le phosphore nucléophile s'additionne au carbone cétonique du groupement CF3CO suivi de la migration du groupe triméthyl- silyle. L'hydrolyse de cet intermédiaire conduit à la formation de l'acide γ-cétophosphonique [46], qui est isolé et caractérisé comme sel de potassium. Les spectres RMN du 31P et 19F, en solution dans le DMSO-d6 montrent la présence de deux tautomères A et B (A:B = 83:17).
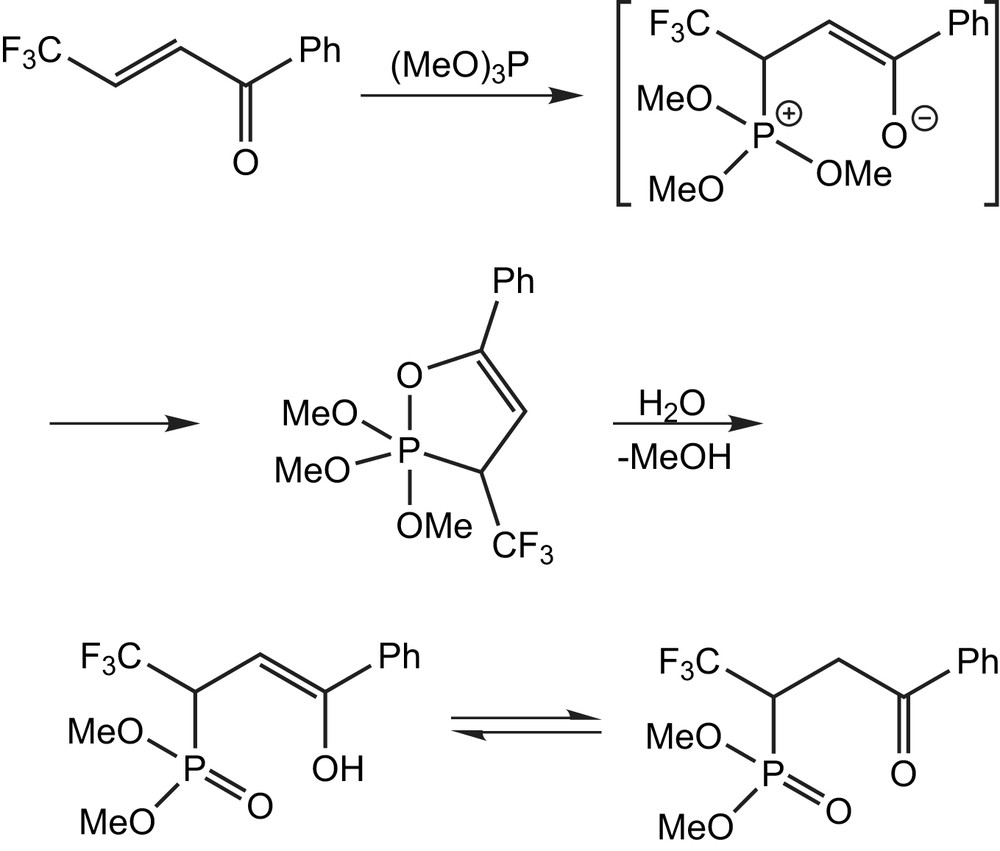
2.3.5 Utilisation des cétones α,β-insaturées
Les cétones α,β-insaturés trifluorométhylées réagissent avec le triméthylphosphite pour former le 1,2λ5σ5-oxaphosphol-4-ène [46], un produit unique avec un rendement presque quantitatif, sous forme de liquide, sensible à l'humidité (Schéma 31). Puisque la cétone existe comme tautomère énolique pur de configuration trans, un seul isomère d'oxaphosphole est obtenu stéréosélectivement par addition [4 + 1] et l'attaque initiale du phosphore sur l'atome de carbone en β est suivie d'une cyclisation. L'addition de l'eau mène à la forme énolique du cétophosphonate résultant (Schéma 31).
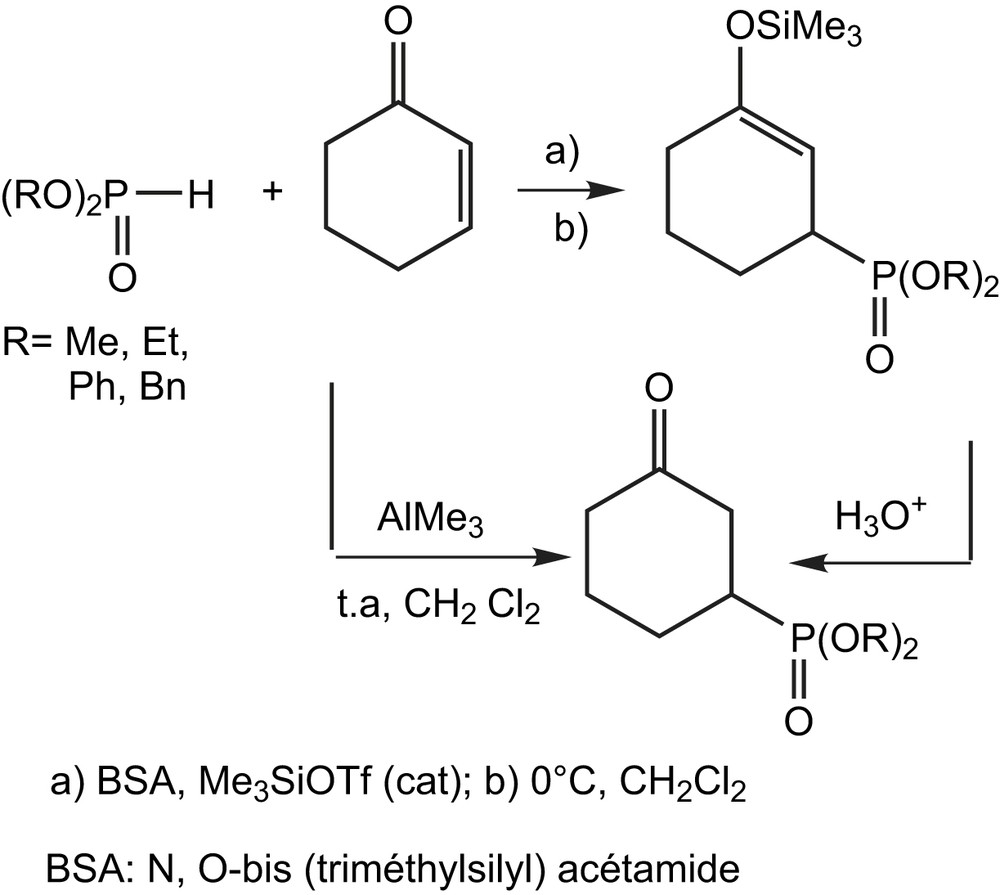
La phosphonosilyation des cétones α,β-insaturées est la méthode la plus utilisée pour la préparation des dialkyl γ-cétophosphonates cycliques [47]. L'utilisation catalytique ou stoechiométrique des acides de Lewis tels que AlMe3 ou Me3SiOTf facilite l'addition conjuguée des dialkyl phosphites (Schéma 32).
2.3.6 Réactions des diénylphosphonates
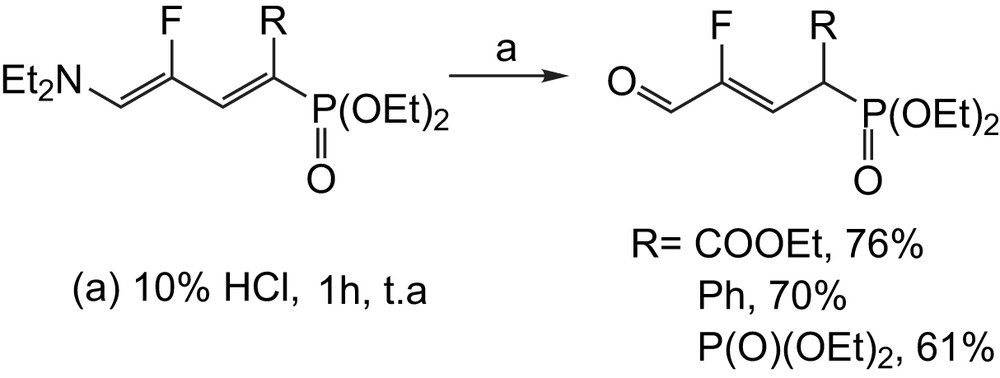
Pour des chaînes plus longues, Arimitsu et coll. [48] ont constaté que l'hydrolyse à température ambiante des 1,3-butadiénylphosphonates par une solution à 10% de HCl, conduit aux oxophosphonates fluorés α,β-insaturés avec de bons rendements (Schéma 33).
2.3.7 Par addition conjuguée énantiosélective
Concernant les additions conjuguées asymmétriques, les phosphonates de vinyle ont été choisis comme accepteurs de Michael bis-activés, en raison de leur accès facile et de leur potentiel pour offrir des intermédiaires synthétiques fonctionnalisés. Sulzer–Mossé et coll. présentent la première addition conjuguée organocatalytique et énantiosélective des aldéhydes aux phosphonates de vinyle [49]. L'évaluation de plusieurs catalyseurs a mené à l'éther silylé (S)-diphénylprolinol (Schéma 34), qui a déjà montré son efficacité pour la réaction de Michael des aldéhydes aux nitrooléfines. Ce catalyseur, qui a été intensivement exploré par Jørgensen dans diverses réactions organocatalytiques [50] et rapporté aussi par Hayashi [51] est aussi utilisé dans cette réaction.

Les γ-gem-phosphonates aldéhydes optiquement actifs sont isolés avec de bons rendements (75–85%) et avec des énantiosélectivities élevés (75–97%). Plus l'aldéhyde est stériquement encombré, plus la stéréoinduction est meilleure. Les meilleurs résultats sont atteints en utilisant le 3,3-diméthylbutyraldéhyde.
2.3.8 Déphosphonylation des β-cétophosphonates
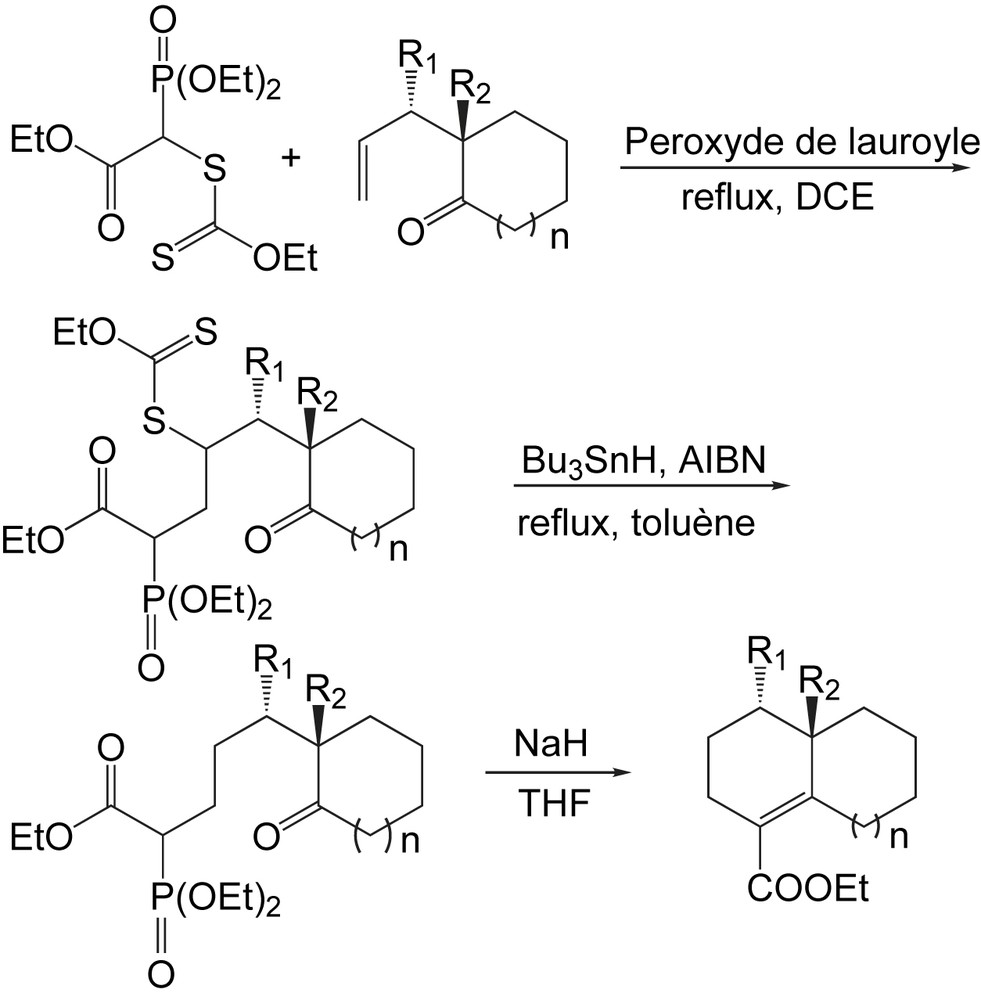
Le chauffage de la 2-allyl-2-méthylcyclohexanone avec un xanthate dérivé du diéthyl 1-(éthoxycarbonyl) méthylphosphonate, en présence de peroxyde de lauroyle comme initiateur dans le 1,2-dichloroéthane, fournit le 6-oxoalkylphosphonate [52] (Schéma 35). Cet exemple illustre l'utilisation des réactions radicalaires dans la synthèse des dialkyl ω-oxoalkylphosphonates. Dans la plupart des cas, la réduction du groupe xanthate est aisément réalisée par traitement avec du Bu3SnH en présence d'AIBN.
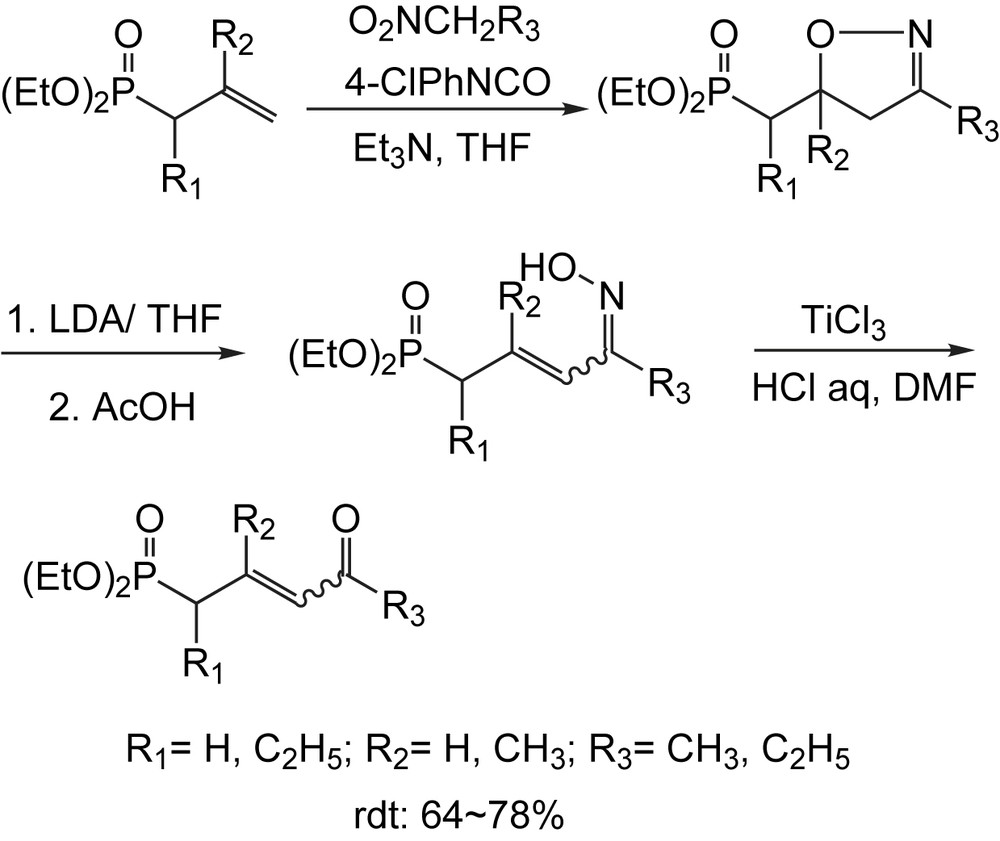
Une longue chaîne polyéthylénique est fréquemment présente dans la structure des composés naturels tels que les rétinoïdes et les caroténoïdes, d'oue l'intérêt considérable pour la construction de phosphonates fonctionnalisé avec de longues chaînes conjuguées. Ce type de précurseurs [53] est illustré par le Schéma 36. La synthèse des 4-oxoalcèn-2-yl-phosphonates a été réalisée via une réaction de cycloaddition avec des phosphonates allyliques et de l'oxyde de nitrile. L'isooxazoline intermédiaire est isolée avec un bon rendement, puis traitée avec le LDA dans le THF et hydrolysée pour donner les γ-phosphonooximes α,β–insaturées. L'oxime ainsi obtenue, est alors soumise à l'action d'une solution de trichlorure de titane dans le DMF en présence d'une solution aqueuse d'acide chlorhydrique pour donner les 4-oxoalcèn-2-yl-phosphonates.
3 Réactivité des oxophosphonates
3.1 Les β-cétophosphonates
3.1.1 Action des imines
La synthèse des α-aminophosphonates est développée par une addition stéréosélective directe des β–cétophosphonates sur une imine activée, en présence d'acides de Lewis chiraux [54]. L'évaluation des différentes imines activées prouve que le N-tosyl-α-imino ester s'additionne aux β-cétophosphonates d'une façon diastéréo- et énantiosélective, lorsque la réaction est catalysée par des complexes chiraux du cuivre (II)-bisoxazoline (Schéma 37).
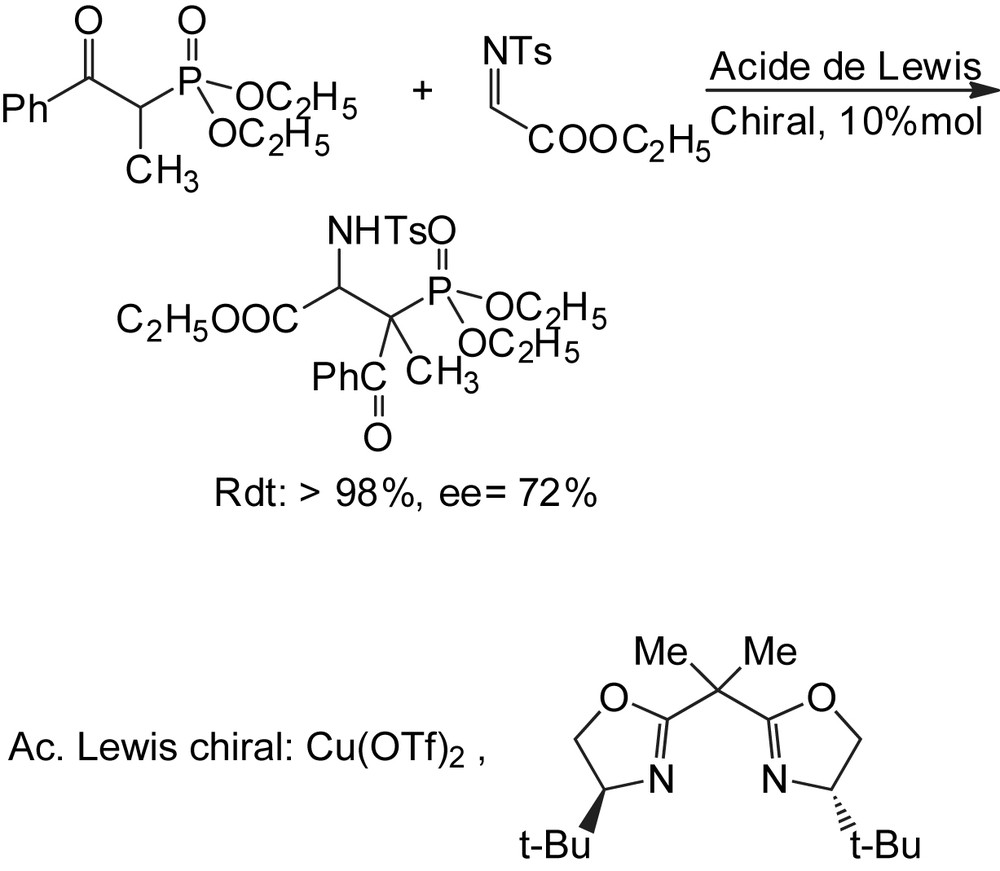
3.1.2 Bioréduction
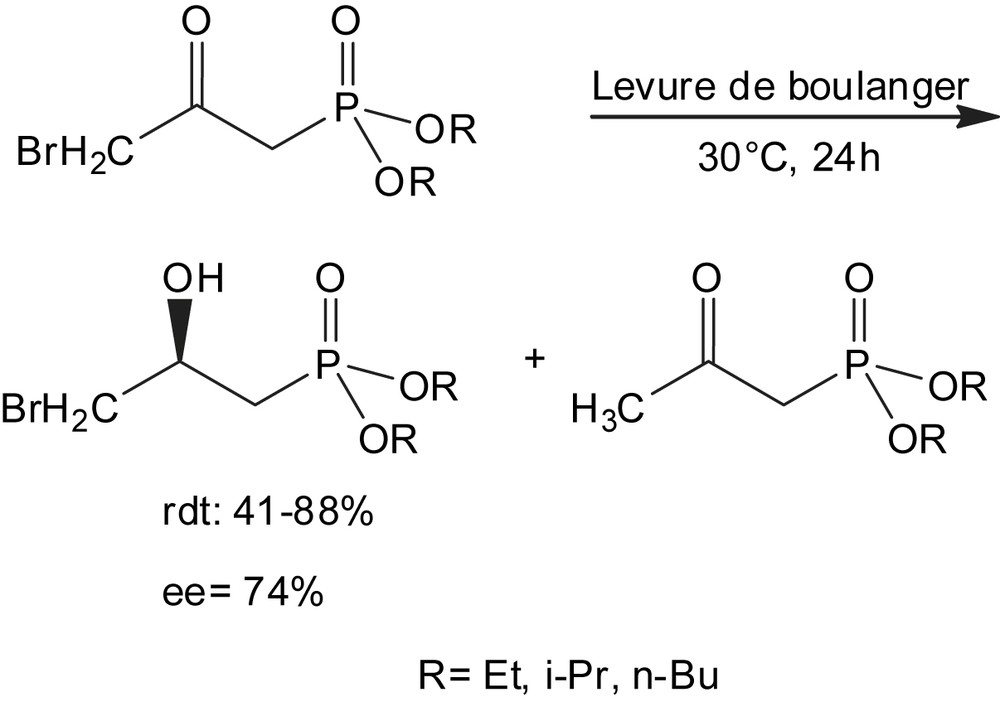
Comme les phosphonates possèdent en général des propriétés biologiques utiles, dont les applications possibles s'étendent de la médecine à l'agriculture, l'évaluation de leur activité biologique exige la disponibilité des composés énantiomériquement purs de configuration définie. Il est donc nécessaire de rechercher de nouvelles méthodes de préparation qui répondent à cet exigence. Dans ce contexte, la réduction asymétrique du diéthyl 2-oxo-alkylphosphonate par la levure du boulanger, qui est un système extrêmement riche contenant des centaines d'enzymes intracellulaires et extracellulaires, a montré une activité catalytique stéréospécifique. L'influence de ce biocatalyseur sur des cétones substituées par un groupe phosphoré a été étudiée en 1995 par Zymanezek-Duda et coll. [55a].
L'efficacité de ce biocatalyseur a incité Yuan et coll. à l'utiliser pour la réduction énantiosélective du 2-céto-3-bromométhyl-phosphonate [55b] (Schéma 38). Le 2-cétopropane-phosphonate a été isolé comme un sous-produit et les rendements en produit désiré varient de 41 à 88%.
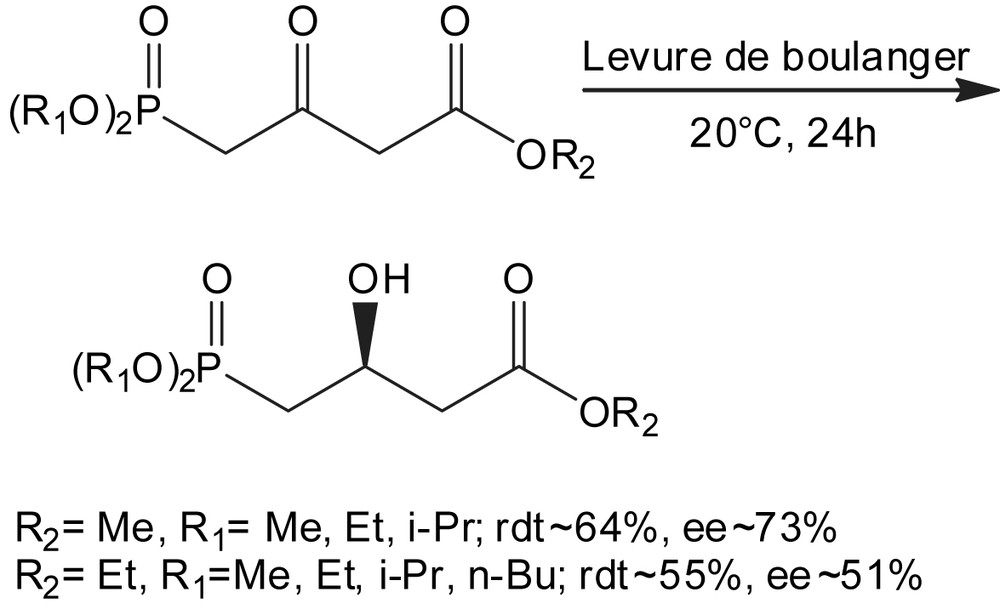
Le comportement bioréducteur de la levure a été étudié avec des dialkyl 4-(dialkylphosphoryl)-3-oxobutanoates ; molécules contenant à la fois des fonctions carboxylate, phosphonate et carbonyle [56]. Dans les mêmes conditions, la stéréosélectivité de la réduction dépend fortement de la structure chimique des 4-phosphoryl-3-oxobutanoates. De plus, la valeur des excès énantiomériques (ee) de cette bioréduction est également influencée par la présence du groupe phosphoryle (Schéma 39).
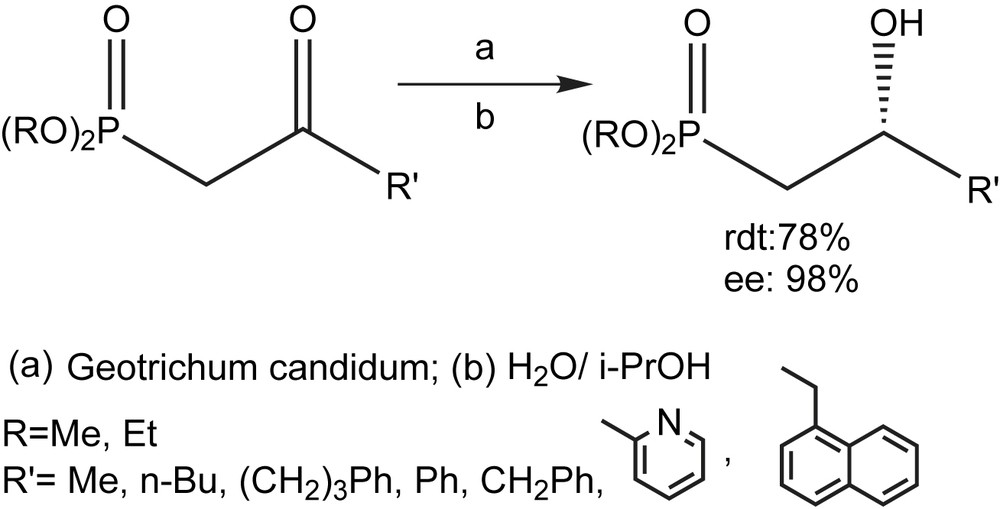
Dans le même contexte, une série de 2-oxoalkyl-phosphonates ont fait l'objet de réduction par une autre levure (Geotrichum candidum) [57]. Cependant, la réduction asymétrique n'a eu lieu qu'avec le diéthyl 2-oxopropanephosphonate pour donner le (+) –(R)-diéthyl 2-hydroxypropanephosphonate avec un ee de 98% (Schéma 40).
La résolution enzymatique des 2-hydroxy-alcanephosphonates racémiques, obtenus par réduction des 2-oxoalcanephosphonates correspondants avec le borohydrure de sodium, constitue une autre voie d'accès. Elle consiste à acétyler les 2-hydroxyalcanephosphonates racémiques par l'acétate de vinyle en présence d'une lipase [57]. Parmi les sept substrats étudiés (Schéma 40), trois seulement (R = Et ; R′ = Ph, R′ = CH2Ph, R′ = 1′-naphtyl) se sont avérés être non réactifs dans les conditions appliquées. Pour le reste, les produits ont été obtenus avec de bons rendements mais avec des excès énantiomériques variés (Schéma 41).
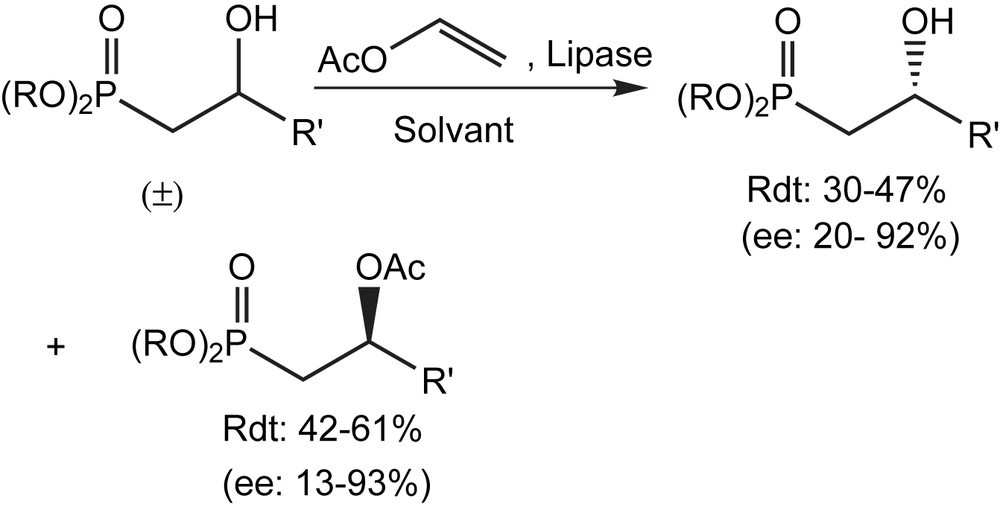
Une autre approche énantiosélective aux δ-hydroxy-β-cétophosphonates est basée sur l'acétylation énantiosélective catalysée par le candida antarctica lipase-B (CALB) et l'hydrolyse énantiosélective catalysée par le candida rugosa lipase (CRL).
Avec l'acétate de vinyle comme agent d'acylation, le rendement de la réaction s'avère fortement dépendant de la structure de l'hydroxyphosphonate (aussi bien les substrats aliphatiques qu'aromatiques ont été employés), de la source de la lipase utilisée (les meilleurs résultats ont été réalisés dans le cas de Candida antartica et de Candida rugosa) ainsi que olu choix du solvant organique. Ce procédé a été également employé pour la résolution des δ-hydroxy-β-cétophosphonates, dont la conversion chimique par des réactions de HWE fournirait des molécules à fonctions multiples, qui pourraient trouver beaucoup d'applications en chimie organique de synthèse [58,59]. En plus, de nombreux produits naturels tels que le « yashabushicétol », possèdent une telle unité de structure dans leur squelette (Schéma 42).
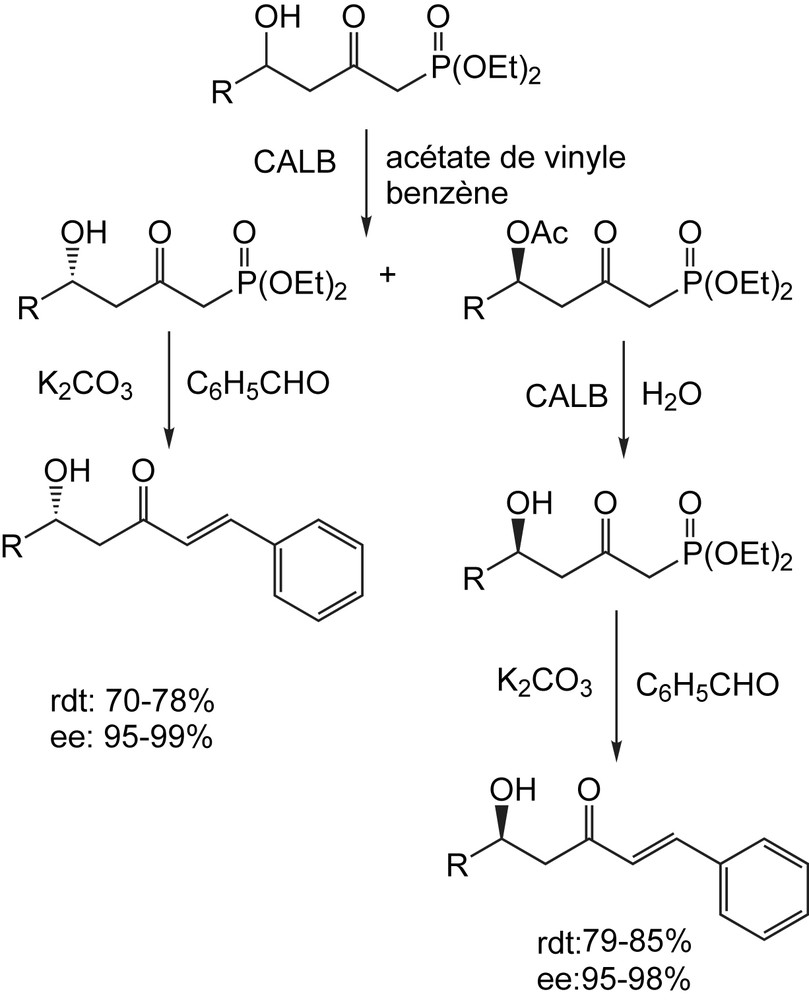
3.1.3 Réduction par le complexe borohydrure de sodium-acide tartrique
La réduction de dimenthyl cétophosphonate avec le borohydrure de sodium dans le THF conduit au (R)-hydroxyphosphonate avec un faible rendement et une faible stéréosélectivité (30–35% de). La réduction dans le THF procède avec un rendement élevé mais la stéréosélectivit demeure faible. Une amélioration de la stéréoselectivité est obtenue par l'utilisation du complexe chiral [borohydrure de sodium avec l'acide-(+)-tartrique (AT) naturel (R,R)] [13–15,60]. Il apparaît que la stéréochimie de la réduction dépend de la configuration absolue de l'acide tartrique. En effet, la réduction avec l'acide tartrique-(R,R) conduit à la formation du (S)-hydroxyphosphonate ; par contre avec l'acide (S,S), c'est le (R)-hydroxyphosphonate qui est isolé (Schéma 43).

3.1.4 Hydrogénation asymétrique
L'hydrogénation asymétrique des cétones prochirales peut être réalisée par des catalyseurs chiraux de ruthénium-diphosphine générés in situ. Des β-cétophosphonates tels que le diméthyl 2-oxoheptylphosphonate et le diéthyl 2-oxophényl phosphonate ont été réduits avec des énantiosélectivités très élevés pour conduire respectivement à l'énantiomère (S) avec un ee de 99%, et à l'énantiomère (R) avec un ee de 98%. Les réductions ont été effectuées à température ambiante ou avoisinant les 50 °C pour le deuxième cas (Schéma 44).
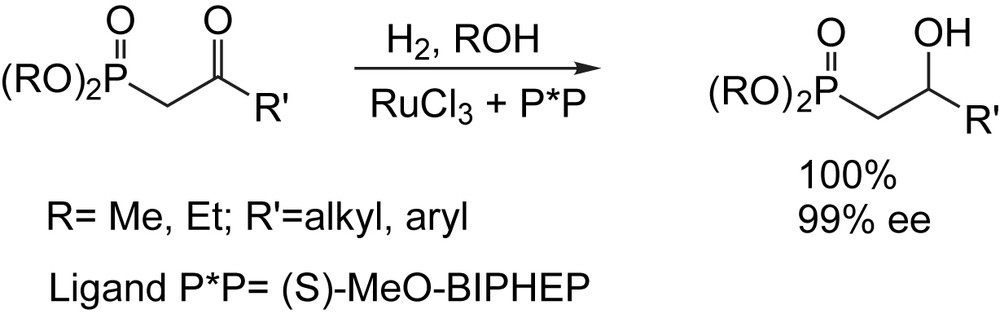
Le RuCl3 anhydre est le précurseur des catalyseurs chiraux de ruthénium, le plus disponible et le moins coûteux. En outre, cette préparation est facile à exécuter et n'exige ni réactions multiétapes, ni isolement des intermédiaires. Elle permet aussi, un criblage particulièrement rapide de nouvelles diphosphines chirales (P*P) [61].
Pour l'hydrogénation catalytique en présence d'oxyde de (S,S)-Bis(2-hydroxypropyl) (phnyl)-phosphane [(S)-MeO-BIPHEP-ruthénium] comme catalyseur, l'oxyde de (2-oxopropyl)-dipénylphosphane a été choisi comme substrat modèle. La conversion en alcool avec un excès énantiomérique supérieur à 98% permet d'appliquer ces conditions aux dicétones (Schéma 45) [62]. Cet exemple indique que l'hydrogénation catalytique des oxydes de β-oxophosphane par les complexes de ruthénium peut présenter une approche générale et commode pour la synthèse énantiosélective des dérivés phosphorés.
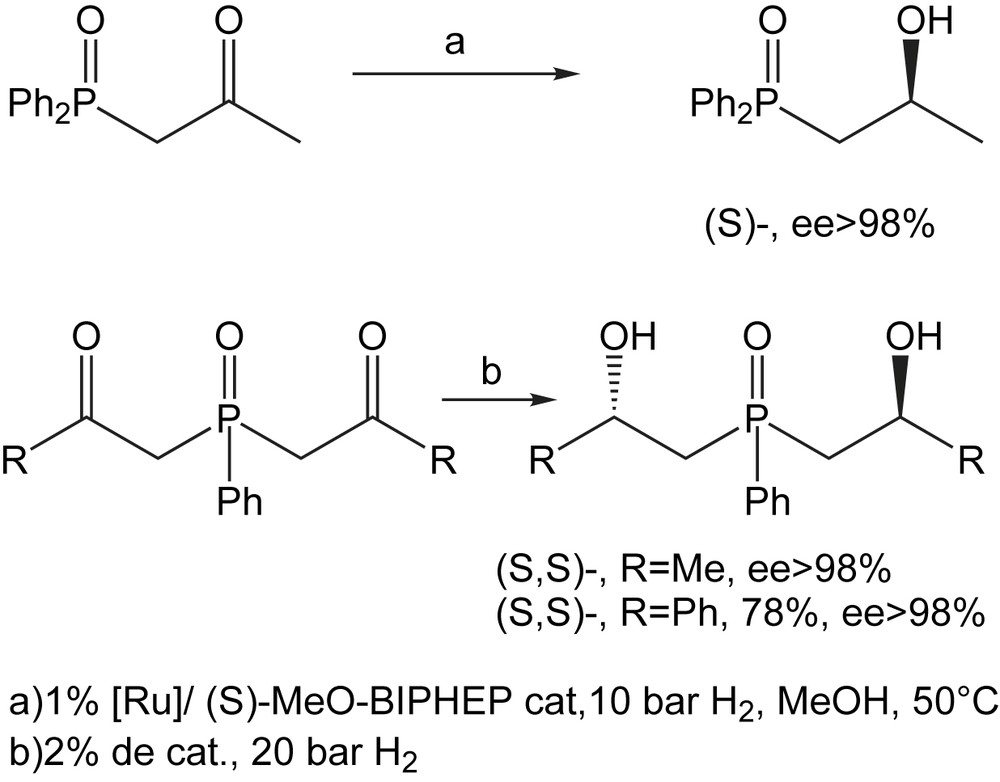
L'extension de l'hydrogénation asymétrique par le BINAP-RuII est une méthode efficace pour la préparation des α-aminophosphonates possédant un groupement hydroxy supplémentaire en position β. Cette méthode [63] utilise l'α-acétamido β-céto-phosphonate racémique pour donner l'hydroxyphosphonate (1R,2R) avec une diastéréosélectivité élevée (syn:anti, 97:3) et une excellente énantiosélectivité (≥98%). L'hydroxyphosphonate obtenu est transformé en (1R,2R) phosphothréonine sous forme d'un énantiomère pur avec un rendement de 92% (Schéma 46).

3.1.5 Réduction ou dibal, et divers hydrures
La synthèse des analogues phosphorés de l'acide (R)-γ-amino-β-hydroxybutyrique (GABOB) a fait l'objet d'une série d'études menées par Ordóňez et coll [64–66]. La préparation efficace des deux énantiomères (S) et (R) du β-cétophosphonate est effectuée par la réduction diastéréosélective avec différents hydrures (NaBH4, LiBH4/LiClO4, Zn(BH4)2). Un bon rendement (97%) et une diastéréosélectivité modérée (72:28) sont obtenus avec l'énantiomère (S) lorsqu'il est réduit par le NaBH4. Les meilleurs résultats de la réduction des diméthyl γ-N,N-bis (α-méthylbenzyl)amino-β-cétophosphonates (S) et (R) en hydroxyphosphonates, sont obtenus par le catécholborane (CB) en présence de LiClO4 (Schéma 47). Cette réaction constitue un exemple de diastéréosélectivité élevée (>99%).

Le remplacement du groupe benzyle par le p-toluènesulfonyle et l'accès facile aux γ-[(N-p-toluènesulfonyl)-β-cétophosphonate, permettent d'évaluer la diastéréosélectivité de la réduction, en opérant dans des conditions différentes avec divers agents réducteurs [67]. Pour cette étude, le choix s'est porté sur l'analogue isopropyle (R = iPr) pour lequel la réduction par le NaBH4 dans le méthanol à 0 °C offre les hydroxyphosphonates (anti et syn) avec d'excellents rendements (99%) et une bonne diastéréosélectivité (81/19) en faveur du diastéréoisomère anti (Schéma 48).

D'autres travaux dans le cadre des analogues phosphorés de l'acide γ-aminobutyrique (GABA) et du GABOB ont été décrits [13,15,60,68]. Une approche efficace pour la synthèse des acides γ-amino-β-hydroxyphosphoniques énantiomériquements purs utilise le dimenthyl (R)- ou (S)-3-chloro-2-hydroxypropyl-phosphonate. Ce dernier peut être transformé de façon quantitative (99%) en (R)-époxyphosphonate par traitement du (S)-dimenthyl 2-hydroxy-3-chloropropylphosphonate avec le K2CO3 dans l'acétonitrile-DMF en présence de l'iodure de potassium. Soumis à l'action de la dibenzylamine et en chauffant à 80 °C, l'acide (R)-hydroxyaminophosphonique optiquement pur (analogue phosphonique du GABOB naturel) est isolé sous forme de cristaux avec 90% de rendement, après hydrogénation et hydrolyse (Schéma 49).
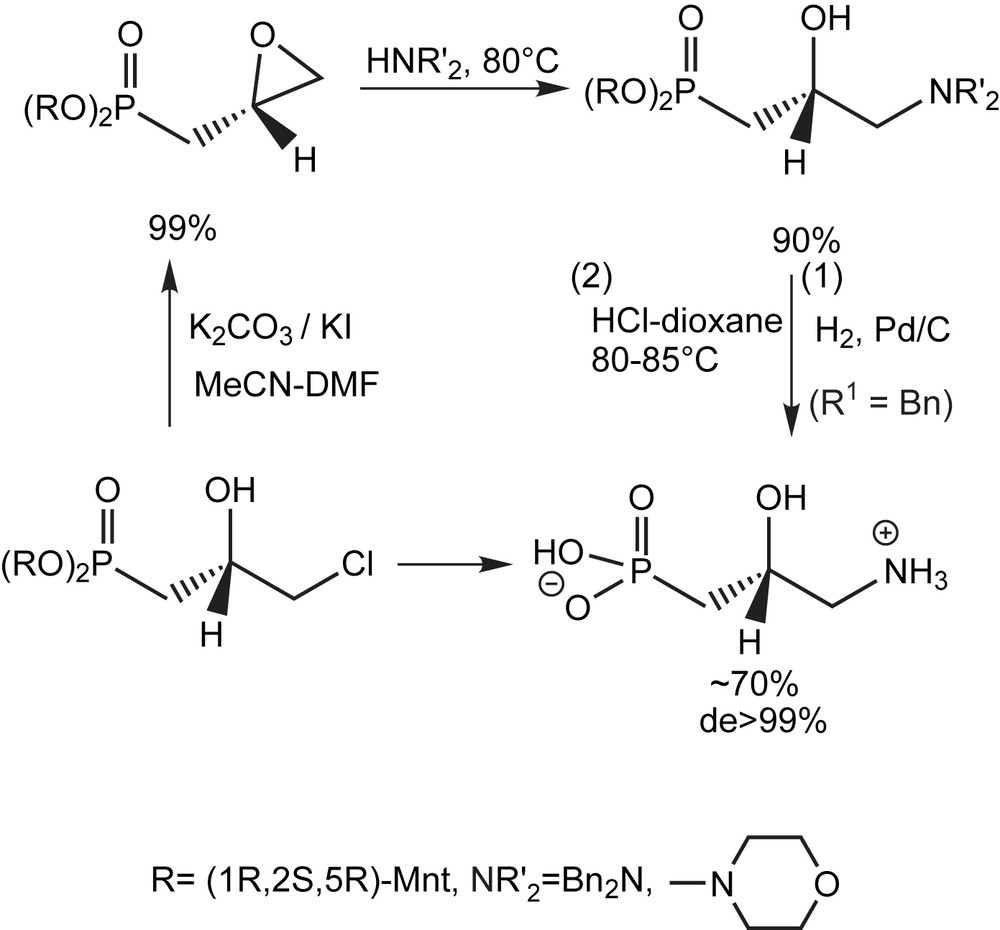
Le (S)- et le (R)-dimentyl-3-chloropropyl-phosphonate sont utilisés pour la synthèse des énantiomères (R)- et (S)- de la phosphonocarnitine (Schéma 50).
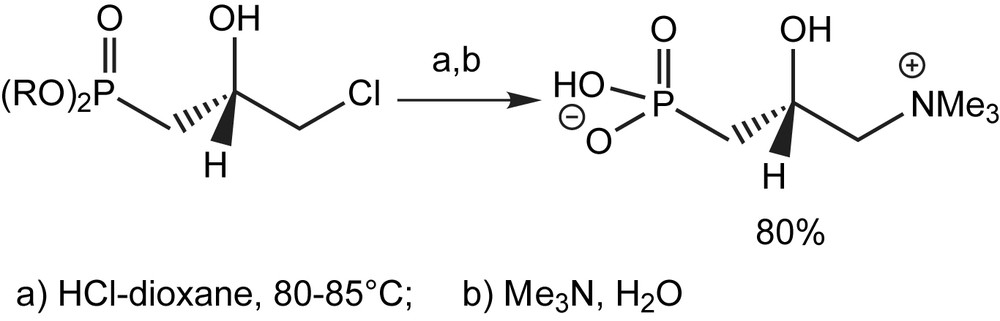
3.1.6 Substitution par les hér-éroatomes
Les sulfures α-phosphorés font partie d'une classe importante de composés utiles en synthèse organique et en médecine. Mikolajczyk et coll. ont décrit une nouvelle synthèse de ces composés [69] par réaction des β-cétophosphonates O-silylés avec le chlorure de sulfanyle. Ainsi, le diéthyl 1-méthyl-2-oxopropylphosphonate a été traité avec le chlorure de triméthylsilyle en présence de la triéthylamine et le dérivé O-silylé formé a été ensuite engagé avec le chlorure de méthylsulfanyle. Le produit sulfanylé correspondant est obtenu avec un rendement se situant entre 36 et 40 % (Schéma 51).

Le diéthyl 3-chloro-2-oxopropylphosphonate peut être aussi utilisé pour la préparation d'un nouveau β-céto-γ-xanthylphosphonate [70] (Schéma 52).
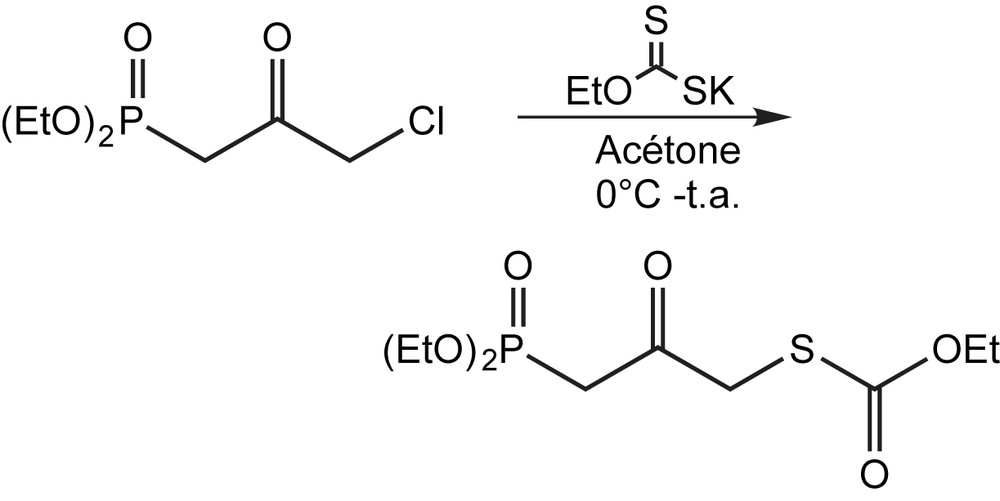
La synthèse de xanthate a été facilement réalisée avec un rendement global de 40% à partir du β-cétophosphonate. Un tel réactif s'avère prometteur pour créer efficacement des liens carbone-carbone aux deux extrémités de cette nouvelle cétone, par un mode ionique et radicalaire.
3.1.7 Préparation des dérivés monofluorés et monochlorés
Les molécules organiques fluorées jouent aussi des rôles importants dans le domaine de la chimie médicinale parce que le remplacement des hydrogènes ou du groupe hydroxyle, par des atomes de fluor améliore souvent le profil biologique du composé parent [71]. Pour cette raison, le développement de méthodes efficaces pour la synthèse des composés monofluorés chiraux a suscité une attention considérable.
Hamashima et coll. se sont concentrés sur la fluorations électrophile des β-cétophosphonates cycliques et acycliques par une catalyse énantiosélective efficace utilisant des complexes chiraux du palladium [72–76] (Schéma 53).

Le système catalytique N-fluorobenzéne-sulfonimide (NFSI) [74,77] s'est avéré efficace pour une variété de substrats comprenant les β-cétophosphonates cycliques et acycliques. Tous les substrats examinés ont été fluorés dans des conditions douces d'une façon fortement énantiosélective (plus de 94% ee). La réaction a lieu en présence d'une quantité aussi faible qu'un mol % du catalyseur, sans affecter ni le rendement chimique, ni l'énantiosélectivité. Quand la réaction s'effectue à 0 °C, l'excès énantiomérique est légèrement amélioré à 97% (Schéma 54). Par rapport aux β-cétophosphonates cycliques, les réactions des substrats acycliques se sont avérées lentes, et les produits de départ n'ont pas été entièrement consommés à 40 °C, même lorsque le catalyseur est utilisé à 10 mol%. Bien que le rendement chimique soit modeste à bon, les valeurs d'ee des produits sont excellentes (94 et 95%, respectivement) (Schéma 55).
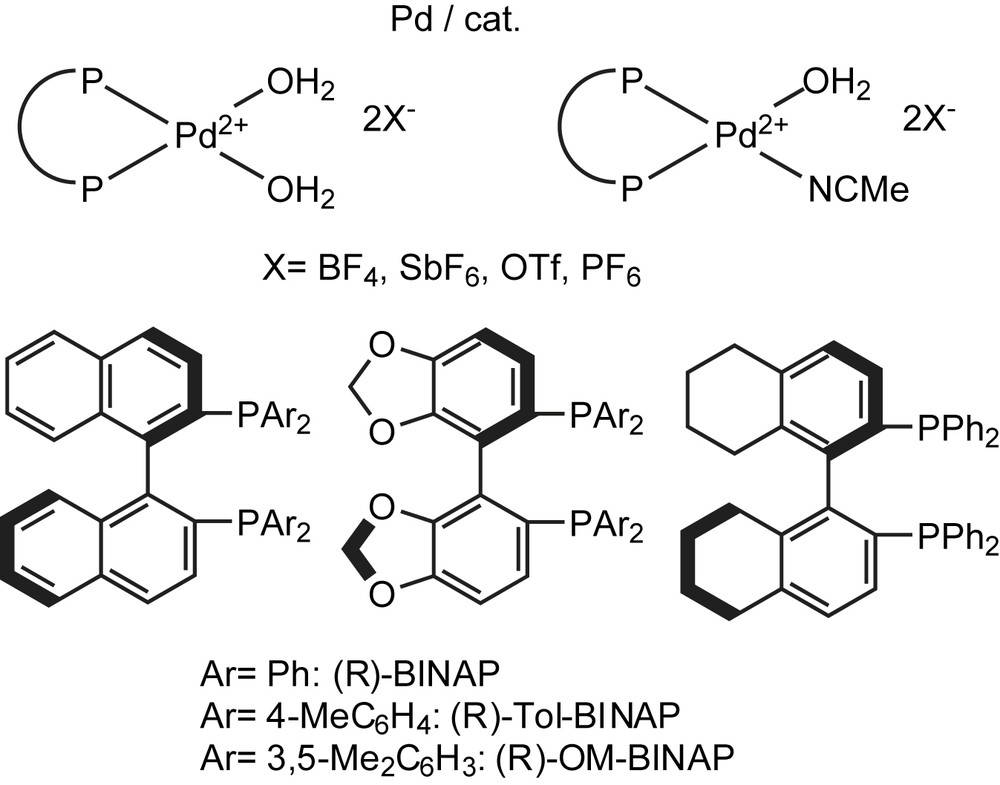
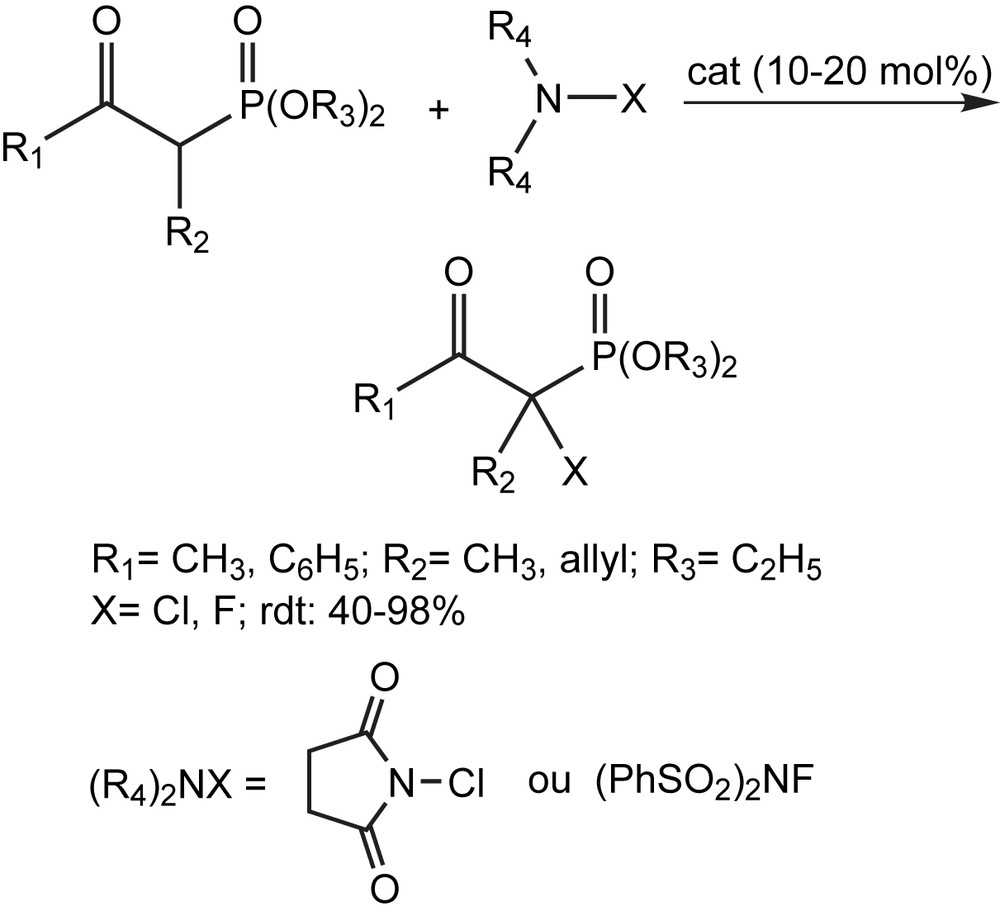
Ces réactions sont commodes étant donné que les précautions spéciales pour exclure l'air et l'humidité sont inutiles. D'autres acides de Lewis chiraux ont été utilisés pour la chloration et la fluoration énantiosélective des β-cétophosphonates [78]. La chloration s'effectue par la N-chlorosuccinimide (NCS) et fournit des rendements élevés et des excès énantiomériques de l'ordre de 94%. Quant à la fluoration, ils utilisent le N-fluorobenzène-sulfonimide (NFSI) pour obtenir les α-fluoro-β-cétophosphonates avec une pureté optique qui atteint les 91% et des rendements modérés à bons (Schéma 55). Le Schéma 56 représente les différents ligands chiraux utilisés dans cette réaction.
3.1.8 Alkylation
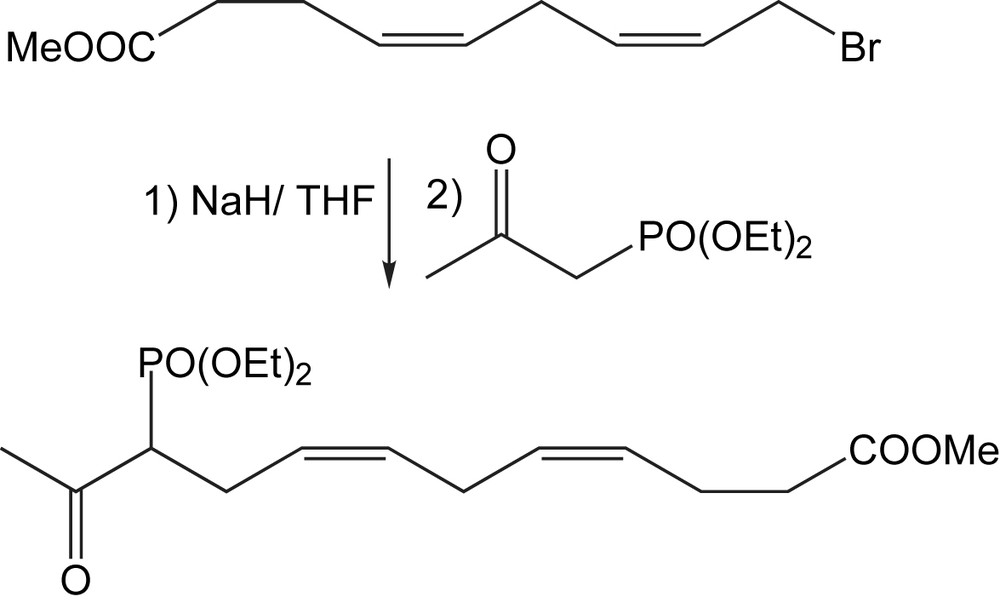
Des cétophosphonates plus complexes sont obtenus avec de bons rendements par l'alkylation des β-cétophosphonates avec un dérivé bromé (Schéma 57) [79]. La condensation du sel de sodium de ce cétophosphonate avec l'isopropylidène-D-glycéraldéhyde selon la réaction de Horner-Emmons, conduit à des énones chirales non racémiques utiles pour la synthèse totale du 17-iso-Levuglandin E4.
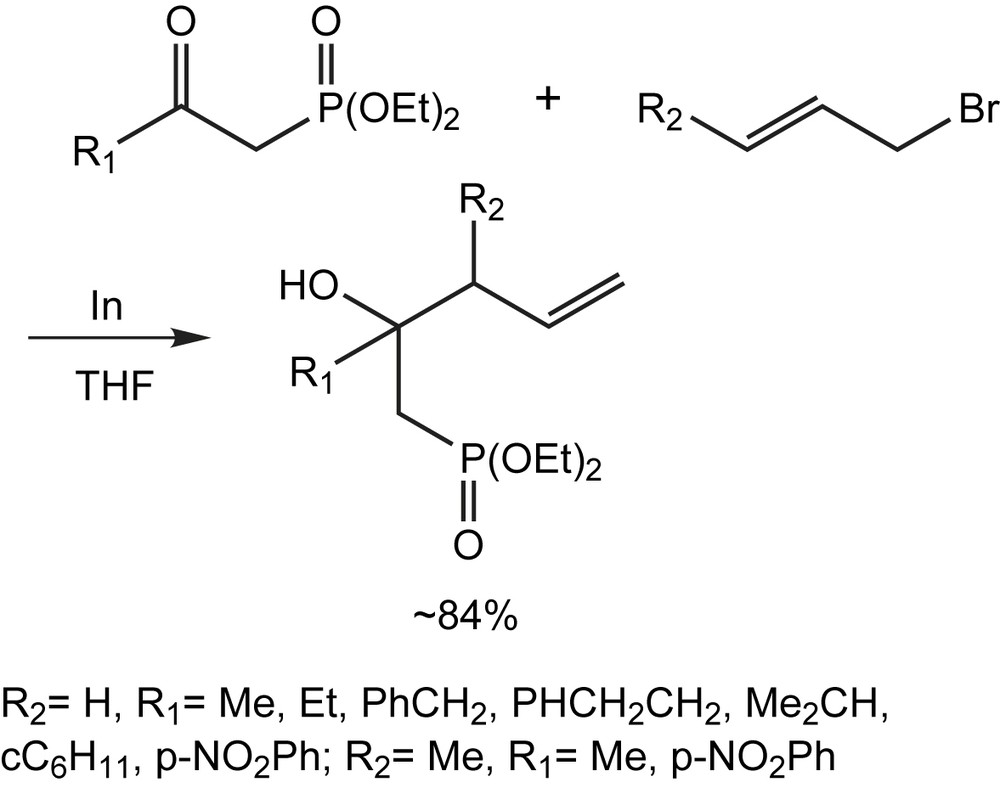
Les dérivés d'allylindium s'additionnent facilement au groupe carbonyle des β-cétophosphonates sans la nécessité d'un acide de Lewis [80]. Plusieurs β-cétophosphonates structurellement variés ont été soumis aux allylations par ce procédé pour produire aux β-hydroxyphosphonates correspondants. Les phosphonates présentant des groupes alkyle, cycloalkyle, hétérocycloalkyle, ainsi que des substituants aromatiques en position β conduisent aux produits désirés sans problème majeur. Les β-cétophosphonates cycliques ne posent également aucune difficulté pour l'addition nucléophile du réactif allylindium (Schéma 58).
Le passage aux homologues supérieurs de β-céto-phosphonates peut être réalisé par une γ-alkylation [81]. Elle consiste à alkyler le dianion du 2-oxo-propylphosphonate diéthylique intermédiaire par l'iodure de métallyle pour donner des produits γ-monoalkylés avec de bons rendements (65–75%) (Schéma 59).

3.1.9 Préparation des cétones α,α’-disubstituées non symétriques
Bien qu'il soit possible d'alkyler sélectivement le carbone γ d'un β-cétophosphonate, afin d'obtenir des rendements plus élevés, l'alkylation est effectuée en deux étapes consécutives. Les atomes de carbone α sont alkylés dans un premier temps, en utilisant le NaH comme base. Pour l'alkylation en γ, une base plus forte telle que le LDA ou le NaHMDS (hexaméthyldisilazane de sodium) est nécessaire. Ces réactions se déroulent à température ambiante, dans le THF, avec une durée de 2–5 h. Aucun produit d'o-alkylation n'a été isolé [82]. Cette méthode offre la possibilité d'accèder aux cétones non symétriques α,α’-disubstituées (Schéma 60).
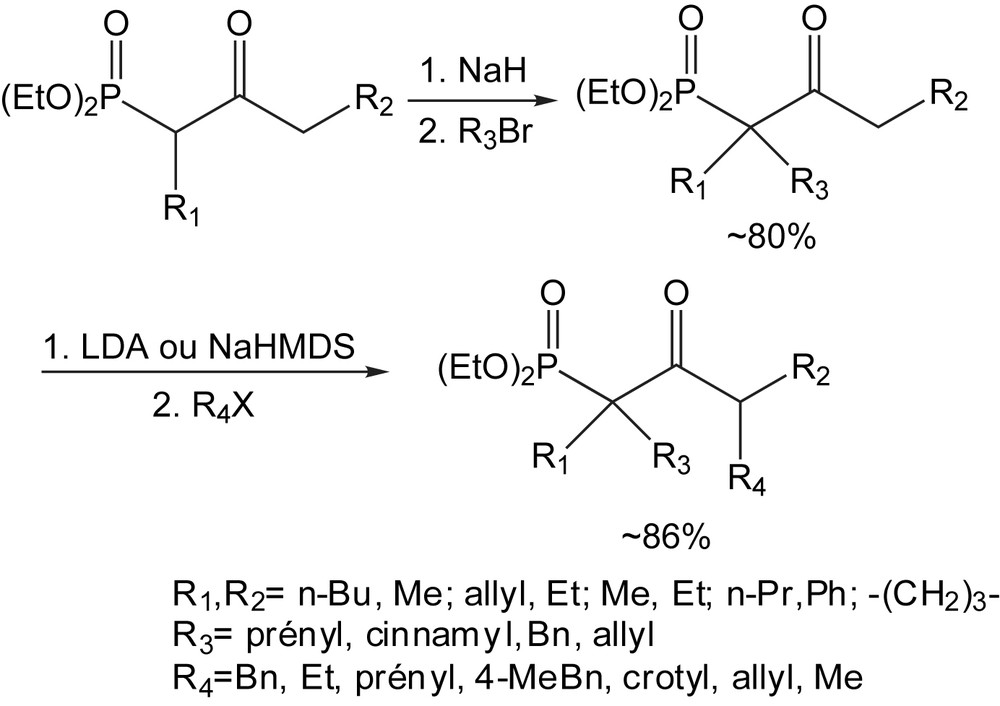
3.1.10 Allylation asymétrique
La condensation des β-cétophosphonates avec le (S)-1-phényléthylamine au reflux du toluène ou du cyclohexane donne les imino/énamino-phosphonates correspondants (Schéma 61). Ces composés existent dans un rapport approximatif de (35/65), alors que le composé avec R = Ph existe seulement sous la forme énamino. L'addition de ces imino/énaminophosphonates bruts aux accepteurs de Michael tels que la phénylvinylsulfone, les acrylates benzyliques et méthyliques, l'acrylonitrile et le méthylvinylcétone se fait dans des conditions neutres.
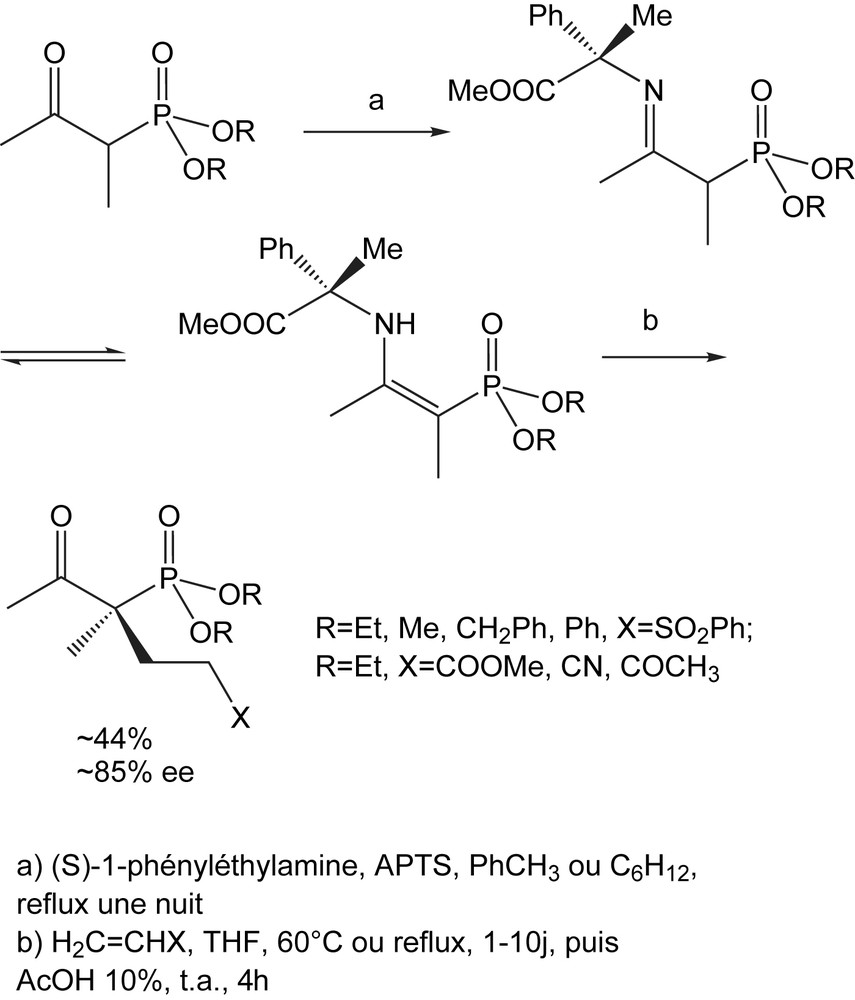
Cette étude montre que la réaction asymétrique sur un énaminophosphonate acyclique avec les accepteurs de Michael non-substitués est faisable avec de bons rendements et des excès énantiomériques élevés, comme pour les dérivés acycliques d'énaminoesters. Elle offre aussi l'accès aux nouveaux β-cétophosphonates chiraux, analogues des β-cétoesters [8].
3.1.11 Amination reductrice
L'amination réductrice du 2-oxoéthylphosphonates constitue un autre accès aux acides 2-aminoéthylphosphoniques [6]. Elle peut être effectuée avec les amines primaires ou secondaires, aliphatiques ou aromatiques ou encore avec l'acétate d'ammonium, avec le borocyanohydrure de sodium (NaBH3CN) en tant qu'agent de réduction. L'hydrolyse de 2-aminoethylphosphonates avec de l'acide chlorhydrique concentré conduit aux acides 2-aminoethylphosphoniques (Schéma 62).
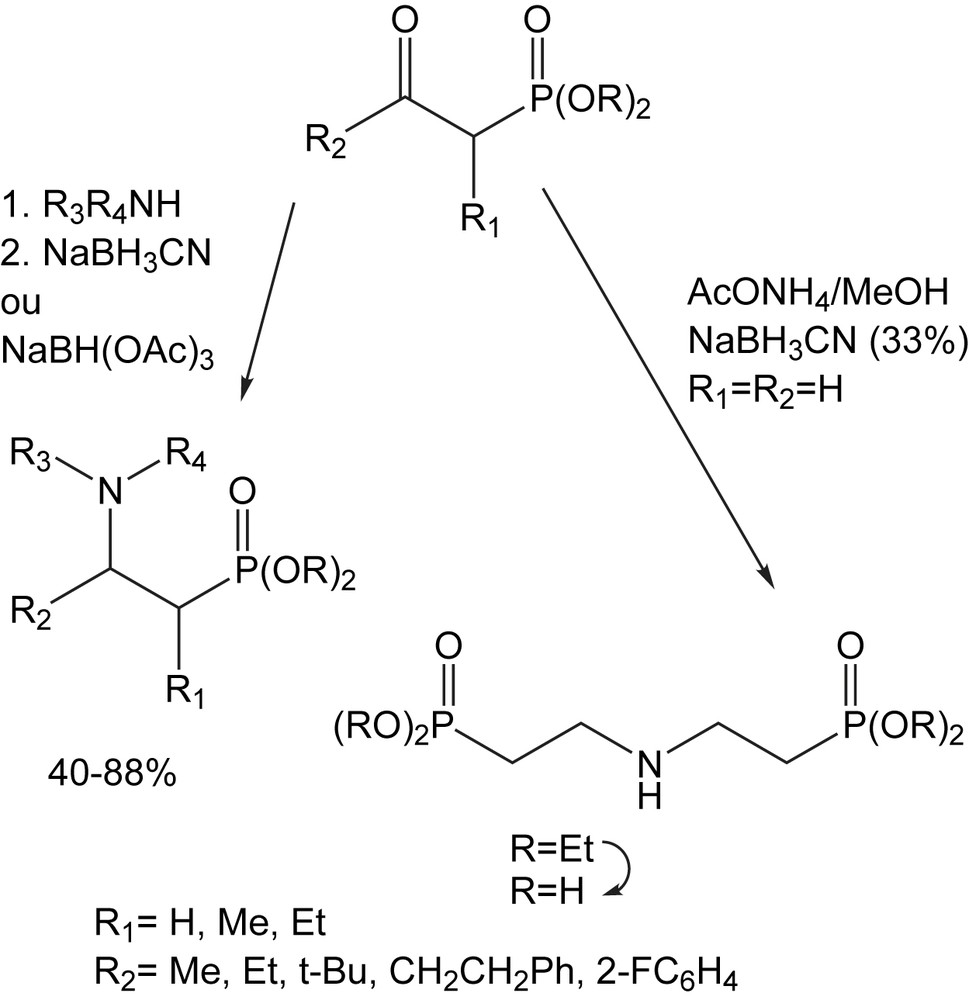
Lorsque l'amination réductrice des β-formyl-phosphonates (R2 = H) est effectuée avec H2/palladium, la réduction et la débenzylation sont observées simultanément. Cette même réduction des β-formyl-phosphonates (R1 = R2 = H) effectuée avec de l'ammoniaque et le NaBH3CN fournit le bis(éthylphosphonate) aminé.
La réaction du 2-oxoalkylphosphonate avec la benzylamine (R3 = H, R4 = Bn) ou le benzhydrylamine (R3 = H, R4 = CHPh2) suivi de la réduction avec le triacétoxyborohydrure et l'hydrolyse acide donne les acides aminoalkylphosphoniques correspondants avec des rendements satisfaisants. Aucune stéréoselectivité n'est observée par l'utilisation des amines optiquements pures.
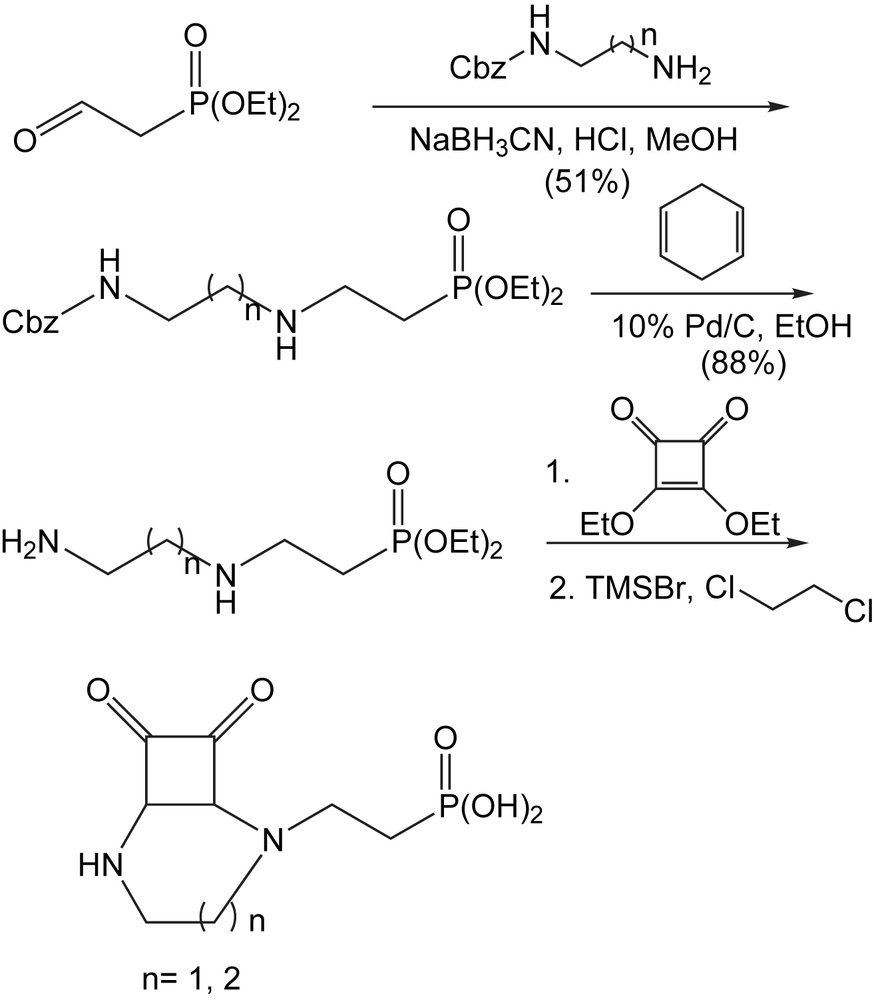
Cette méthodologie a été employée pour la synthèse de l'acide [2-(8,9-dioxo-2,6-diazabicyclo [5.2.0] non-1(7)-én-2-yl) éthyl] phosphonique, qui a été identifié comme antagoniste efficace de NMDA pour le traitement des désordres neurologiques. La combinaison de la diamine Cbz-protégée avec le diéthyl (2-oxoéthyl) phosphonate dans les conditions d'amination réductrice suivie de l'hydrogénation catalytique fournit la diamine correspondante (Schéma 63). La réaction de cette dernière avec le 3,4-diéthoxy-3-cyclobutène-1,2-dione suivi de la déprotection de l'ester avec le bromotriméthylsilane conduit à l'acide dioxo diazabicyclo phophonique [6].
3.1.12 Réduction des oximes
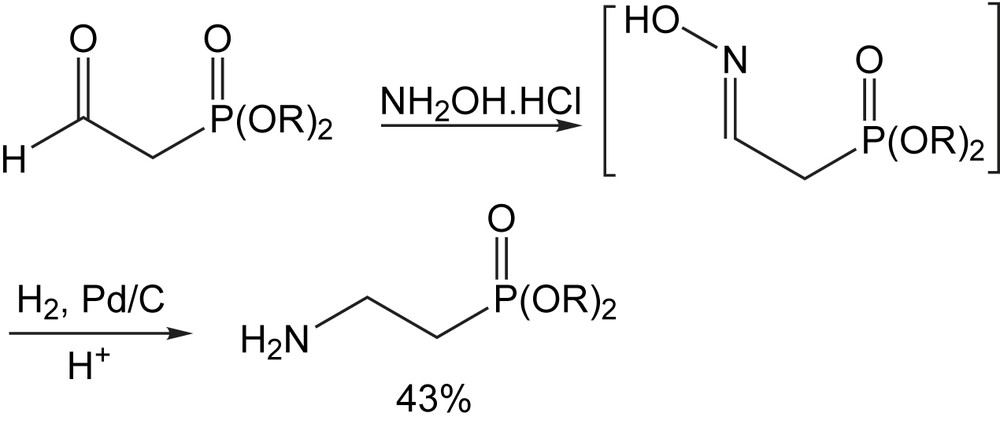
Les β-aminophosphonates peuvent également être préparés, par une simple réduction d'oximes. Le phosphonoacétaldéhyde diéthylique peut être converti en β-aminoéthylphosphonate (AEP) biologiquement important, par l'addition du chlorhydrate d'hydroxylamine suivie d'hydrogénation par le palladium sur carbone dans un mélange anhydride acétique/acide acétique glacial (Schéma 64) [6].
3.1.13 Synthèses des hétérocycles
3.1.13.1 Phosphono-β-lactames monocycliques
Les cétènes possédant des hétéroatomes comme substituants ont été développés et appliqués avec succès pour la synthèse des β-lactames fonctionnalisés. Cependant, les réactions avec les phosphonocétènes ont été, la plupart du temps, limitées à quelques réactions électrophiles.
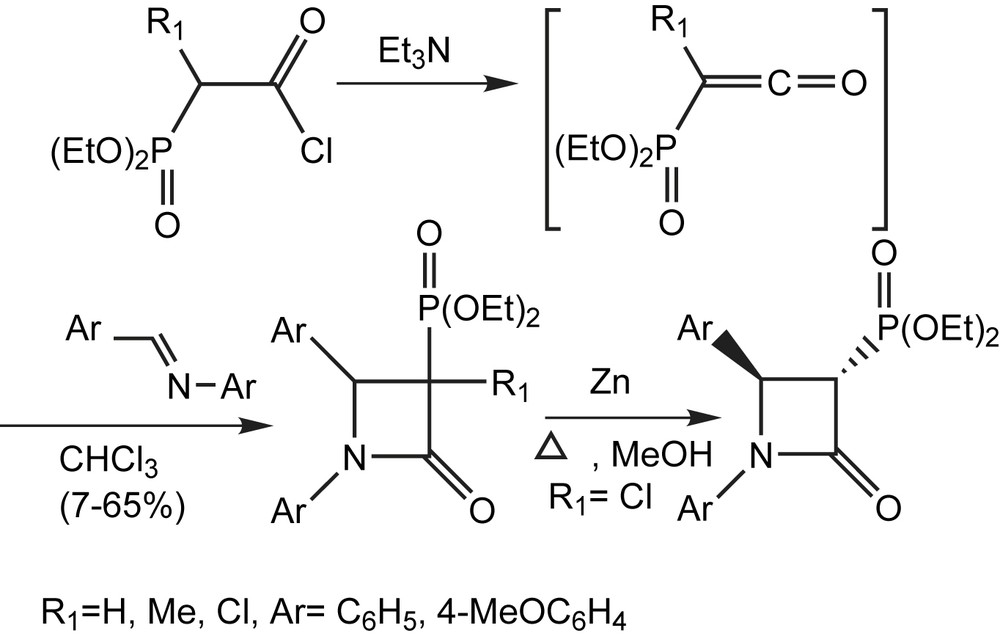
La cycloaddition a été employée une fois seulement, pour la construction de phosphono-β-lactames mono-cycliques. En présence d'un excès de benzyl-idèneaniline, les cétènes mènent aux cycloadduits avec des rendements de 7–65%. Les (diéthylphosphono) cétènes méthyliques et chlorés sont générés in situ à partir des chlorures d'acides correspondants et de la triéthylamine. La stéréochimie des β-lactames n'a pas pu être déterminée. Cependant, après le déplacement réductif de l'atome de chlore, il s'avère que ces β-lactames sont des isomères trans (Schéma 65) [83].
3.1.13.2 Phosphonopyrrolidines
Les azotures sont généralement réduits avec des phosphines en amines correspondantes par l'hydrolyse des iminophosphoranes correspondants (réaction de Staudinger). Cependant, en présence d'un groupe carbonyle et en l'absence de H2O (aucune hydrolyse ne peut se produire), l'iminophosphorane mène aux imines cycliques par l'intermédiaire d'une réaction d'aza-Wittig intramoléculaire. L'alkylation du phosphonate avec un iodoazide [83] fournit un précurseur approprié pour la cyclisation en phosphono-2-pyrrolidine et en pipéridine (Schéma 66).

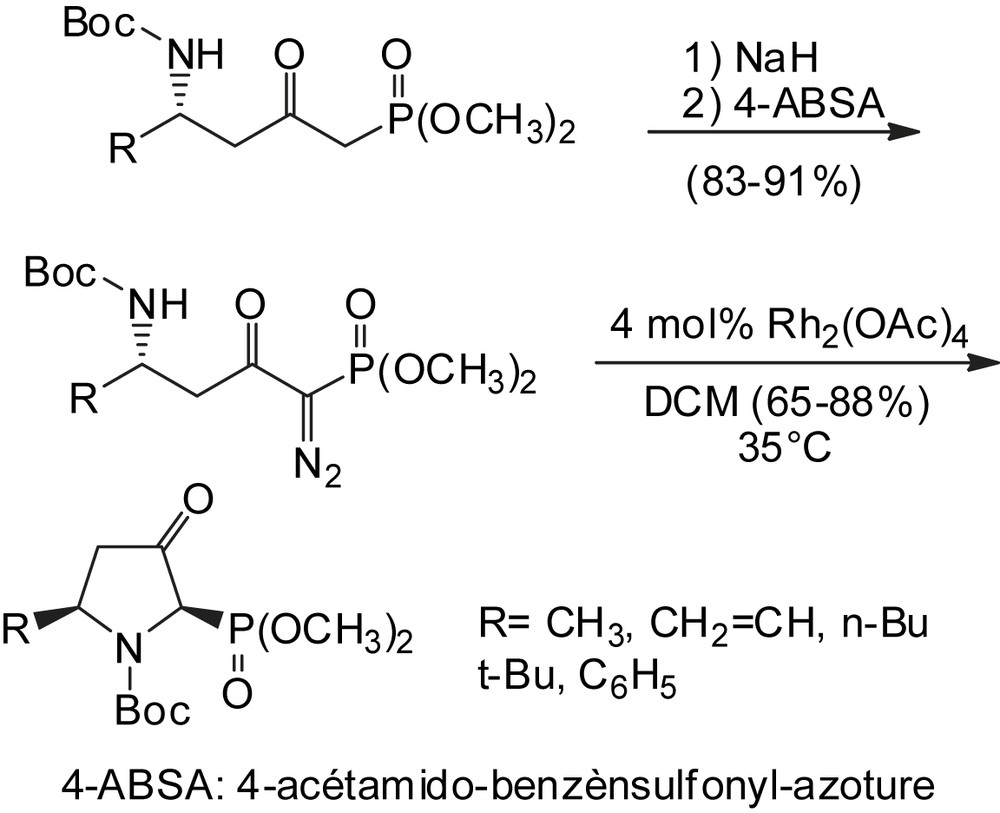
Davis et coll. présentent une nouvelle méthodologie pour la synthèse asymétrique des pyrrolidines 2-phosphonates-cis substituées [10,84], considérées comme d'importants substituts de proline. Elles sont obtenues à partir des N-sulfinyl δ-amino β-cétophosphonates. Le remplacement du groupe sulfinyl par le groupe Boc, suivi de la conversion en dérivé diazo conduit au produit cis. Cette méthode s'avère être fortement stéréosélective (Schéma 67).
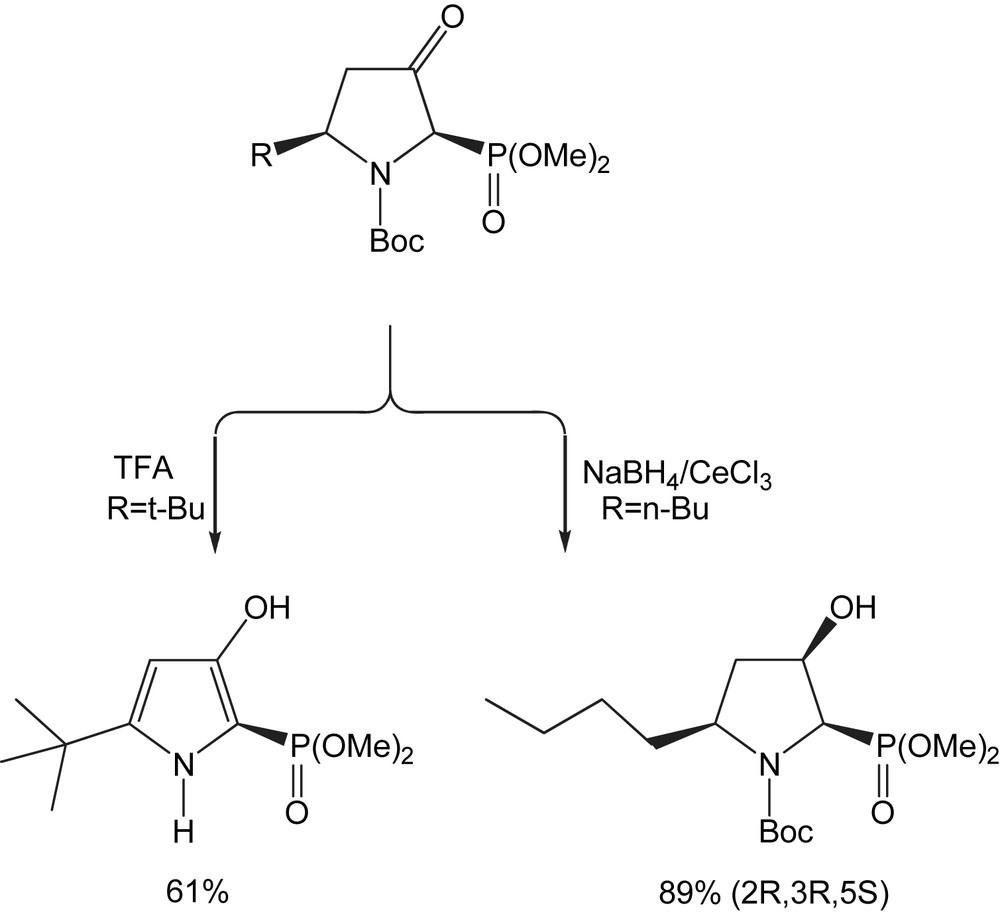
La chimie de ces nouveaux 3-oxo-pyrrolidine phosphonates a été très peu étudiée. Le traitement de la pyrrolidine-(2R,5R)-(−) avec (R = t-Bu) pendant 24 h, à température ambiante, par le TFA dans le DCM en présence de gel de silice, conduit au diméthyl 5-tert-butyl-3-hydroxy-1H-pyrrole 2-phosphonate avec 61% de rendement (Schéma 68). Ces composés peuvent être des « briques de construction » pour la génération de bibliothèque combinatoire. Dans le cas de la pyrrolidine-(2R,5S)-(−) avec (R = n-Bu), la réduction de Luche (NaBH4/CeCl3) donne l'alcool-(2R,3R,5S)-(−) comme isomère unique avec un rendement de 89%. L'addition d'hydrure s'additionne du côté le moins gêné, pour donner les pyrrolidines phosphonates cis 2,3,5-trisubstitués [84].
3.1.13.3 Les furylphosphonates
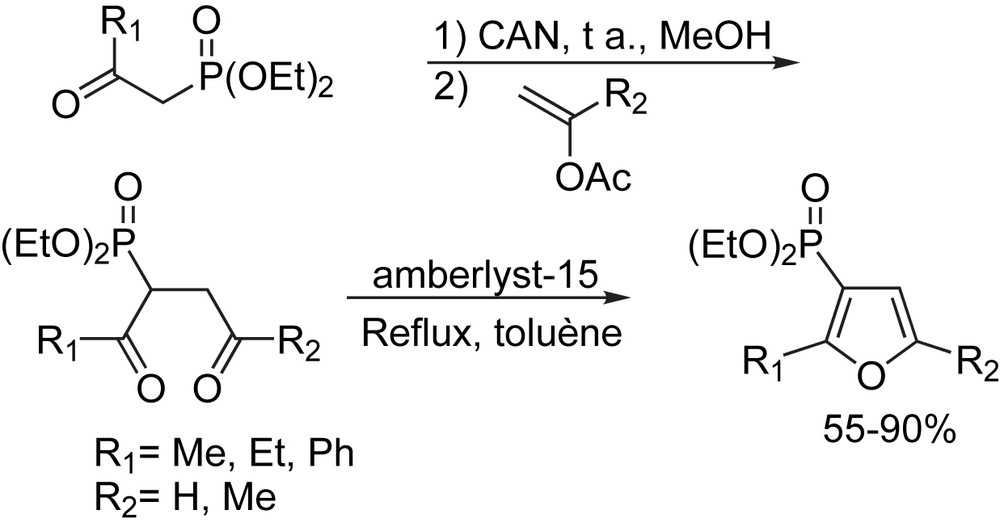
Les 3-furylphosphonates diéthyliques sont préparés en bons rendements (55–90%) par une procédure en deux étapes: l'addition oxydante du nitrate d'ammonium cérique (CAN) aux acétates vinyliques suivie d'une réaction de cyclisation de Cercueil–Knorr en condition acide (Schéma 69) [85].
3.1.13.4 Les furazanopipérazines
Les dérivés furazano [3,4] pipérazines sont utilisés en tant qu'intermédiaires dans la synthèse organique et comme composés biologiquement actifs. Cependant, leur préparation ainsi que l'étude de leurs propriétés chimiques, n'ont pas été étudiées en juste proportion.
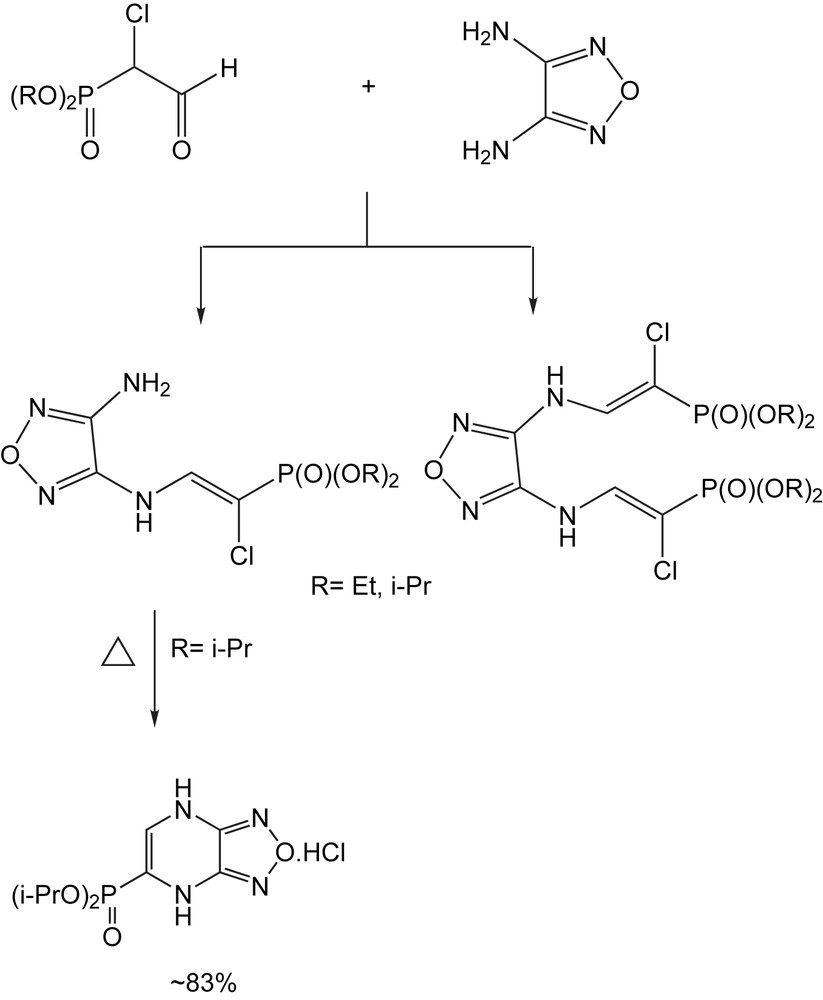
Les travaux dans ce domaine [86] montrent que le diéthoxyphosphorylphényl-α-chloroacétaldéhyde réagit avec le diaminofurazane dans un solvant polaire (acétonitrile) pour donner un hémiaminal (R = H). La même réaction conduite dans l'éthanol donne plutôt le O,N-acétal (R = Et) (Schéma 70), dont la structure est confirmée par diffractions des rayons X.

La synthèse des furazanopipérazines C-phosphorylées s'effectue par la condensation du phosphorylmonochloroacétaldéhyde avec le 3,4-diaminofurazane. Elle a lieu au reflux d'éthanol et mène à la formation de la monoénamine et de la bis(énamine) dépendant du rapport des réactifs. Au reflux olu xylène, l'énamine subit l'hétérocyclisation au moyen d'une substitution nucléophile intramoléculaire pour donner le chlorhydrate, selon le Schéma 71.
3.1.13.5 Dérivés du thiophène
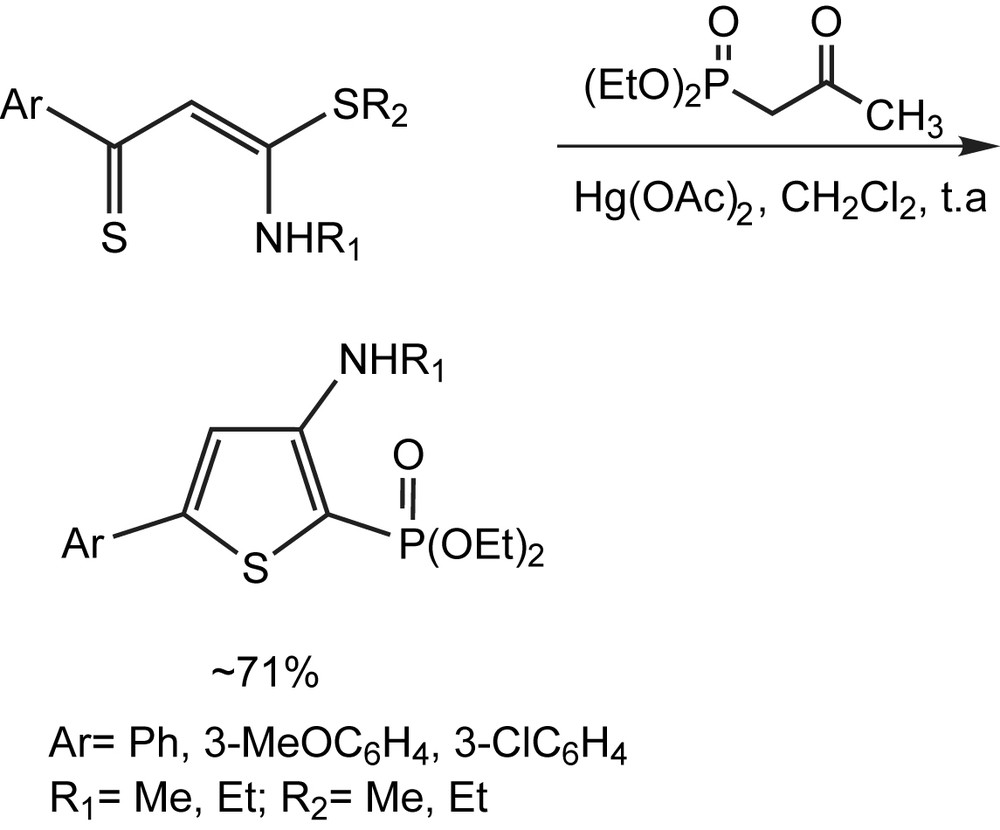
Les dérivés du thiophène différemment substitués ont été décrits [87] par une méthode de synthèse pratique, engageant le diéthyl (2-oxopropyl) phosphonate et N,S-thioaroylcétène en présence d'acétate de mercure dans le CH2Cl2 à température ambiante (Schéma 72).
3.1.13.6 Dérivés du pyrrole
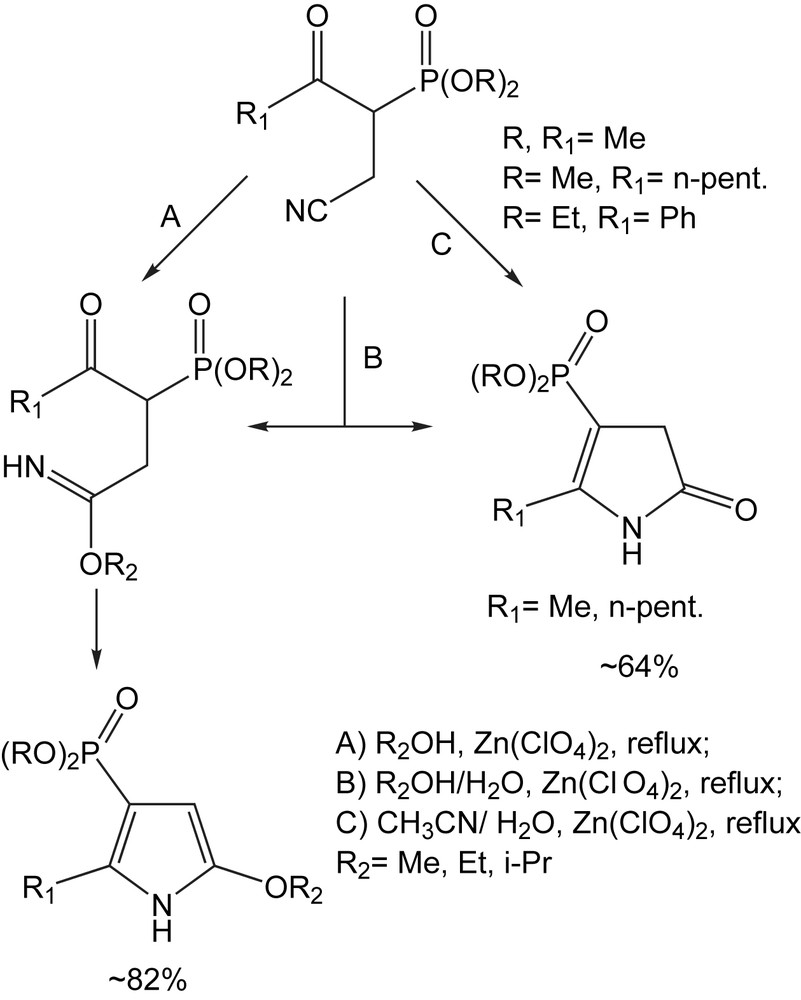
Les azahétérocycles phosphonylés sont une classe importante de composés, avec un potentiel biologique important en tant qu'isostères d'acides aminés à conformation restreinte. De tels hétérocycles peuvent être obtenus à partir des α-cyanométhyl-β-cétophosphonates, qui réagissent aussi bien avec les alcools qu'avec l'eau, en présence de Zn(ClO4)2, pour donner les 5-alkoxypyrroles et pyrrolin-2-ones [88]. Avec les alcools, les 5-alkoxypyrrole-3-phosphonates sont obtenus avec des rendements de 72–89%. Dans l'eau-alcool ou dans l'eau, les mêmes réactions fournissent les pyrrolin-2-ones avec des rendements de 82–85% (Schéma 73).
3.1.13.7 Pipéridin-2-ones
L'introduction d'un groupe attracteur d'électrons tel que le phosphonate, en position β d'une cétimine, présente plusieurs avantages principaux dans la réaction d'aza-annelation. Le déplacement de l'équilibre tautomérique à partir de la cétimine jusqu'à la fonction β-énamino augmente de manière significative le rendement de la réaction et la sélectivité. Cette stratégie est utilisée pour la synthèse des pipéridinones phosphonylés, sous forme de diastéréoisomères (78/22) selon un processus en deux étapes (Schéma 74) [83].
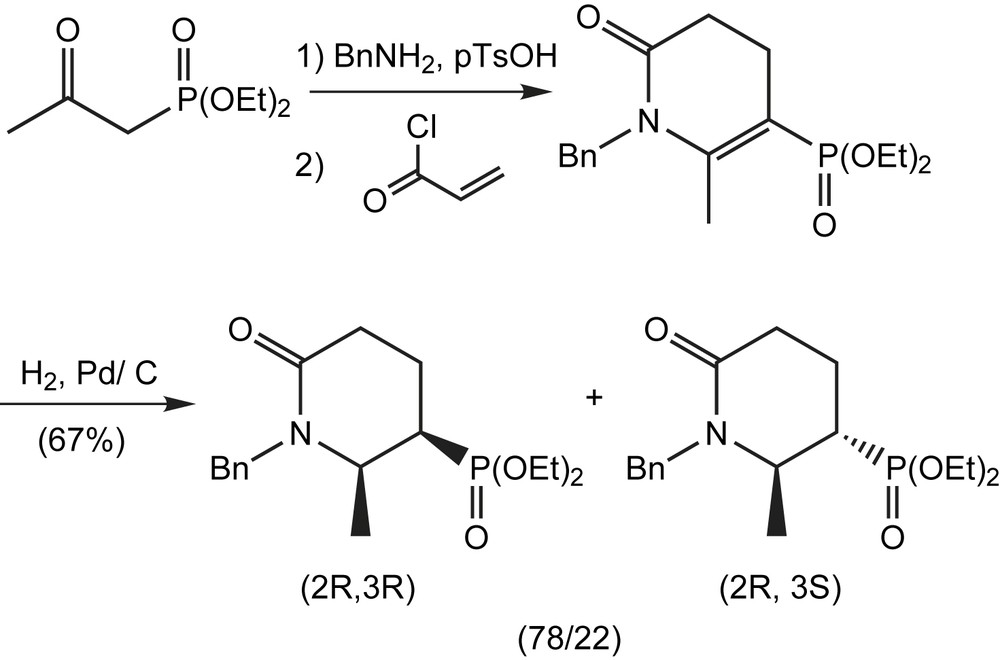
3.1.13.8 Pipéridines substituées
Les pipéridines 2,6-disubstituées sont connus pour posséder une large gamme d'activités biologiques (nécrotique, insecticide, antibactérien, antifongique, anti-HIV). L'intérêt pour ce motif pipéridinique a suscité une nouvelle méthode de synthèse asymétrique du trans-1,2,5,6-tétrahydropyridine énaminone 2,6-disubstituée, à partir des N-sulfinyl δ-amino β-cétophosphonates [89]. L'énaminone formée, en raison de son instabilité, est traitée avec HCl 4 N dans le dioxane puis avec Cbz-Cl/DMAP pour donner le (2R)-5-(diméthoxyphosphoryl) 2,3-dihydropyridone avec un rendement de 55% (Schéma 75).
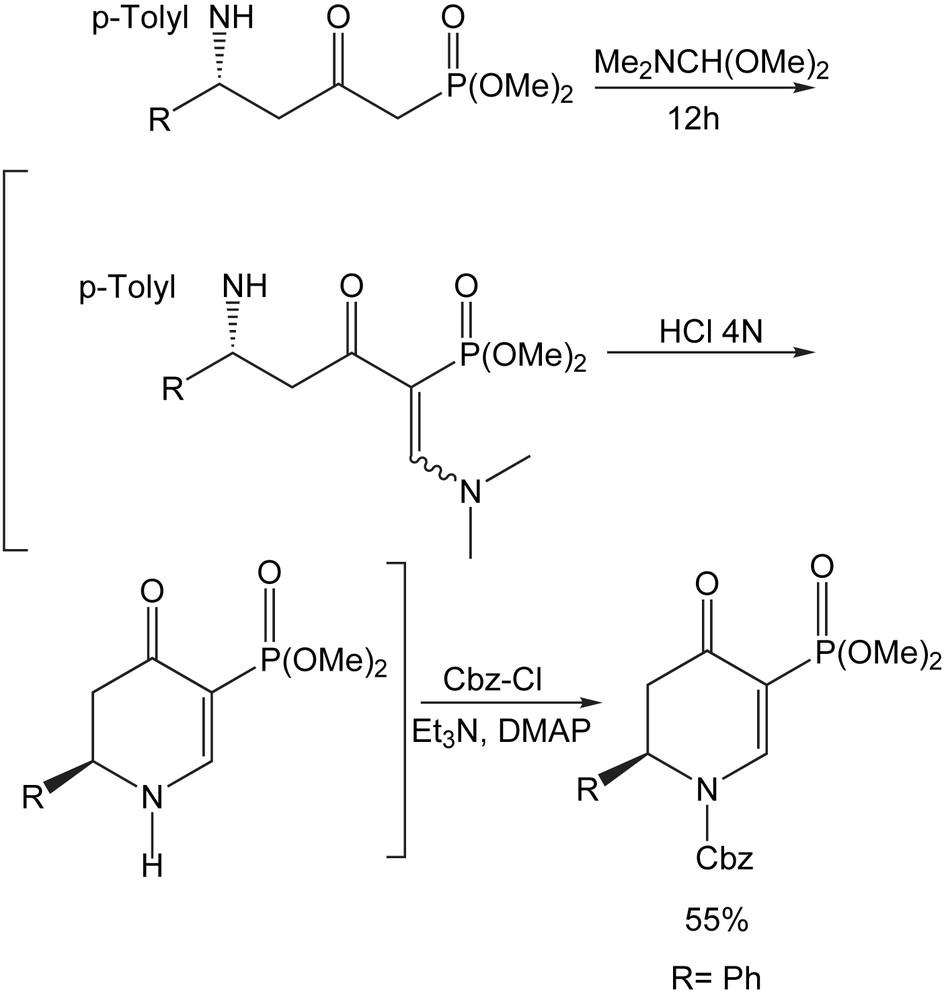
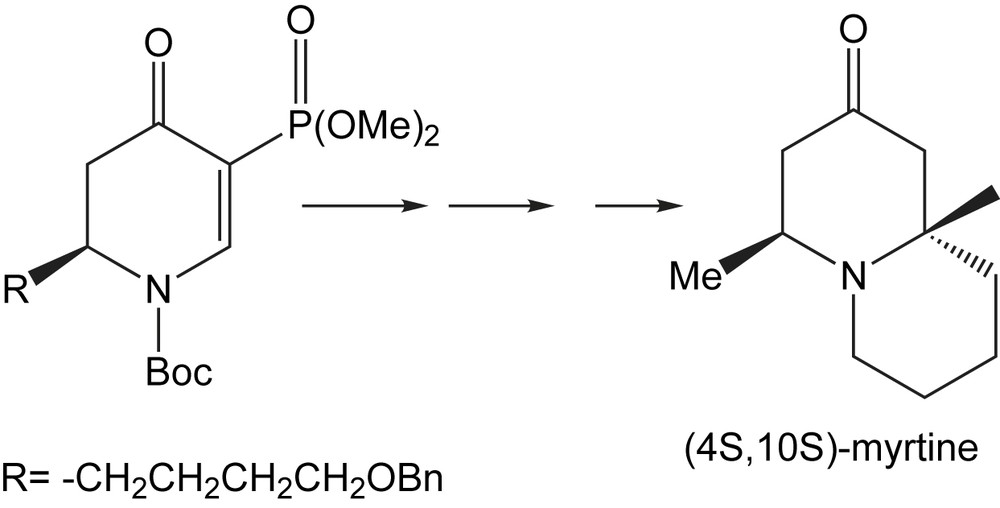
En outre, les pipéridines servent aussi comme modules de base à la synthèse d'alcaloïdes plus complexes, y compris les systèmes cycliques d'indolizidine et quinolizidine [90], qui présentent une large gamme d'activités biologiques.
L'utilité de cette nouvelle méthodologie est illustrée par la synthèse totale du (4S,10S)-(−)-myrtine, l'isomère non naturel de la myrtine qui n'a été jamais préparé (Schéma 76).
3.1.13.9 Dérivés de la pyridine
L'accès aux pyridines fonctionnelles est possible en traitant le diéthyl-1-acétyl-4-(diméthylamino)-3-fluoro-(1E,3Z)-butadiényl-phosphonate [48] par une solution aqueuse de NH3 à 25%. Cette opération se déroule à température modérée (70 °C), pendant 2 h, et permet d'isoler le produit final avec un rendement de 60% (Schéma 77).

3.1.13.10 Dérivés de quinoléine
Le groupe phosphonate présente des propriétés électroniques semblables aux groupes CO ou SO et possède une bonne affinité avec les ions magnésium qui jouent un rôle fondamental dans les réductions par les modèles du NADH.
Dans le but de synthétiser le modèle annelé dans la série des quinoliénes, le diéthyl (2-oxopropyl)-phosphonate est condensé avec l'imine dérivée de 2-amino-4,5-diméthoxybenzaldéhyde selon la modification de Borsche de la réaction de Friedlander [91]. La quaternisation suivie de réduction régiosélective-1,4 conduit au modèle désiré avec un rendement global de 48% (Schéma 78). Le réactif résultant a été utilisé avec succès, dans la réduction de benzoylformate méthylique en présence des ions du Mg2+ fournissant le mandélate méthylique correspondant.
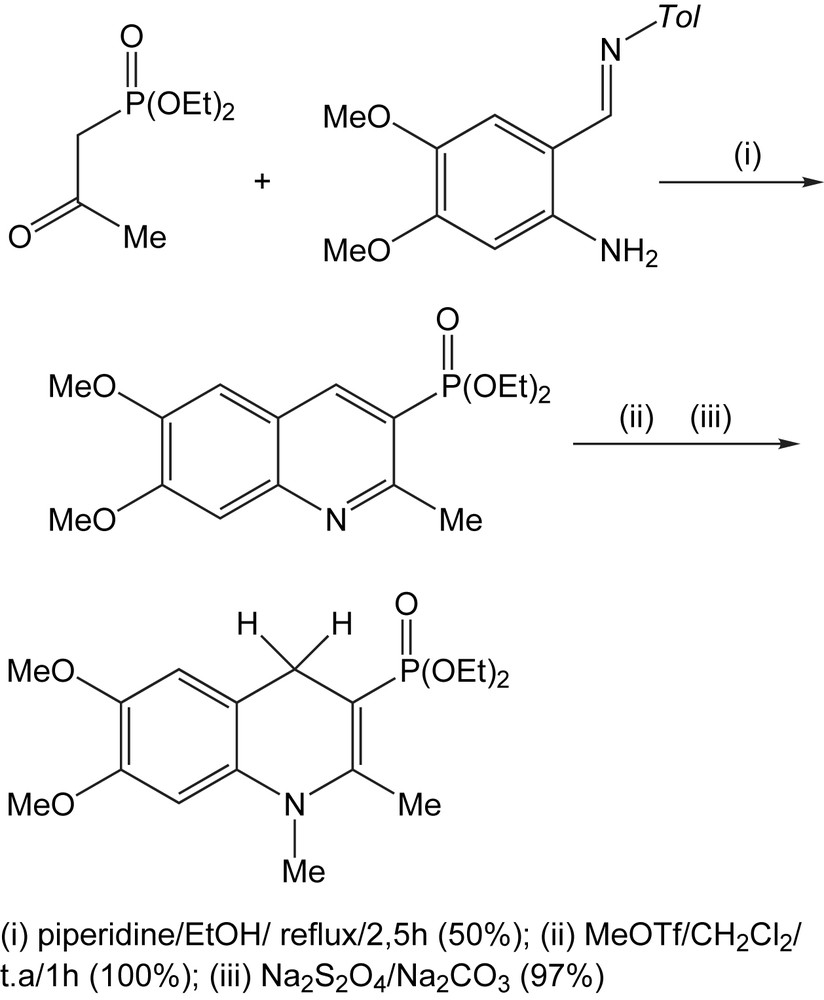
3.1.14 Réactions de cycloaddition
L'accès aux phosphonates bicycliques [92] a été réalisé par une réaction de cycloaddition [2 + 2] entre des phosphonates vinyliques cycliques et des cétènes. Dans cette séquence, le produit intermédiaire peut subir une fragmentation pour donner le carbocycle à 7 chaînons substitué par un groupe phosphoré. Ces cycles fonctionnalisés existent dans un certain nombre de produits naturels dont la préparation est assez difficile (Schéma 79).
Les phosphonates d'énones s'avèrent d'excellents diénophiles pour la réaction de Diels-Alder. Catalysées par les acides de Lewis, elles donnent des polycycles contenant un groupement phosphonate. Parmi les acides de Lewis étudiés (AlCl3, ZnCl2, SnCl4, BF3.Et2O, TiCl4, FeCl3) pour la réaction du piperylène, le chlorure stannique s'est avéré particulièrement efficace en termes de quantité exigée, de taux de conversion et de rendement en cycloadduit. Les réactions de Diels-Alder de ces phosphonates d'énones avec une variété de 1,3-diènes ont été efficacement effectués (Schéma 80) [93].

En termes de stéréosélectivité, l'addition suit uniformément le principe cis et l'addition endo en ce qui concerne la cétone, suggérant la plus grande importance de la cétone sur le groupe phosphonate pour diriger l'addition endo. Il est également remarquable que dans presque tous les cas étudiés, la réaction de Diels-Alder implique la double liaison activée. La seule exception concerne le cyclopenta-1,3-ène où l'addition se produit exclusivement sur la double liaison la moins réactive. Ces diènes ont été aussi utilisés avec la cyclohex-2-ènone phosphonate, pour donner des cycloadduits aux alentours de 85%.
3.1.15 Utilisation des composés diazo et réactif de Bestmann-Ohira
Une amélioration ingénieuse et élégante de la préparation des alcynes est présentée avec l'utilisation du diméthyl 1-diazo-2-oxopropylphosphonate (Schéma 81) [94]. Ce réactif permet l'addition aux aldéhydes dans des conditions douces évitant l'utilisation des bases fortes à des températures très basses. Différents aldéhydes ont été soumis à la transformation avec succès.

Des composés variés tels que le 2,3-O-isopropylidène-D-glycéraldéhyde ou l'aldéhyde de Garner conduisent aux alcynes par le biais de cette méthode (Schéma 82) [95].

L'homologation d'un carbone des aldéhydes aux alcynes est réalisée à température ambiante, par le réactif d'Ohira en présence de K2CO3 dans le MeOH. Ce réactif est employé avec succès dans la synthèse des dérivés du dioxalane [96] du glycol-1-ynitol d'éthynyle [97], et des composés naturels [7] (Schéma 83).
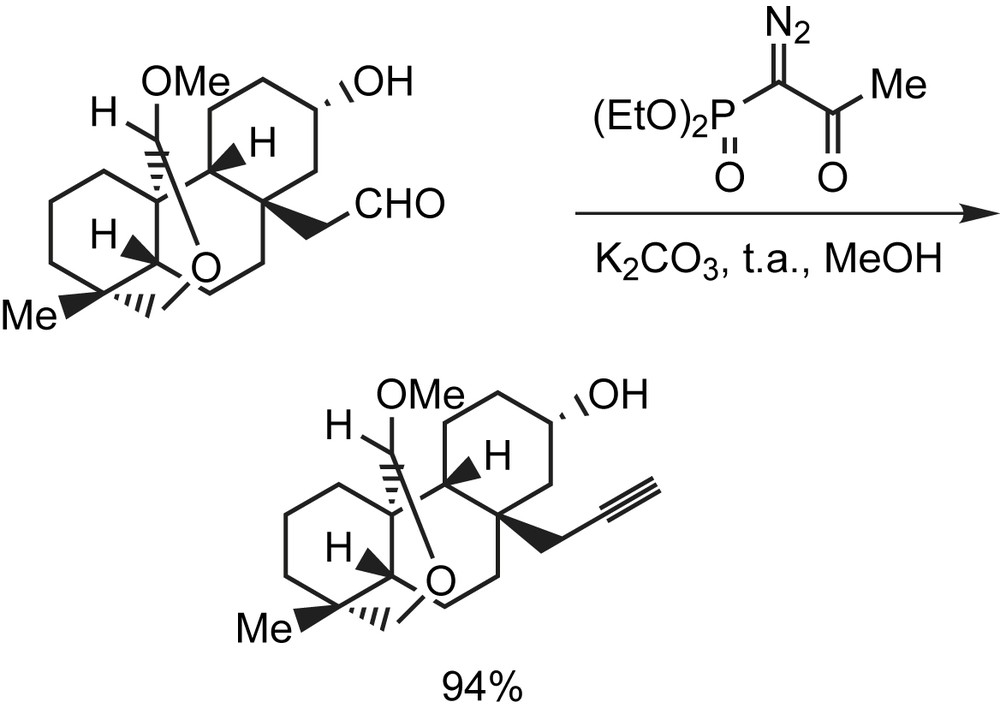
Le plan rétrosynthétique établi, pour la synthèse du diastéréomère (9S,18R)-seco de l'acide du cyclamenol A (un inhibiteur naturel d'adhérence de leucocyte aux cellules endothéliales) [98], fait intervenir un couplage stéréosélectif de l'iodure de vinyle à un alcyne, au moyen d'une réaction de Sonogashira. La synthèse de cet iodure fait intervenir le réactif de Bestmann-Ohira (RBO) avec une lactone pour donner l'alcyne correspondant. Ce dernier sera transformé en molécule cible par un procédé en deux étapes (Schéma 84). Divers analogues du cyclamenol A sont aisément accessibles avec cette stratégie.
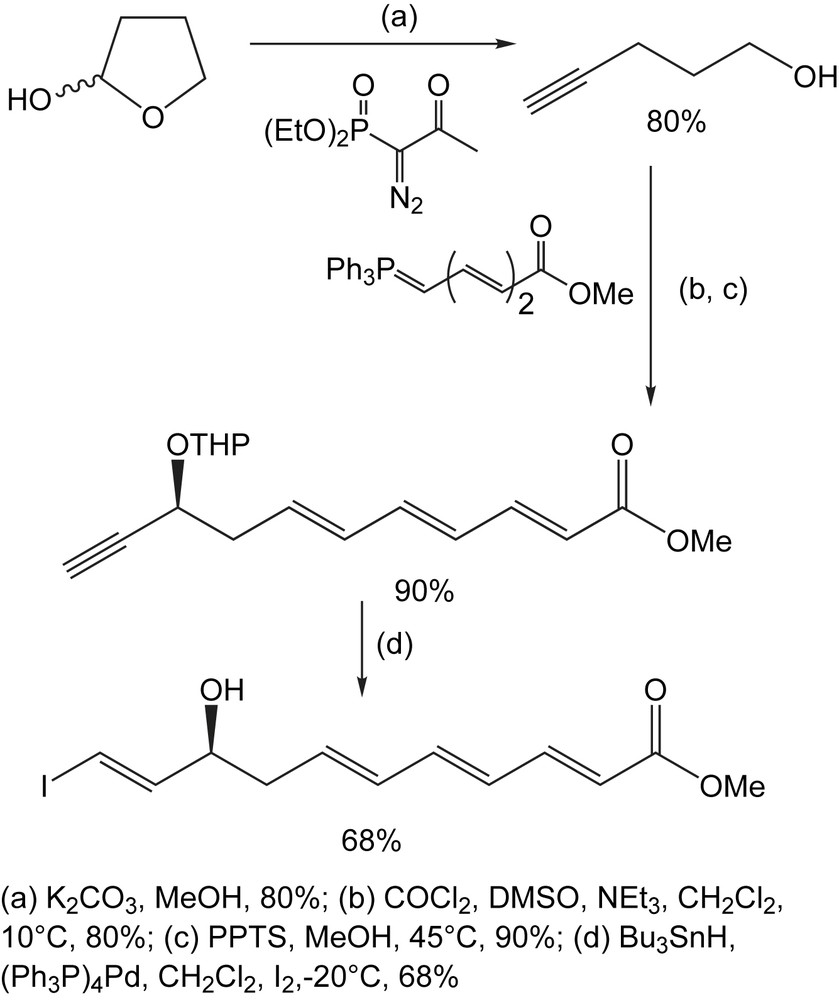
Un autre exemple de l'utilisation de ce réactif est illustré par la synthèse de la cyclopentanone énantiomériquement pure, nécessaire pour accèder aux noyaux tétracycliques de tigliane [99], présents dans un certain nombre de composés biologiquement actifs tels que le phorbol, un protecteur puissant contre les tumeurs.
La lactone énantiopure (1S,5R)-(−)-oxa-bicyclo[3.3.0] oct-6-én-3-one sert de produit de départ. Cette séquence (Schéma 85) comporte la préparation initiale de l' hydroxyester TBS-protégé, suivie de l'hydrogénation catalytique de la double liaison et la réduction de l’ester par le DIBAL pour fournir l'aldéhyde. Cet aldéhyde est efficacement converti en alcyne correspondant, par traitement avec le diméthyl (1-diazo-2-oxopropyl) phosphonate (RBO) et le K2CO3 dans le méthanol. Après déplacement du groupement TBS et l'oxydation de Swern, la cyclopentanone (S)-(−)-2-(2-propynyl) énantiopure est isolée. Il convient de noter que toutes les tentatives de convertion directe du lactol en 2-propargylcyclopentanol par l'utilisation du diméthyl (1-diazo-2-oxoprpyl) phosphonate ou encore, par le TMS-diazométhane de lithium ont complètement échoué.
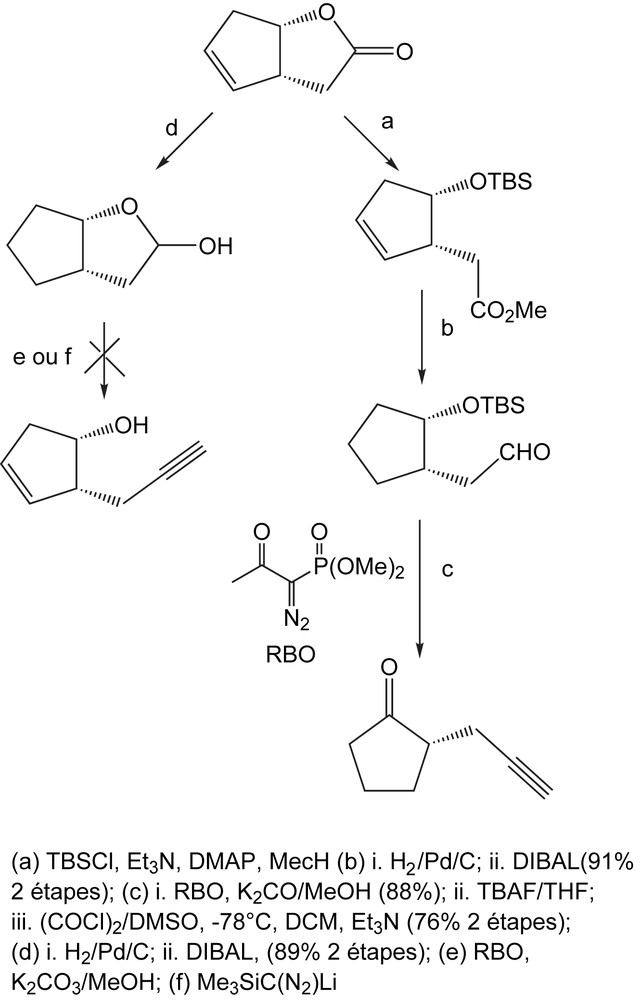
3.1.16 Réaction d'insertion
La réaction d'insertion de liaison C-H des composés α-diazocarbonylés catalysée par le Rh (II) est bien connue pour la construction de divers carbocycles à cinq chaînons. Soumis à une catalyse au rhodium (II) et au reflux de toluène, l'éther triméthylsilyloxy donne principalement des α-phosphono-δ-lactones [100], par un réarrangement de Wolff du métallocarbène intermédiaire (Schéma 86).
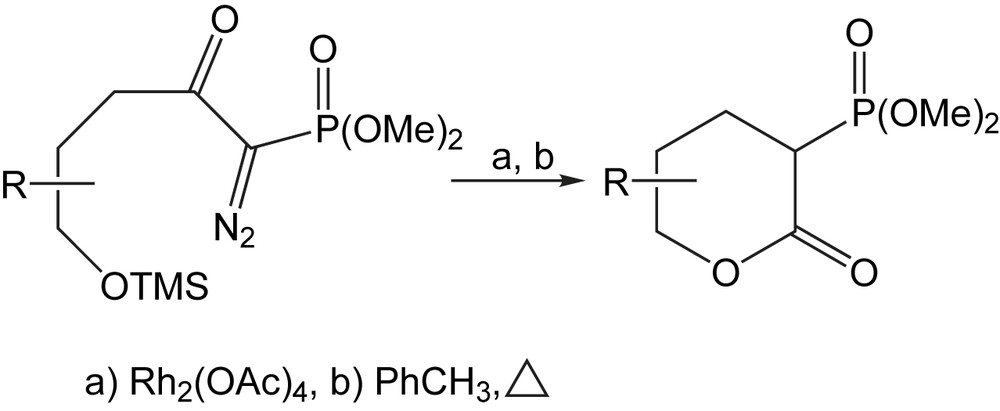
En appliquant les mêmes conditions de réaction à certains composés analogues aux α-diazo-carbonylés présentant une fonction éther tert-butyldiméthylsilyloxy, la réaction d'insertion ne se produit pas et mène aux cyclopentanones. Les composés diazo de départ sont facilement préparés, en deux étapes, à partir de (S)-méthyl-3,4-tert-butyldiméthylsilyloxy butanoate de méthyl connu ou des esters aisément disponibles (Schéma 87) [100].

Dans les mêmes conditions, le composé diazo dans lequel R1 = R2 = H, donne la cyclopentinone avec des rendements de 62% à côté d'une petite quantité de la δ-lactone (9%) (Schéma 88). Ces résultats ont démontré que l'emplacement du groupe silyloxy présente un effet connu pour favoriser l'insertion des métallocarbènes dans la liaison C-H adjacente. La réaction n'est pas sensiblement influencée par la nature des groupes alkyles substituant l'atome de silicium. Eneffet, le remplacement du groupe triméthylsilyloxy par le tert-butyldiméthylsilyloxy ne modifie pas le résultat de la réaction, probablement parce que la proximité de la liaison C5-H vicinal cause un gêne stérique pour empécher la réaction d'insertion d'avoir lieu.
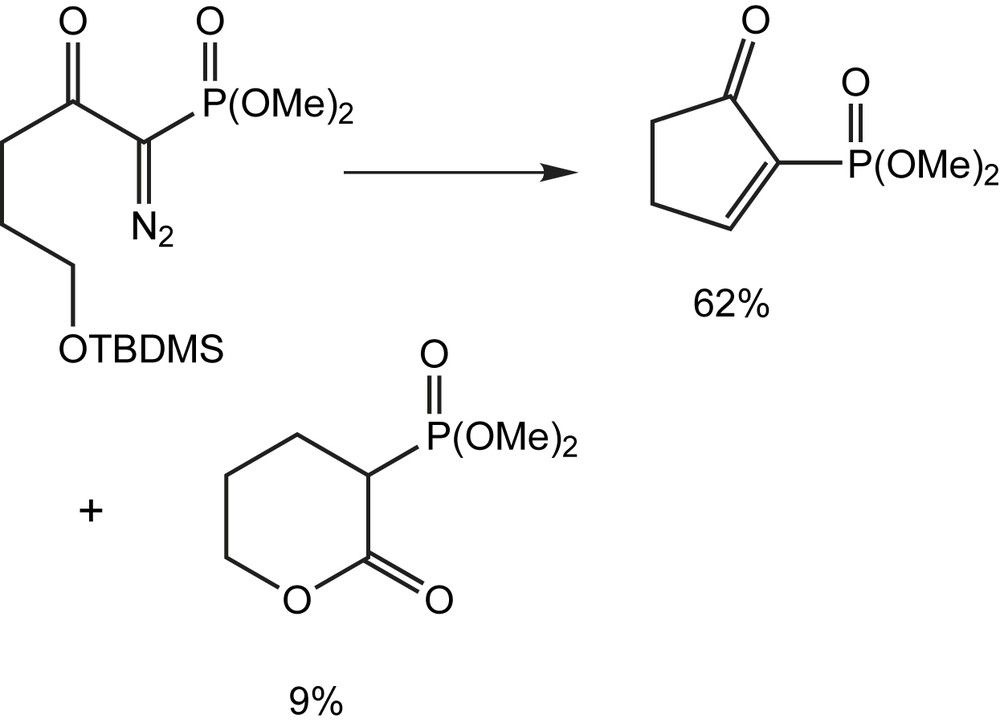
La présence d'un groupe gem-diméthyle en position δ augmente l'encombrement de la liaison C-H, en position ω adjacente, et empêche ainsi la réaction d'insertion d'avoir lieu. Le composé diazo ne donne pas la cyclopentènone prévue mais, l'acétal de cétène silylé avec le rendement de 72%, selon un réarragement de Wolff [101].
3.1.17 Utilisation des carbanions dans les réactions de Horner-Emmons
En plus de leurs applications diverses, les β-cétophosphonates sont le plus souvent employés dans les réactions d'oléfinations de Horner-Wadsworth-Emmons (HWE), une modification de la réaction de Wittig.
3.1.17.1 Réaction de déphosphonylation
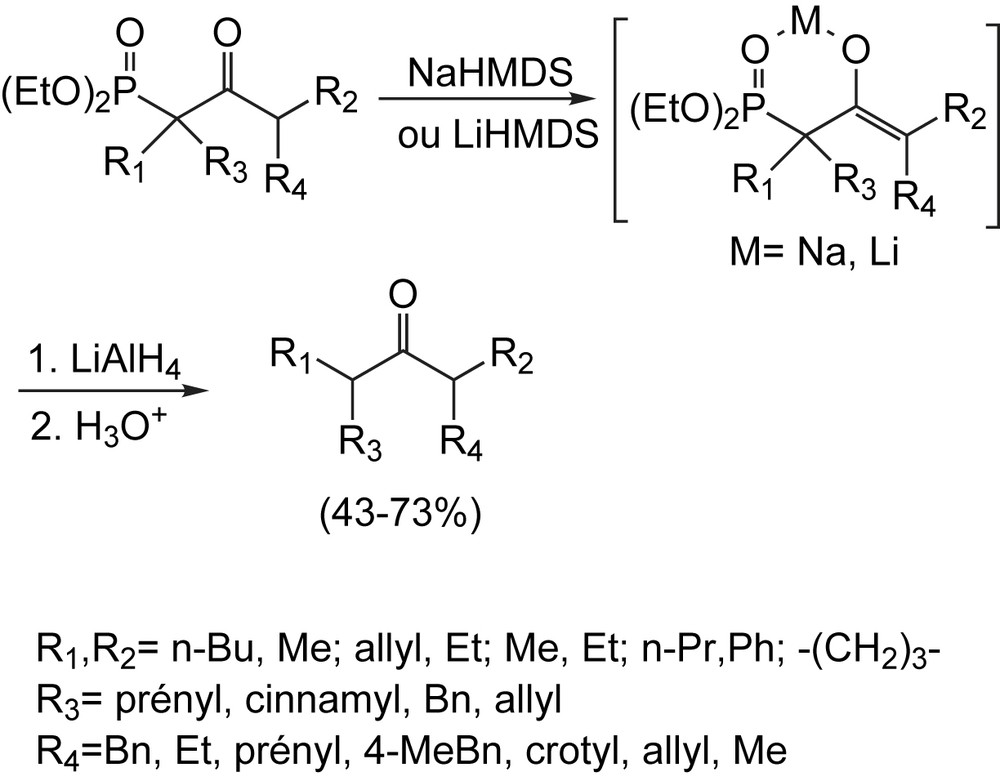
La déphosphonylation des précurseurs résultants conduit aux cétones correspondantes (Schéma 89) par l'action des énolates de sodium ou de lithium avec LAH (hydrure de lithium et d'aluminium) suivie d'un traitement acide. Ce procédé de déphosphonylation des β-cétophosphonates constitue un complément pour les méthodes déjà existantes pour l'alkylation régiospécifique des cétones non symétriques et fournit un accès facile à de nouvelles cétones [80].
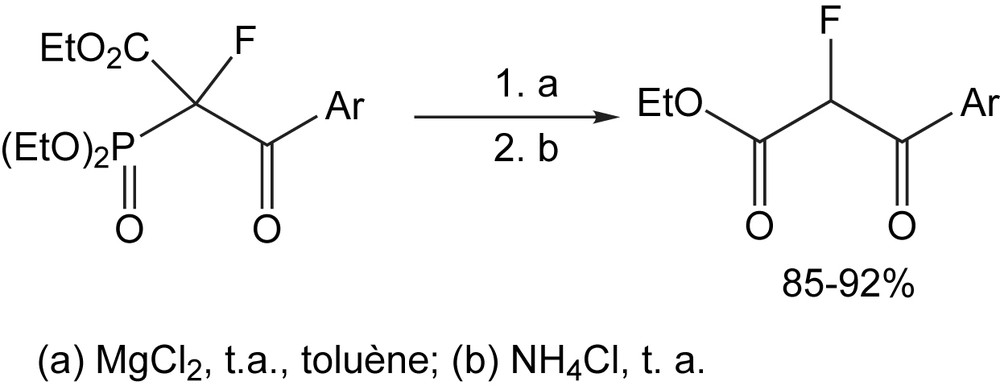
Une illustration claire des avantages de cette méthode synthétique est fournie par la conversion des diéthyl 1-fluoro-1-(éthoxycarbonyl)-2-aryl-2-oxothylphosphonates en α-fluoro-β-cétoesters correspondants. En raison de la nature sensible de la liaison P-C, la réaction évolue facilement à température ambiante par le clivage de la liaison P-C du diéthyl 1-fluoro-1-(éthoxycarbonyl)-2-aryl-2-oxoéthylphosphonates (aisément obtenu par la réaction de 1-fluoro-1-(éthoxycarbonyl) diéthylique avec des chlorures d'acide carboxylique aromatique) induit par MgCl2 (Schéma 90) [102].
3.1.17.2 A partir des diéthyl 1-(phénylsulfonyl)-éthylphosphonate
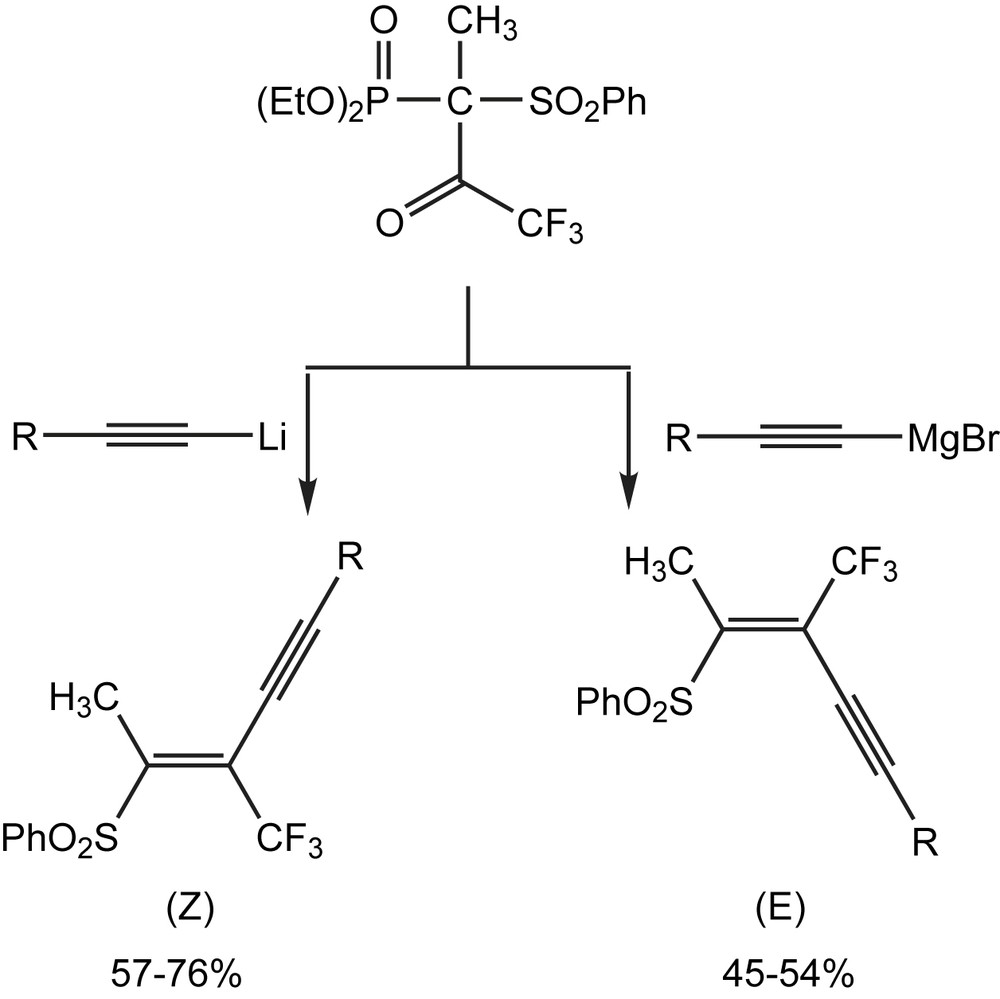
La synthèse one-pot stéréocontrollée des trifluorométhyl (E)- ou (Z)-ényne sulfones [103] s'effectue par l'attaque des trifluoroacyl-phosphonates par des acètylènures de lithium suivie de l'élimination de l'anion phosphate avec des rendements de 57–76%. Alors que le traitement du même phosphonate par des réactifs de Grignard acétyléniques donne plutôt le trifluorométhyl (E)-ynényl sulfones avec des rendements moyens (45–54%) (Schéma 91).
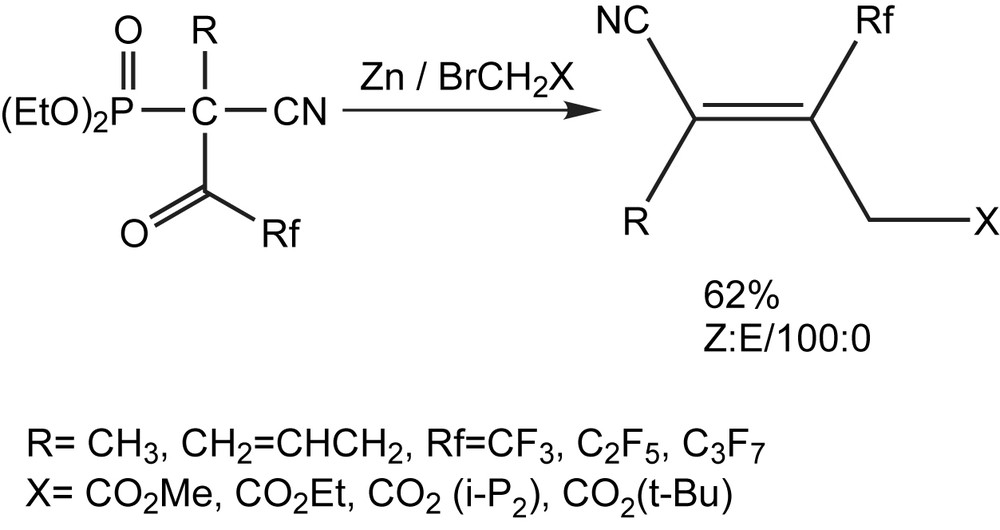
Après les sulfones, les esters β,γ-insaturés (Z)-γ-cyano-β-perfluoroalkylés, peuvent être obtenus stéréosélectivement en trois étapes, avec des rendements de 50–71% [104]. Cette méthode est commode, pour la préparation des dérivés fluorés, qui sont des intermédiaires utiles dans la synthèse des composés biologiquements actifs. Elle est basée sur l'action d'un organozincique sur des phosphonates, suivie de l'élimination de l'anion acide phosphonique (Schéma 92).
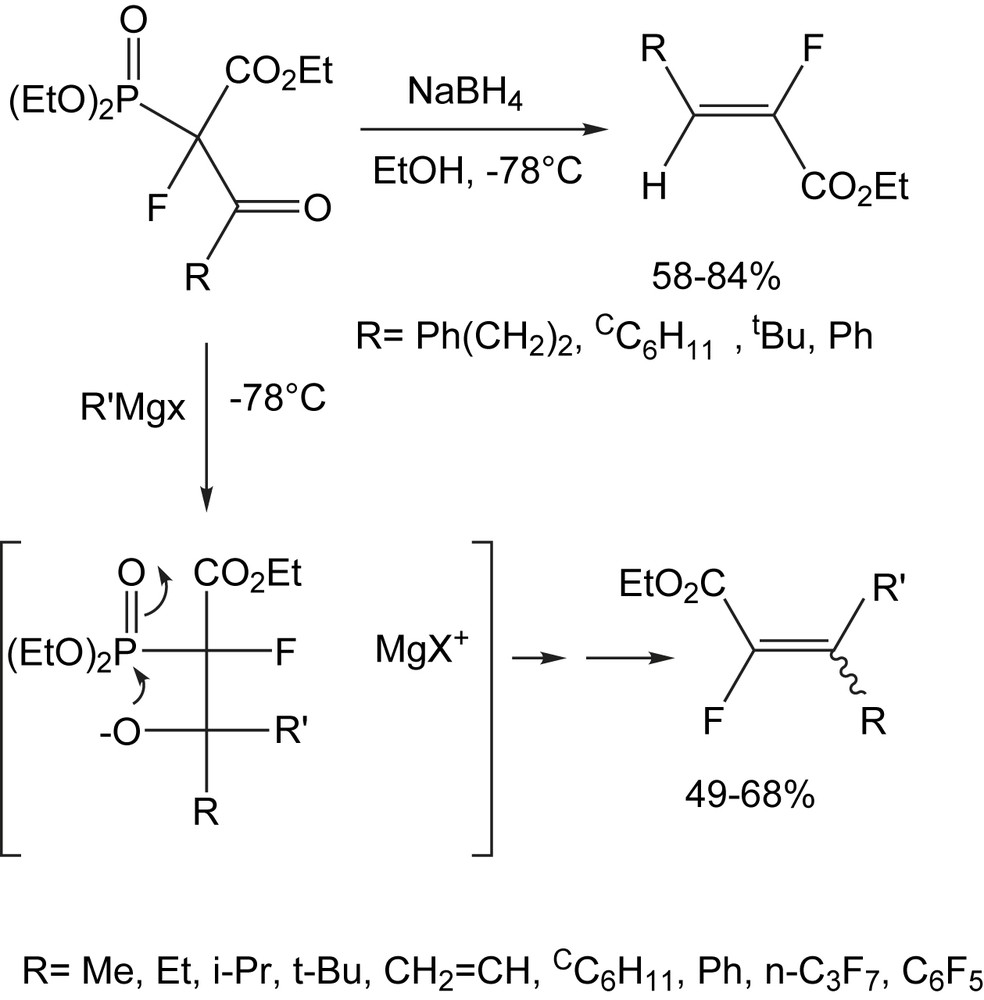
Les diesters α,β-insaturés α-fluorés peuvent être préparés par une voie directe (Schéma 93). La méthode est applicable aux alkyls primaires, secondaires, et tertiaires, alcényles, aryles, cyclohexyles, et réactifs de Grignard perfluorés [19]. Dans la plupart des réactions des cyano magnésiens étudiées, l'isomère E est exlusivement obtenu ; c'est le cas pour R = Me, i-Pr, t-Bu, et cyclohexyle. L'isomère E prédomine également (96%) pour R = n-Pr. Cependant, cette tendance est inversée en faveur de l'isomère Z, dans les cas ou R = Ph et CH2CH et le rapport (E/Z = 0:100) pour R = n-C3F7. Le borohydrure de sodium joue aussi le rôle de nucléophile et il est ajouté à la solution du phosphonate dans EtOH à −78 °C, pour donner les esters α,β-insaturés α-fluorés de configuration Z comme produit unique avec des rendements autour de 83% (Schéma 93).
Une autre application concerne la préparation des allylsilanes, qui sont particulièrement intéressants lorsqu'ils sont porteurs d'un groupement carbonyle en position β car ils réagissent aussi bien avec les nucléophiles qu'avec les électrophiles. Leur préparation utilise une réaction de Horner-Emmons entre divers aldéhydes et β-cétophosphonates silylés (obtenus par alkylation des β-céto-phosphonates) selon les deux méthodes [105] décrites dans le Schéma 94. Dans tous les cas, c'est l'isomère Z qui est obtenu majoritairement.

L'extension aux aldéhydes optiquement actifs tel que le D-glycéraldéhyde acétonide, permet l'accès à de nouveaux β-cétoallylsilanes chiraux avec une très bonne diastéréosélectivité (95/5). Ces derniers peuvent être soumis à une amination pour donner des β-amino cétones optiquement actifs avec un très bon excès diastéréomérique.
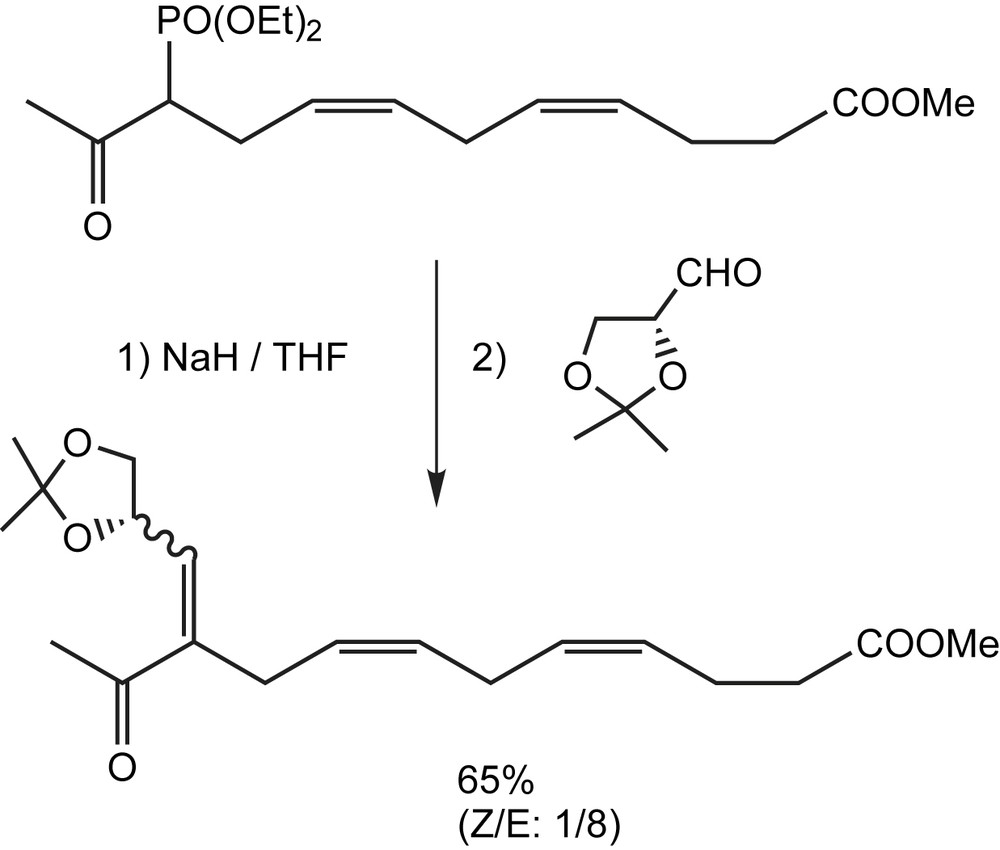
La condensation de Horner-Emmons du sel de sodium du cétophosphonate avec l'isopropylidène D-glycéraldéhyde produit des énones chirales non racémiques E et Z (8/1) avec un rendement de 65% (Schéma 95). Il est intéressant de noter que le rapport plus élevé d'énones E/Z est semblable à celui de la synthèse de leukotriènes LGE2, probablement dû à la chaîne latérale plus longue et plus rigide [77].
3.2 Les α-cétophosphonates
3.2.1 Transformation en hydroxyphosphonates
La réduction des α-cétophosphonates par les alkylphosphines, comme la triméthylphosphine ou encore la diphényl méthylphosphine [106], conduit dans des conditions douces, aux α-hydroxy-phosphonates correspondants avec d'excellents rendements (Schéma 96). Les différentes phosphines utilisées, montrent que les substituants aliphatiques sur la phosphine sont nécessaires pour que la réduction puisse se produire.
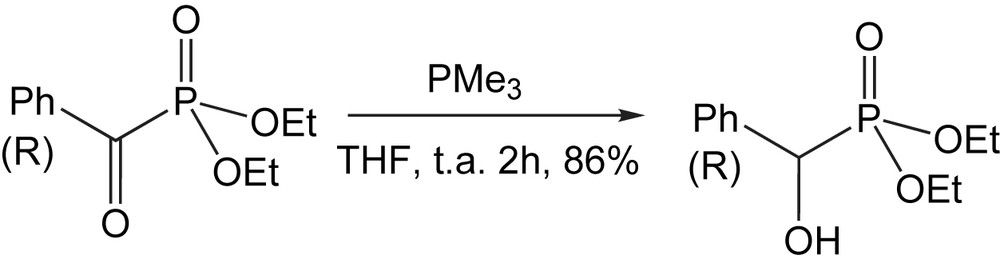
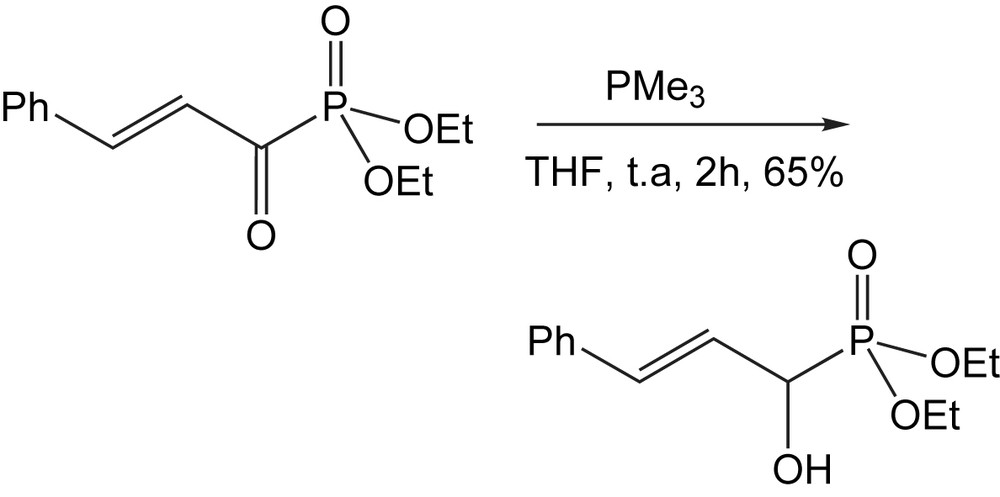
Il convient de noter que dans le cas des α-céto-phosphonates β,γ-insaturés, la réduction aura lieu sur le carbonyle avec de bons rendements et la double liaison carbone-carbone (Schéma 97) reste inchangée dans ces conditions de réaction.
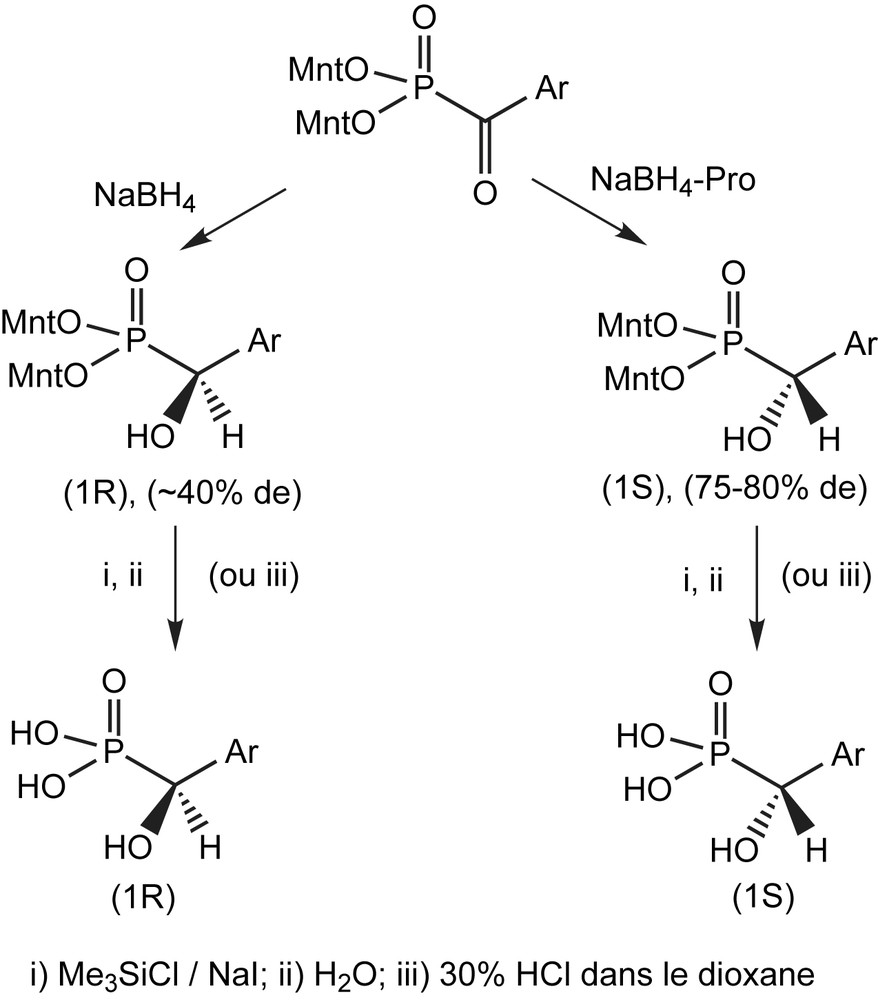
Une étude récente utilise le complexe chiral NaBH4-l-proline, pour la réduction des dimenthyl α-cétophosphonates (§3.2.2) [40]. La réduction par le borohydrure de sodium entraîne déjà une induction asymétrique sur l'atome de carbone en α, avec un faible excès de l'énantiomère R, après hydrolyse en l'hydroxyphosphonate correspondant. A cause de la double induction asymétrique, la réduction par le complexe chiral NaBH4-l-proline (Pro), conduit à une pureté énantiomérique élevée, après hydrolyse. Les hydroxyphosphonates obtenus sont isolés sous forme de diastéréoisomères purs et sont transformés en acide hydroxyphosphonique optiquement actifs par hydrolyse. La configuration R d'un d'entre eux a été déterminée par diffraction des rayons X (Schéma 98).
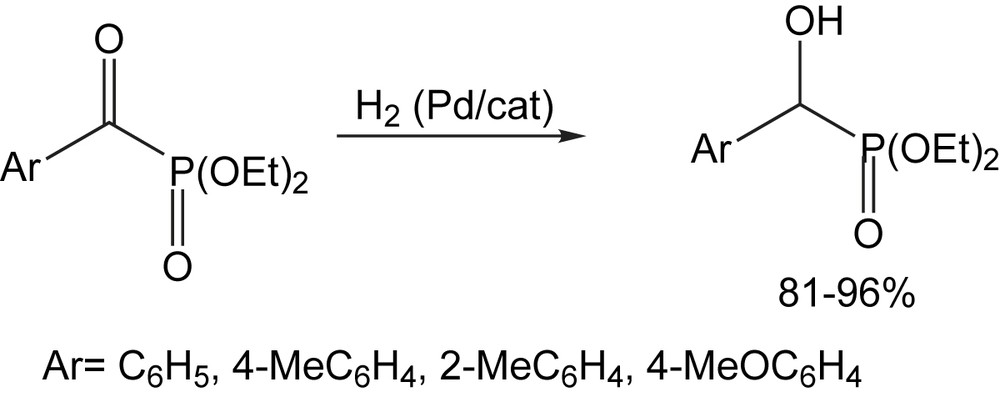
Concernant l'hydrogénation catalytique par l'hydrogène moléculaire, et à l'exception des échecs en présence de Nickel de Raney, la littérature ne dispose pas d'exemple concernant les α-oxophosphonates. Elle a été appliquée uniquement dans le cas de 1-benzoyl-oxyalcènylphosphonates. Des composés modèles tels que les esters diéthyliques des acides benzoyl-phosphoniques substitués et le diéthyl acétyl-phosphonate ont été sujet à cette hydrogènation moléculaire. Les réactions ont été effectuées à la pression d'hydrogène de 1 at en présence de palladium, dans le méthanol ou éthanol [107]. Avec 5–10 mol% de palladium sur carbone, le diéthyl benzoylphosphonate est facilement hydrogéné dans le méthanol à ébullition. Pour une conversion à 100%, le rendement en diéthyl [hydroxy(phényl)méthyl]phosphonate atteint 96% en 1,5 h. A température ambiante, la conversion atteint 92% en 1 h, mais la sélectivité du processus est considérablement réduite, et le rendement en produit désiré ne dépasse pas les 81% (Schéma 99).
Pour les biocatalyseurs, cinq espèces différents des micro-organismes, à savoir, Rhodotorula rubra, Rhodotorula glutinis, Cladosportum sp., Verticillium sp., et la levure du boulanger, peuvent être utiles pour la réduction énantiosélective d'une variété de 1-oxoalkylphosphonates diéthyliques (rdt : 62% et 99% ee). Afin d'éviter la décomposition du substrat, les bio-réductions ont été effectuées dans des conditions anhydres [108], employant des cellules lyophilisées immobilisées sur la célite R630. Le choix de cette dernière assure la protection du biocatalyseur contre la toxicité du solvant tout en maintenant des conditions anhydres (Schéma 100).
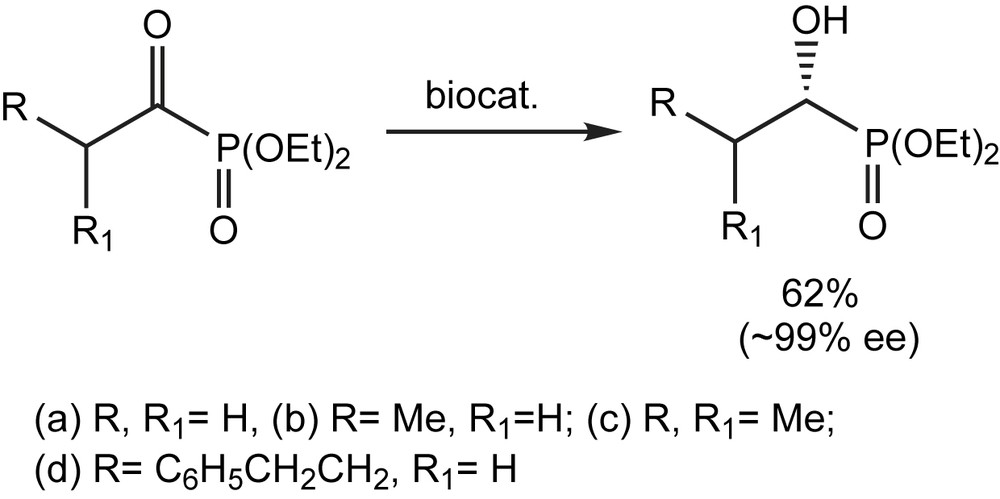
3.2.2 Transformation en α-phosphonoénamides
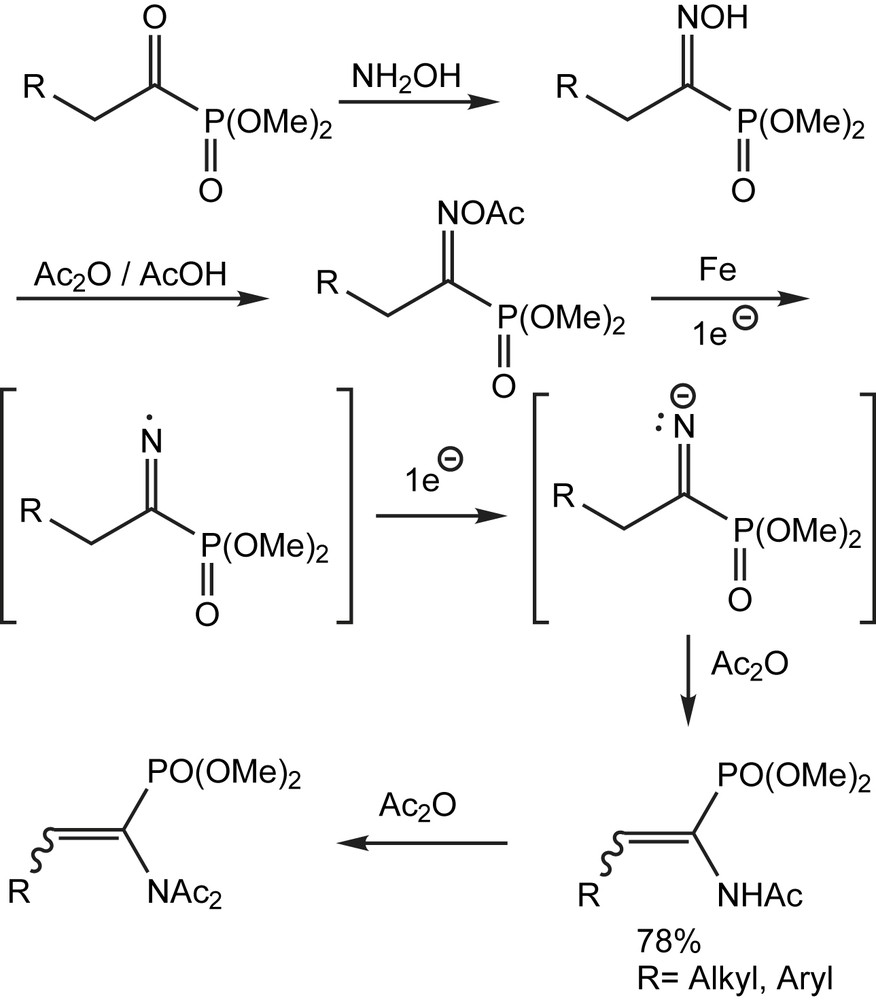
Les diverses applications des phosphonates d'acyle s'étendent aussi à la préparation d'énamides. Ces derniers sont décrits par une nouvelle voie pratique et assez directe [109] illustrée par le Schéma 101. Ainsi, la réduction de l'acétate d'oxime dérivé du phosphonate d'acyle, par transfert d'électron à partir du fer métallique aurait comme conséquence la rupture de la faible liaison N-O et la formation d'un radical iminyle. Un deuxième transfert d'électron mènerait alors à l'anion iminyle, qui serait alors capturé par l'anhydride acétique pour donner l'énamide désiré.
3.2.3 Préparation des hydrazones α-phosphonylées N-fluoroalkylées
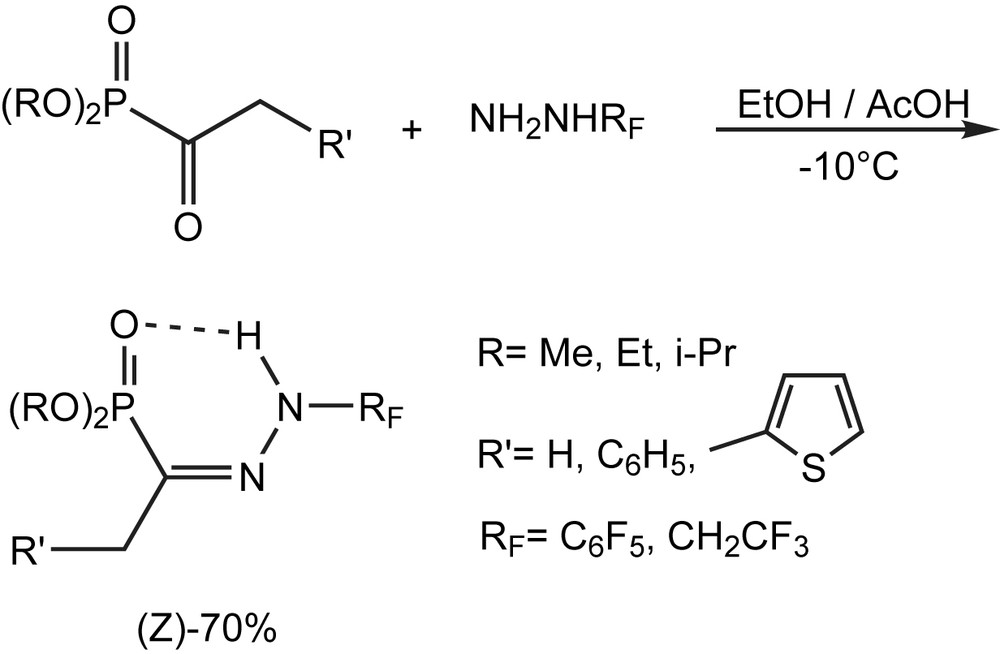
L'action des hydrazines fluoroalkylées sur les α-cétophosphonates conduit aux hydrazones α-phosphonylées N-fluoroalkylées (Schéma 102), qui présentent un intérêt dans la synthèse des phosphopyrazoles. [110]. L'étude spectroscopique montre que la seule configuration obtenue est Z, à cause de l'établissement d'une liaison hydrogène intramoléculaire qui stabilise la structure.
3.2.4 Préparation de cyclopentanones
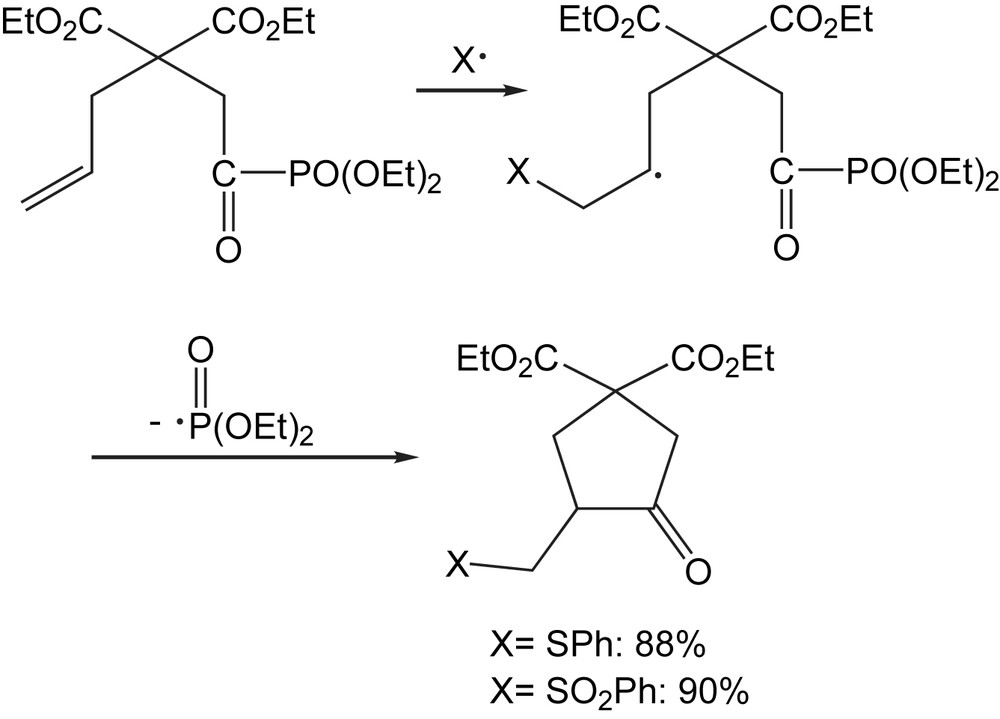
La cyclisation radicalaire des acylphosphonates en présence d'hexaméthyldiétain dans le benzène forment les cyclopentanones isolées avec un rendement de 91%, sans formation du produit de réduction [111]. La facilité de cette β-fragmentation dépend généralement de la nature de la liaison π formée et la longueur de la liaison σ cassée. Plus la liaison π formée est forte, plus la fragmentation-β serait facile (Schéma 103).
3.2.5 Couplage avec les aldéhydes et les cétones
Le couplage des aldéhydes et des cétones avec les acylphosphonates se fait en présence des lanthanides comme le samarium ou le SmI2 selon une méthode indirecte [112]. Elle est constituée de trois étapes différentes comme indiqué dans Schéma 104 : i) réaction des acylphosphonates avec des aldéhydes pour former des acyloxy-phosphonates; ii) élimination réductrice des acides carboxylique, suivie de condensation avec des cétones; et iii) oléfination de Horner-Emmons.
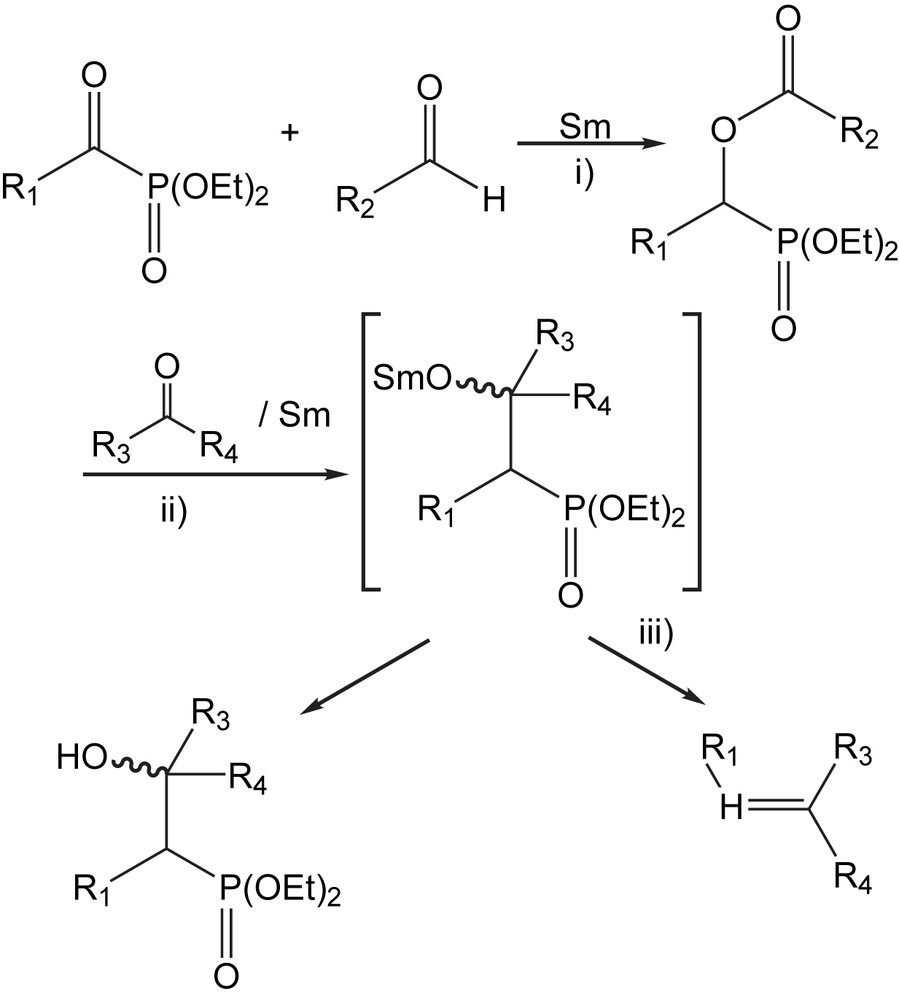
Les acyloxyphosphonates formés subissent une élimination réductrice en présence de SmI2 pour donner les β-hydroxyphosphonates diéthyliques correspondants avec des rendements raisonnables (40–64%). Les β-hydroxyphosphonates peuvent être convertis en oléfines directement avec des bases (réaction de Horner-Wadsworth-Emmons). Néanmoins toutes les tentatives pour produire les oléfines directement à partir des acyloxy-phosphonates et les composés carbonylés, avec un excès de SmI2 dans des solvants polaires, ont échoué même à des températures élevées.
Cette étude révèle que les deux premières réactions fonctionnent bien et produisent les β-hydroxyphosphonates avec de bons rendements (Schéma 105).
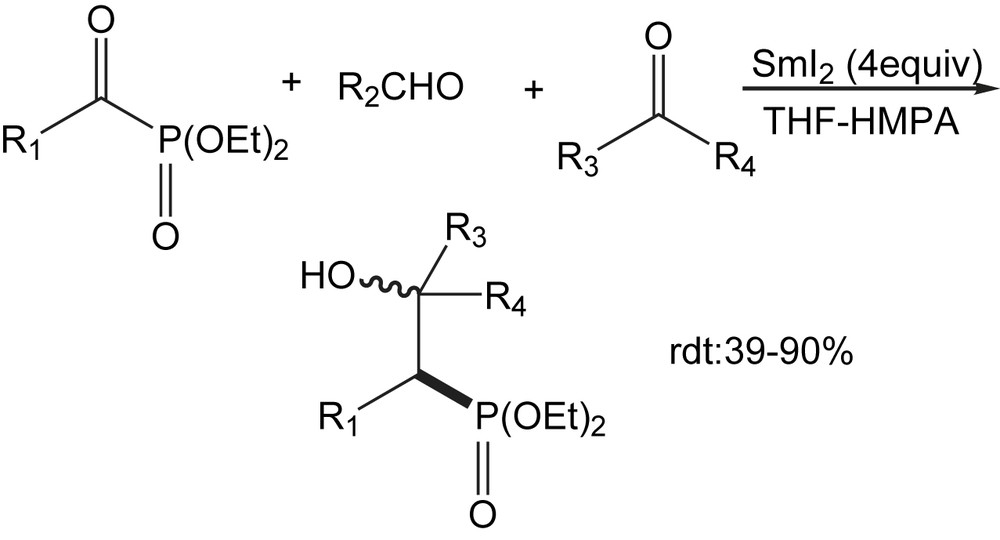
La cyclohexanone représente l'exemple d'une cétone cyclique utilisée . Par contre, une grande variété d'aldéhydes (saturé et insaturé, à chaîne acyclique ou aromatique) a été testée.
3.2.6 Réarrangement [3,3] sigmatropique
L'alkylation des α-cétophosphonates en α de la fonction carbonyle, est possible grâce à un processus en deux étapes qui commence par une O-alkylation et se termine par un réarrangement [3,3] sigmatropique. La première étape est stéréospécifique et conduit exclusivement à l'isomère E [113,114] (Schéma 106).
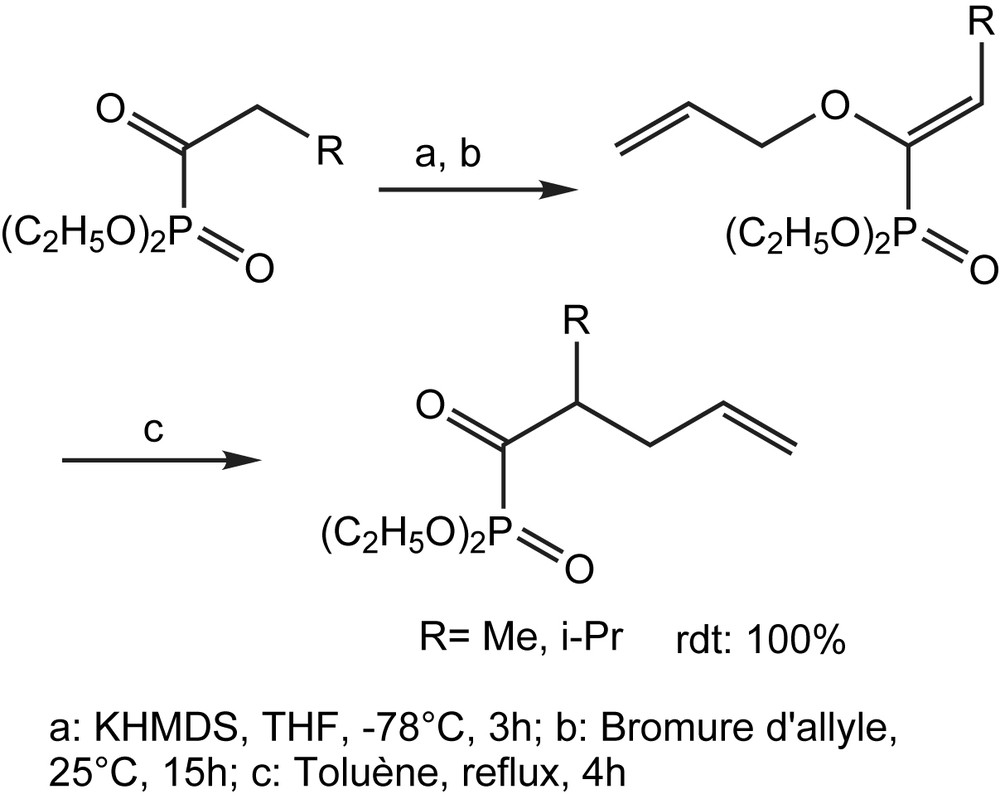
3.2.7 Synthèse de dérivés 1,1-difluorés
La conversion des 1-oxoarylphosphonates par le trifluorure de (diéthylamino)sulfur (DAST) conduit au 1,1-difluoroarylphosphonates avec de bons rendements (Schéma 107). Ces dérivés difluorés peuvent être transformés à leur tour en bisphosphonates, de nouveaux inhibiteurs de la phosphoglycérate kinase (3-PGK). Ces bisphosphonates exigent l'utilisation de produits de départ bifonctionnels ayant une réactivité électrophile sur deux sites. Cette condition est réuni edans les chlorures 3- et 4-(chlorométhyl)benzoyle [115].

3.2.8 Synthèse des acides carboxyliques α-chlorés
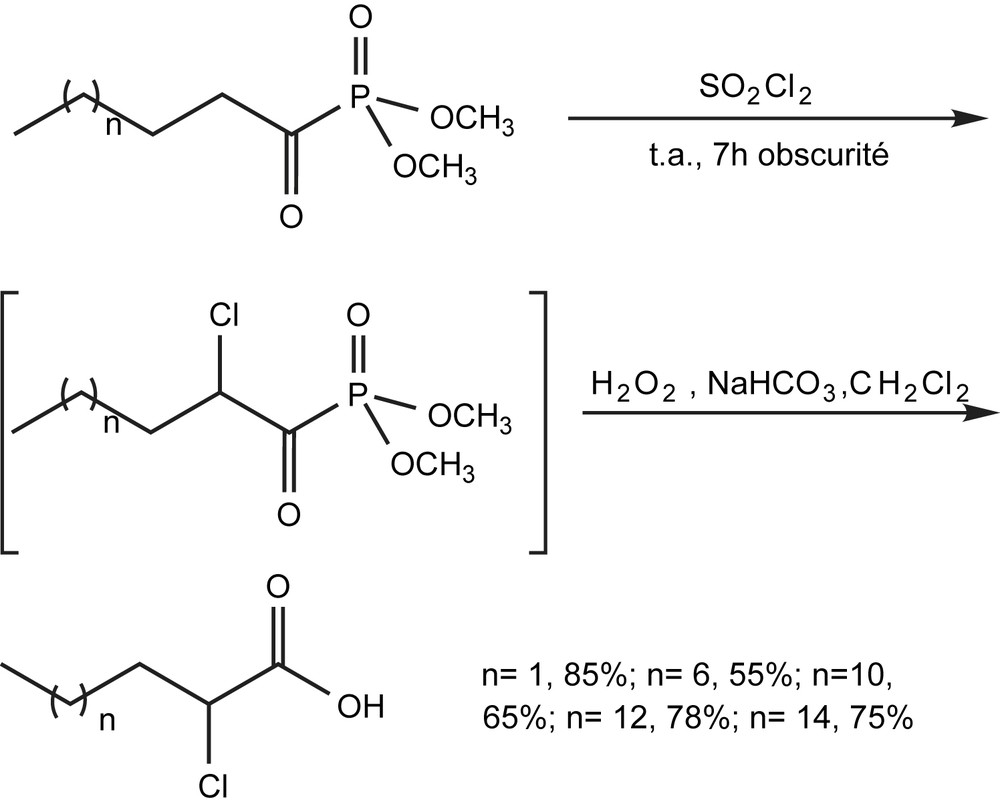
Les acides carboxyliques α-chlorés ont fait l'objet de plusieurs études, en raison de leur importance dans plusieurs domaines, en particulier enagrochimie. La chloration des α-acylphosphonates [116] par le chlorure de sulfuryle, puis l'hydrolyse par le peroxyde d'hydrogène et le bicarbonate de sodium constitue une nouvelle méthode d'accès à ces produits. Elle se déroule à température ambiante sans aucune compétition (Schéma 108).
3.2.9 Synthèse des carbocycles insaturés substitués par un groupement phosphoré
Les α-acylphosphonates β,γ-insaturés peuvent être utilisés comme précurseurs pour la synthèse de carbocycles insaturés de petite taille. Dans la séquence décrite par le Schéma 109, l'irradiation de l'intermédiaire diazo mène à la formation d'un carbène. Ce dernier subit à son tour une réaction intramoléculaire pour donner les cyclopropènes phosphonates [117]. Ces derniers pourront être des diènophiles intéressants dans des réactions de Diels-Alder.
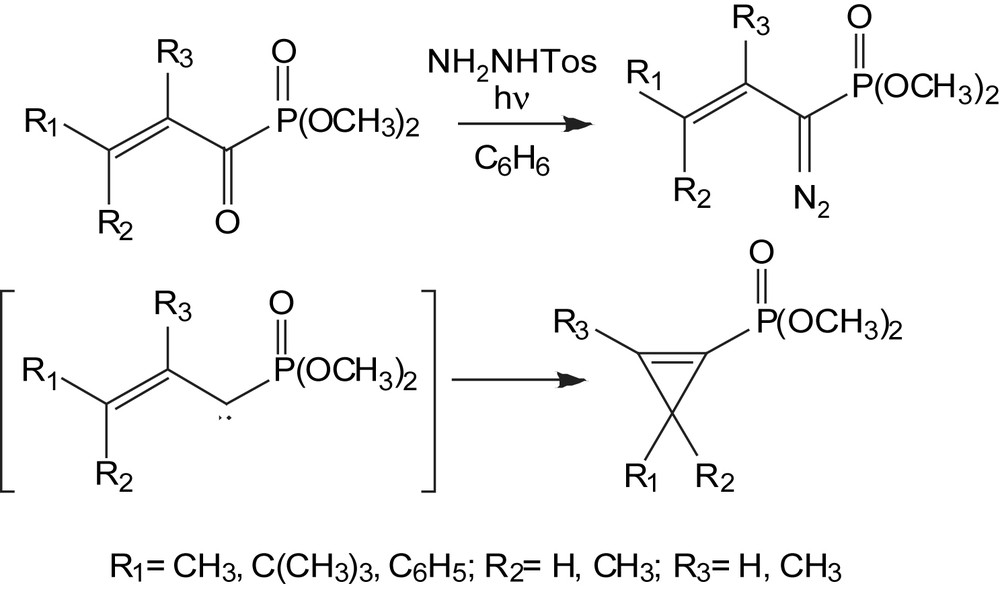
3.2.10 Préparation des acides phosphoniques monoesters α,β-insaturés
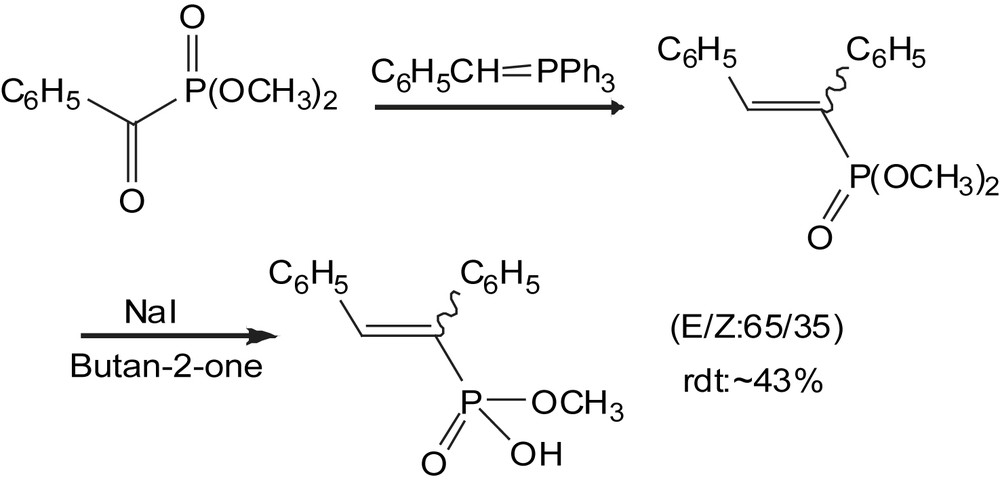
Les α-cétophosphonates sont aussi utilisés comme précurseurs pour la synthèse des acides phosphoniques monoesters α,β-insaturés [118]. Ils sont obtenus par action du diméthyl benzoyl-phosphonate sur le bromure du benzyltriphénylphosphonium. Après séparation, les deux isomères sont monohydrolysés séparément au reflux en utilisant du NaI dans la butan-2-one (Schéma 110).
3.2.11 Action d'allylindium
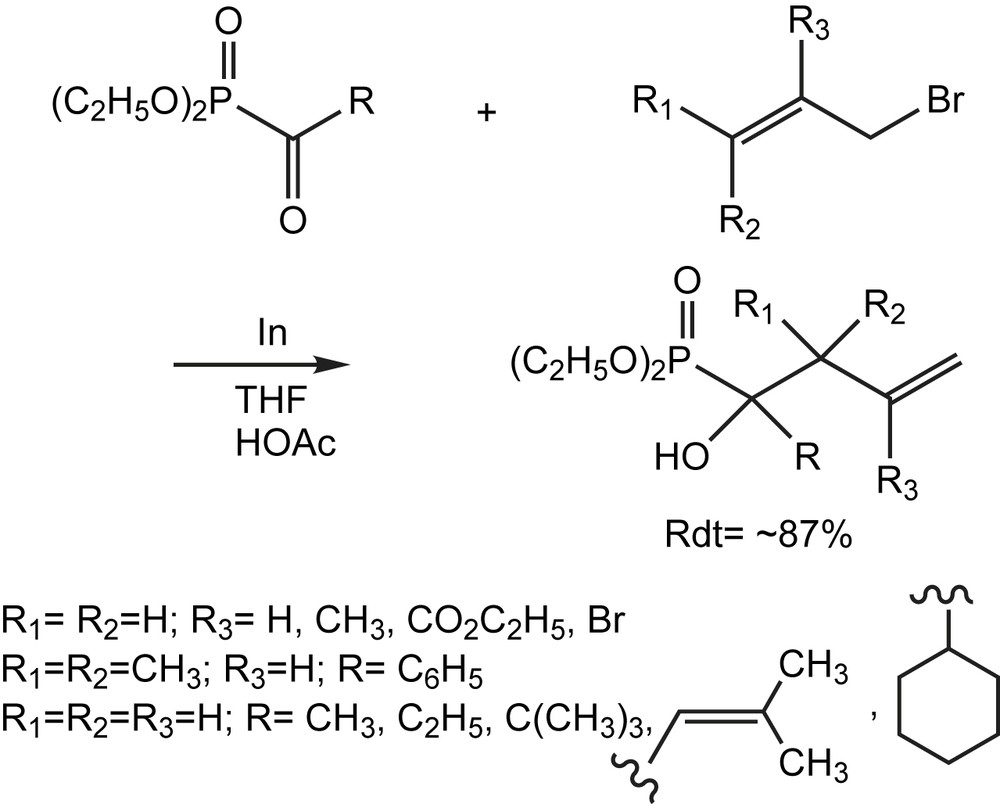
Lors des études sur le métabolisme des isoprénoïdes, des phosphonates analogues de farnésyl pyrophosphate ont été utilisés. Ceci a suscité l'intérêt pour la synthèse des α-hydroxy-phosphonates substitués. Comme la synthèse des α-hydroxyphosphonates tertiaires est peu connue, l'addition d'un allylindium sur des acylphosphonates en présence d'acide acétique [119] a été développée avec succès (Schéma 111).
3.2.12 Synthèse énantiosélective des α-hydroxy-phosphonates
Concernant la réaction aldol croisée des cétones activées, les progrès récents dans la catalyse asymétrique par la proline ont montré que le glyoxylate est un bon substrat pour ce type de réaction. Ces résultats ont été exploités pour la synthèse asymétrique d'α-hydroxyphosphonates optiquements actifs [120]. En raison de la réactivité des α-cétophosphonates envers les attaques nucléophiles et de l'élimination du groupe phosphonate, ils peuvent être considérés comme équivalents synthétiques des chlorures d'acide (Schéma 112).

3.2.13 Estérification de l'inuline
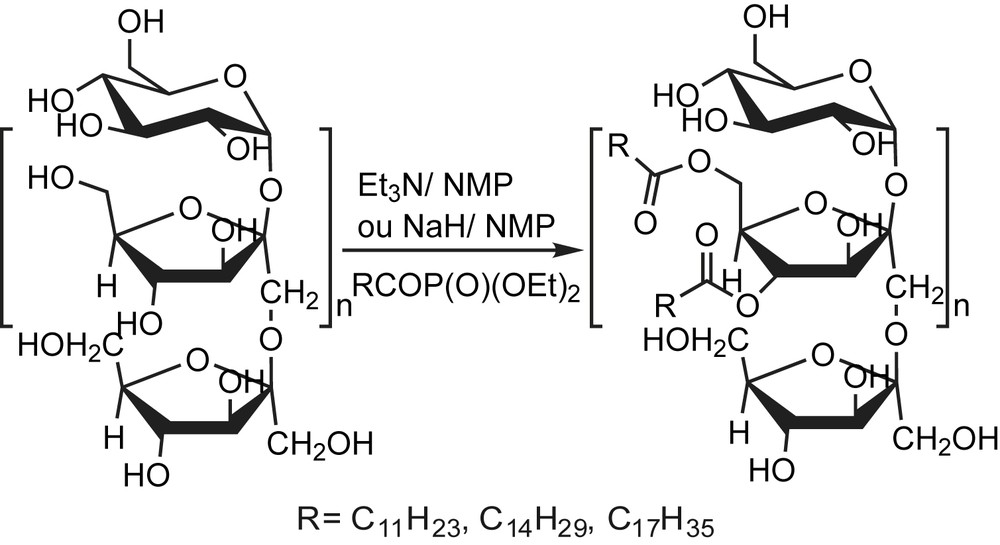
L'inuline, polysaccharide extrait de la chicorée, possède une structure non ramifiée de polyfructose qui, une fois greffée avec des chaînes acyles hydrophobes, peut agir en tant que stabilisant. Des propriétés tensioactives très intéressantes sont attribuées aux dérivés carbamoylés d'inuline, qui ont montré d'excellentes stabilités d'émulsion même dans des concentrations élevées en électrolyte.
Les différentes méthodes d'estérification utilisées ne sont pas appropriées pour le développement de processus industriellement, dû à la faible réactivité des esters.
Les phosphonates d'acyle montrent un caractère ambivalent dans leur comportement vis-à-vis des nucléophiles, puisqu'ils peuvent agir en tant que cétones envers des nucléophiles tels que l'hydroxylamine, et les amines. Cependant, envers la plupart des nucléophiles, ils agissent en tant que dérivés d'acides carboxyliques activés, avec le groupe phosphoryle agissent comme groupe partant. Pour l'estérification de l'inuline, cette propriété acylante est exploitée [121] (Schéma 113).
3.2.14 Préparation d'analogues phosphonylés de proline
Une préparation à grande échelle de l'analogue racémique de la proline phosphonylée [83] est réalisée avec un rendement de 50%. Elle se fait par le biais de la transformation de l'acylphosphonate (obtenu par l'action du chlorure de chlorobutyryle sur le trialkylphosphite) en oxime. La réduction de cette dernière par le zinc et l'acide formique, suivie de la cyclisation donne la proline correspondante (Schéma 114).
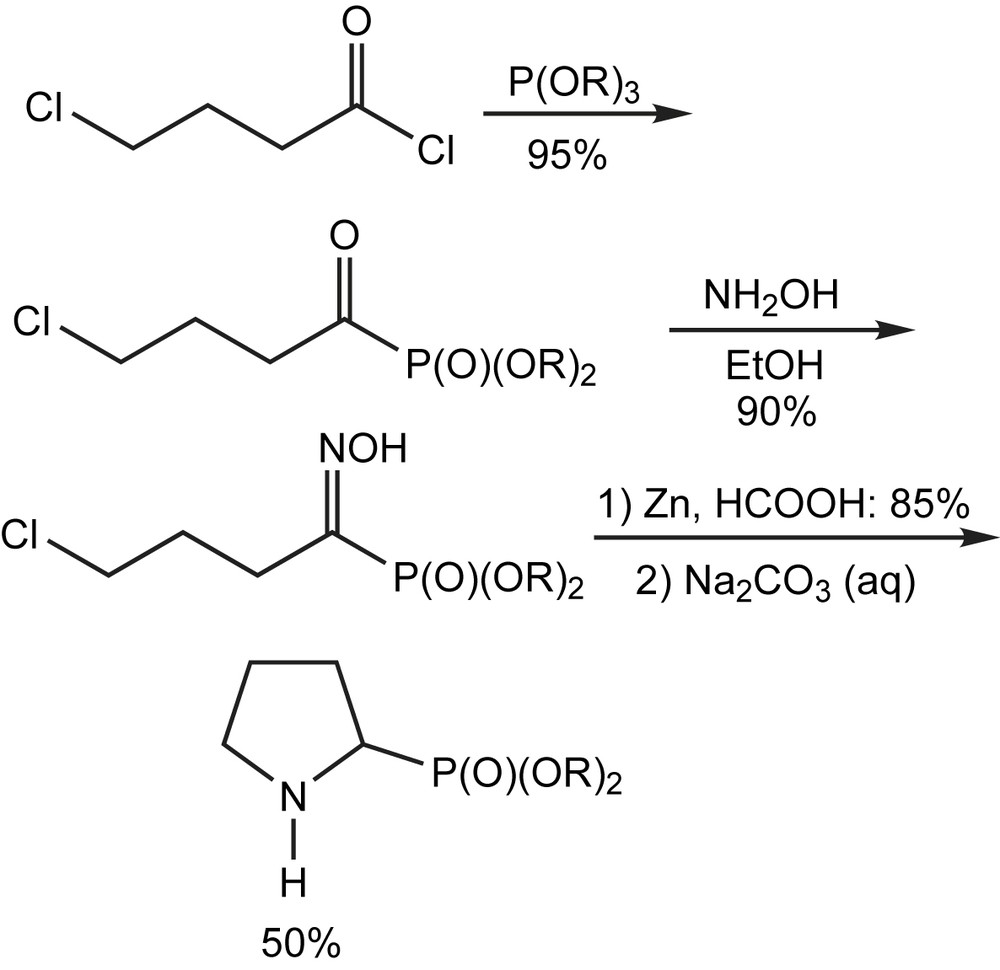
3.2.15 Préparation des anions d'acyle
Les acylphosphonates sont des précurseurs efficaces d'anion acyle, générés par l'action des ions cyanure [122], via un réarrangement phosphonate-phosphate. Ces intermédiaires réagissent aisément avec les aldéhydes pour fournir des produits benzoïnes croisés. De cette façon, il est possible de synthétiser une variété de benzoïnes portant des substituants (R1 – R2) aromatique-aromatique, aromatique-aliphatique, et aliphatique-aromatique (Schéma 115).
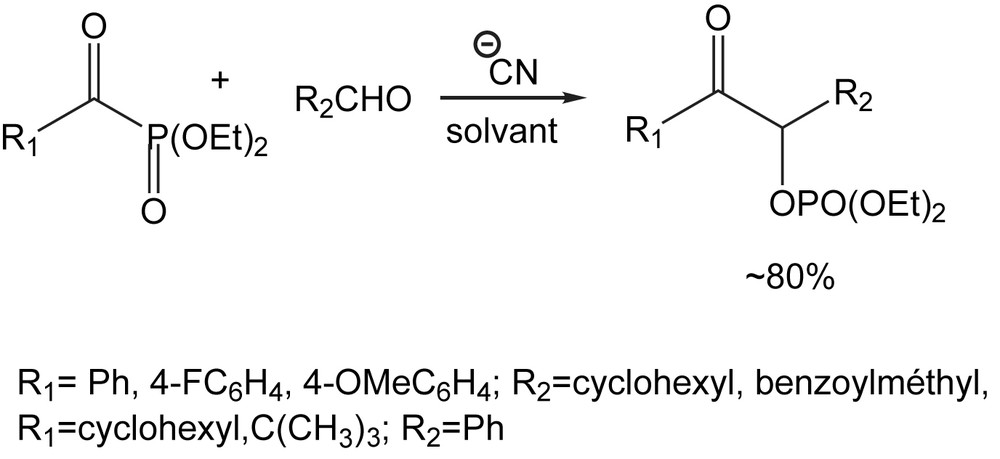
Cette nature carbanionique des intermédiaires générés par le réarrangement phosphonate-phosphate, est exploitée pour la cyanosilylation de divers alkyl- et arylphosphonates [123]. La réaction a lieu dans des conditions modérées en termes de température et temps de réaction et fonctionne sans catalyseur. L'hydrolyse acide des triméthylsilyloxycyanophosphonates mène aux α-hydroxy-α-cyanophosphonates avec des rendements élevés (Schéma 116).
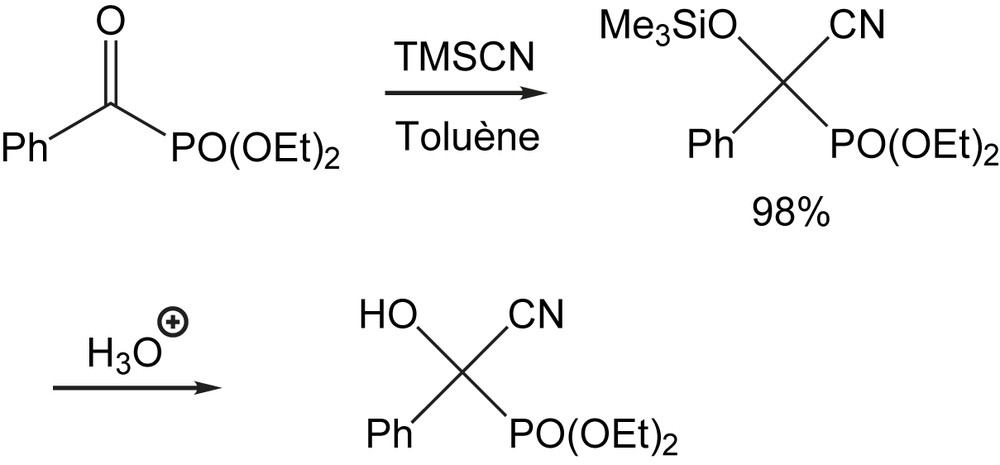
La protonation des équivalents d'anions acyles [124] fournit les cyanohydrines O-phosphates correspondantes avec de bons rendements. Ces cyanophosphates sont des précurseurs très interessants pour la synthèse de divers composés d'intérêt tels que les α-hydroxy β-amino acides et diols.
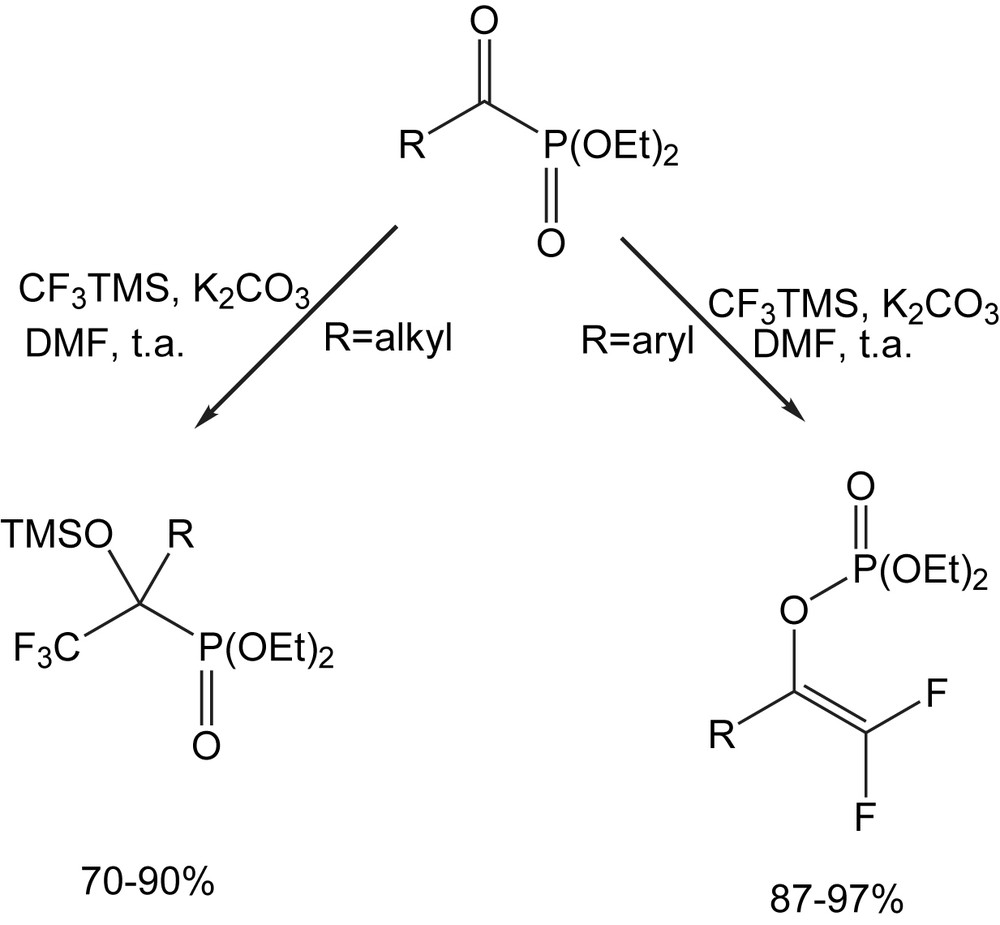
La trifluorométhylation par l'agent CF3TMS constitue [125] une extension à cette étude. Les phosphonates d'acyle réagissent facilement avec le CF3TMS en présence de K2CO3 dans le DMF à température ambiante, pour donner le 1-alkyl-2,2,2-trifluoro-1-triméthylsilyloxyéthyl-phosphonate avec 70–90% de rendement. L'emploi des benzoylphosphonates comme produits de départ conduit avec de bons rendements aux 1-aryldifluoroéthényl-phosphates (87–97%) (Schéma 117).
3.2.16 Accès aux bis-phosphonates
Les applications des α-cétophosphonates s'étendent aussi à la préparation des bis-phosphonates chimiquement et enzymatiquement stables et qui sont généralement employés dans le traitement des diverses maladies montrant un métabolisme anormal de calcium, comme par exemple les dérivés du 1-hydroxyméthylène-1,1-bis-phosphonates (HMBP). De nouvelles méthodes de synthèse de ces monométhylesters sont décrites dans la littérature [126,127], à partir d'un α-cétophosphonate silylé, comme le montre le Schéma 118.
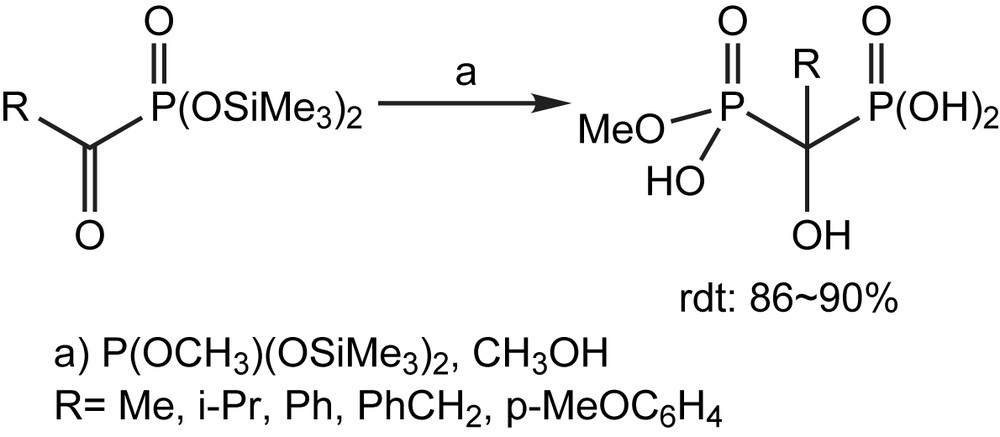
Les α-cétophosphonates sont obtenus à partir de différents chlorures d'acide aliphatiques ou aromatiques. Néanmoins afin d'éviter la formation d'énol, une méthode alternative employant le tris (triméthylsilyl) phosphite a dû être considérée comme dans le cas du chlorure de phénylacétyle par exemple.
3.2.17 Accès aux imines α,β-insaturés
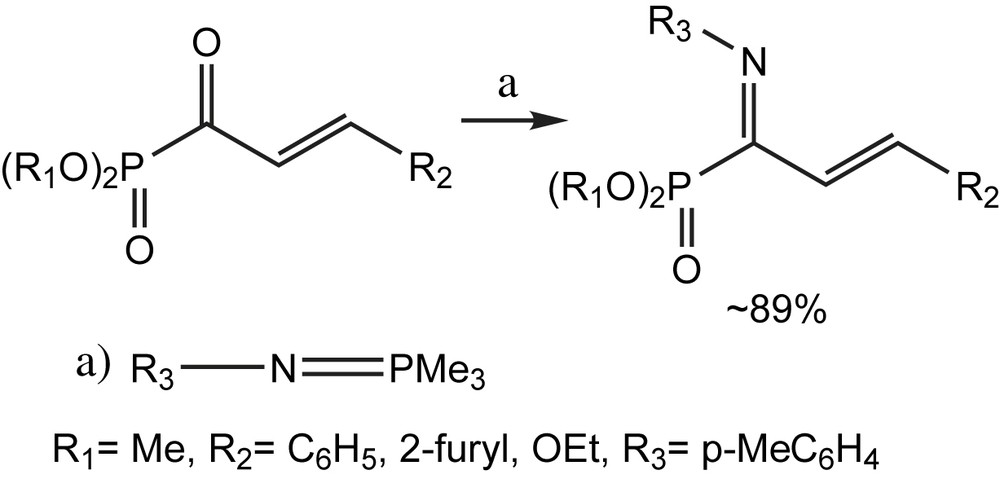
Les α-cétophosphonates β,γ-insaturés peuvent être engagés pour la synthèse efficace des imines α,β-insaturées via une réaction d'aza-Wittig [128] (Schéma 119). La réduction sélective-1,2 de tel azadiènes fournit les α-aminophosphonates β,γ-insaturés, analogues phosphorylés des vinylglycines, qui sont hydrogénés pour conduire aux α-amino-phosphonates saturés.
3.2.18 Cycloadditions
La réaction d'hétéro Diels-Alder à demande électronique inverse des alcènes riches en électrons avec les dérivés carbonylés α,β-insaturés tels que les acylphosphonates, catalysée par les complexes bis(oxazoline)Cu(II) (Schéma 120) est une méthode élégante pour la préparation des dihydropyranes énantiomériquement enrichis [27].
Après l'évaluation systématique de l'effet du catalyseur, il semblerait que ce dernier active les acylphosphonates par complexation entre les groupements CO et PO vicinaux et les rend, par conséquent, susceptibles de subir facilement des réactions de cycloadditions avec des alcènes riches en électrons (Schéma 121).
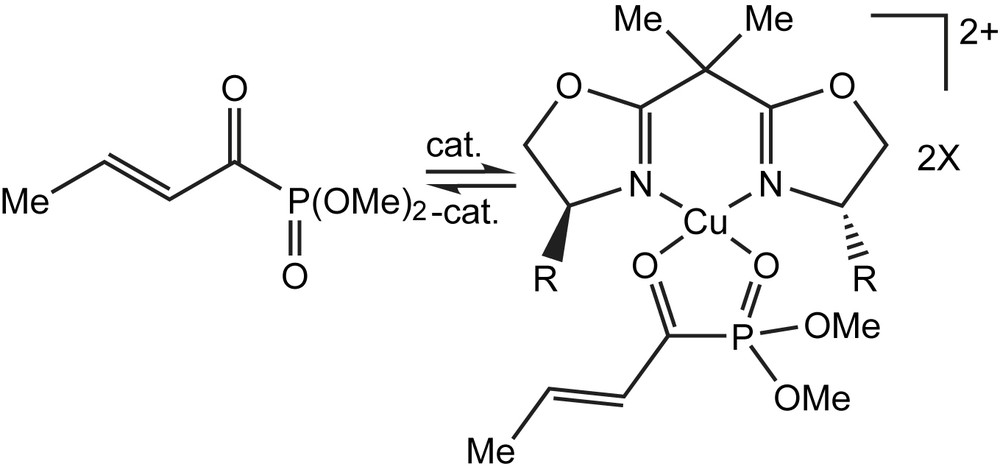
Un certain nombre de phosphonates d'acyle β,γ-insaturés ont été utilisés avec succès comme hétérodiènes. Une large gamme de substrats, testés en particulier ceux substitués par des groupements phényl-, isopropyl-, et éthoxy, ont été mis en réaction avec l'éthylvinyléther et conduisent aux dihydropyranes avec des rendements élevés (85%) via des réactions diastéréo-(de > 99/1) et énantiosélective (92%). La réaction du diméthyl α-crotonylphosphonate avec une variété d'éthers d'énols incluant le dihydrofuranne et le dihydropyrane conduit aux cycloadduits mono- et bicycliques, avec respectivement, de bons à d'excellents rapports endo/exo (>99/1) et des énantiosélectivités élevés (95%) pour chaque isomère (Schéma 122). Ces cycloadduits et leurs dérivés tétrahydropyranes sont des sous-unités structurales dans de nombreux produits naturels.
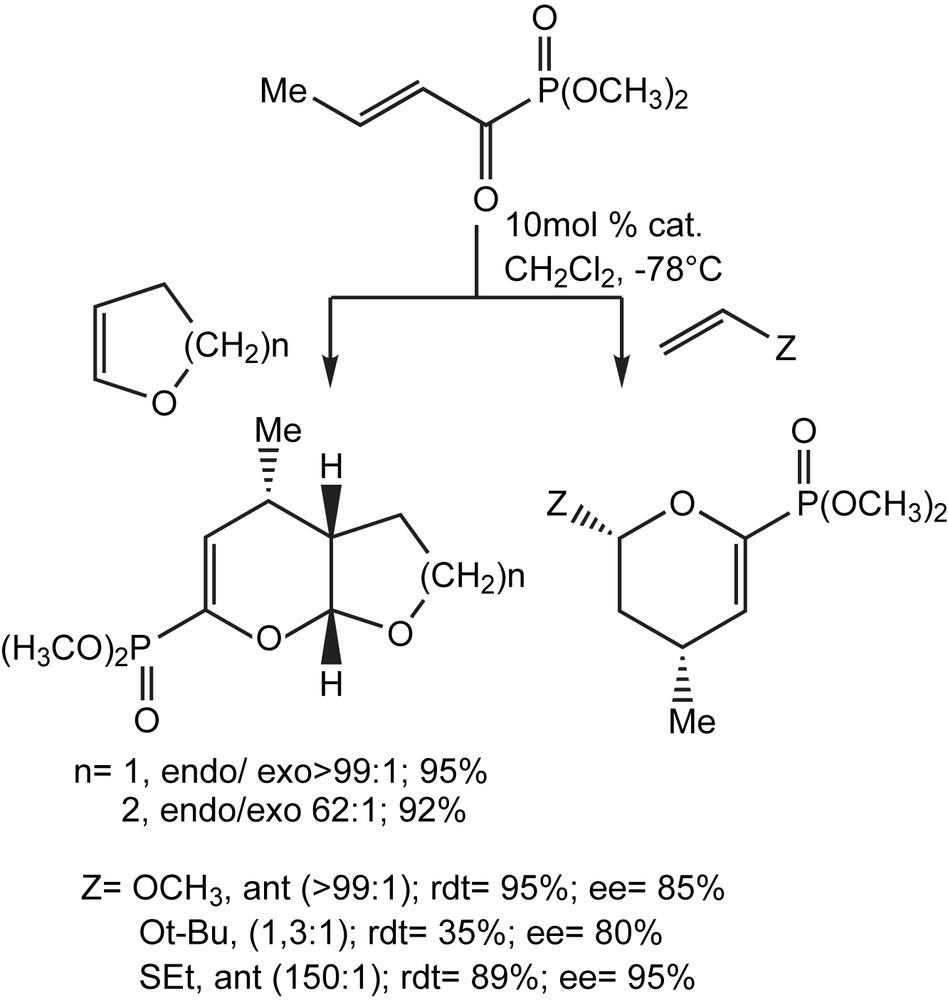
L'examen d’ une série d'acides de Lewis (Sc(OTf)3, Yb(OTf)3, BiCl3, TiCl4, et SnCl4), en utilisant le cyclopentadiène, cyclohexadiène, dihydrofuranne et dihydropyrane, comme diénes, constitue un autre exemple de cycloaddition à demande inverse d'électrons qui utilise les α-cétophosphonates α,β-insaturés [129]. Les produits hétéro Diels-Alder sont obtenus avec de bons rendements et une sélectivité endo très élevée (il semblerait que la taille du groupe phosphonate possède une influence sur le rapport des produits formés).
Les résultats de la réaction du diéthyl α-crotonylphosphonate avec le cyclopentadiène en absence d'acide de Lewis ont été intriguants, puisqu'ils mènent à la formation des produits hétéro Diels-Alder, à côté des cycloadduits normalement prévus (Schéma 123). Le rapport des produits est fortement en faveur de cycloadduit excepté le cas où la réaction a été faite dans l'eau comme solvant. Dans ce dernier et comparé aux solvants aprotiques non polaires, le rapport endo/exo est plus élevé pour le cycloadduit.

Les cycloadduits de Diels-Alder semblent être initiallement formés avec le cyclopentadiéne, pour subir un réarrangement de Claisen [3 + 3] en présence d'un acide de Lewis (Schéma 123).
3.2.19 Actions des trialkylphosphites
La réaction des dialkyl 2-acétoxy et 2-benzyloxy-benzoylphosphonates avec les trialkylphosphites (R = Me, Et, i-Pr) a lieu habituellement par la formation initiale des intermédiaires anioniques qui, en absence d'électrophiles, subit la coupure de la liaison C-O en α, pour donner des carbènes intermédiaires. D'ailleurs, des réactions d'insertion intramoléculaires peuvent se produire et mènent à la formation de nouveaux produits de cyclisation. Le schéma réactionnel (Schéma 124) représente ce processus de cyclisation.
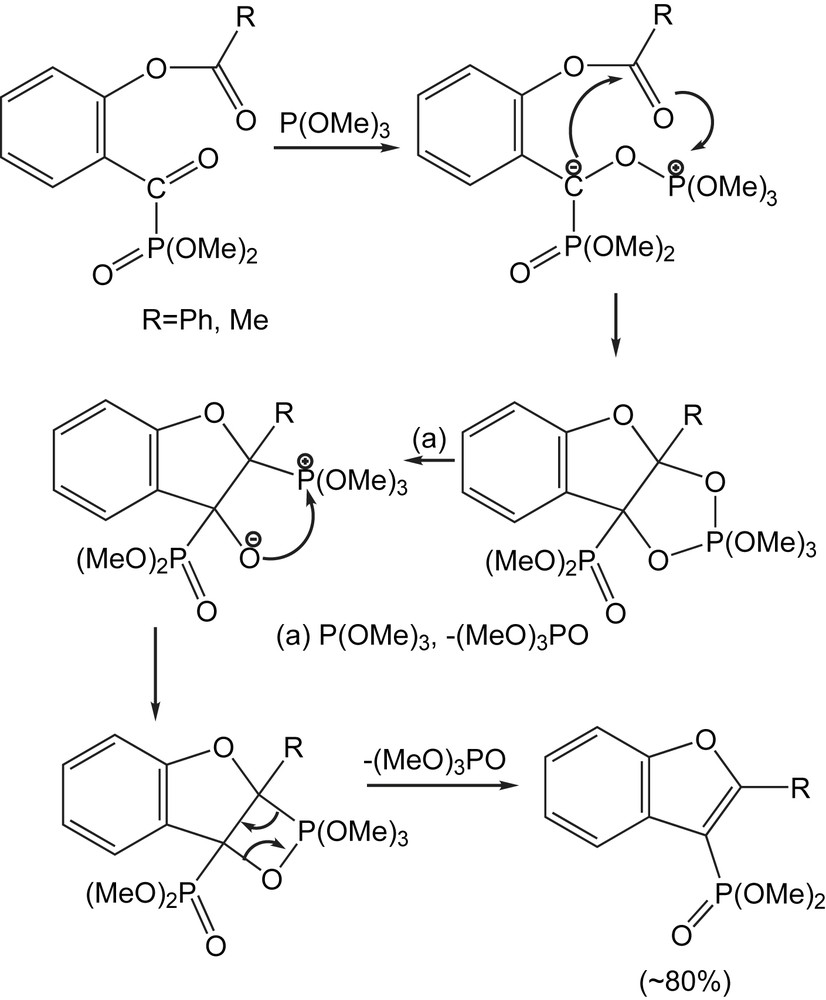
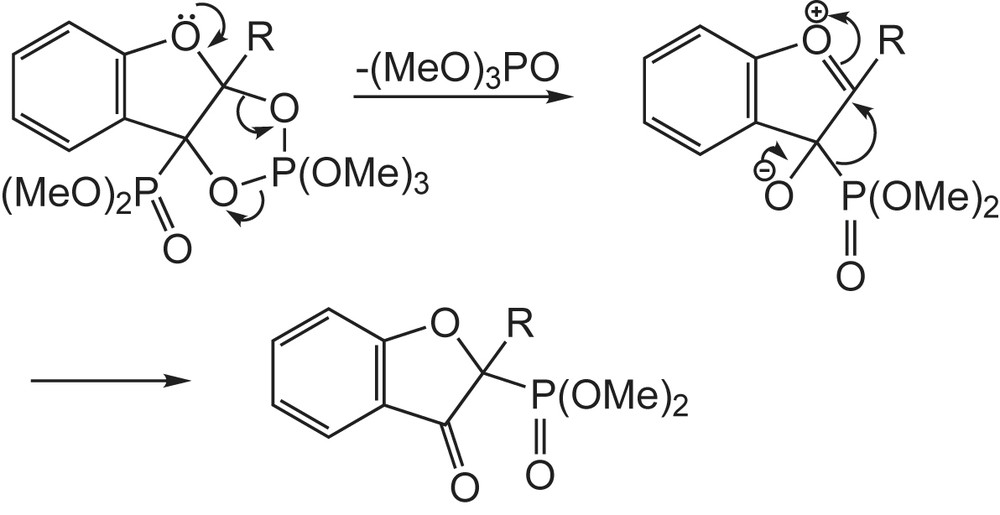
L'intermédiaire, obtenu par l'attaque du phosphite conduit au dioxaphospholane. En présence d'une deuxième molécule de phosphite, il en résulte une ouverture du cycle avec perte de triméthylphosphate. L'intermédiaire obtenu se réarrange avec la perte d'une nouvelle molécule de triméthylphosphate pour donner les benzofuran-3-yl-phosphonates (Schéma 24).
En l'absence d'excès de triméthylphosphite, le dioxaphospholane initiallement formé perd une molécule de phosphate triméthylique et subit alors un nouveau réarrangement (Schéma 125) pour donner les β-cétophosphonates [130].
3.3 Réactions des autres cétophosphonates
3.3.1 Préparation des hydroxyesters cycliques
L'action des carbanions sur des formyl-phosphonates [46], conduit après protection et cyclisation aux hydroxyesters cycliques avec des rendements globaux modestes (Schéma 126).

3.3.2 Préparation des γ-hydroxycycloalkyl-phosphonates
La catalyse par la levure de boulanger a aussi été utilisée pour réduire des systèmes cycliques tels que le (3-oxo-1-cycloalcényl) phosphonate [131] pour obtenir les dialkyl (3-hydroxy-1-alcényl) phosphonates correspondants, notamment dans le cas d'ènones cycliques à six et sept chaînons où les excès énantiomériques ont été de bons (n = 2) à modérés (n = 3) (Schéma 127).
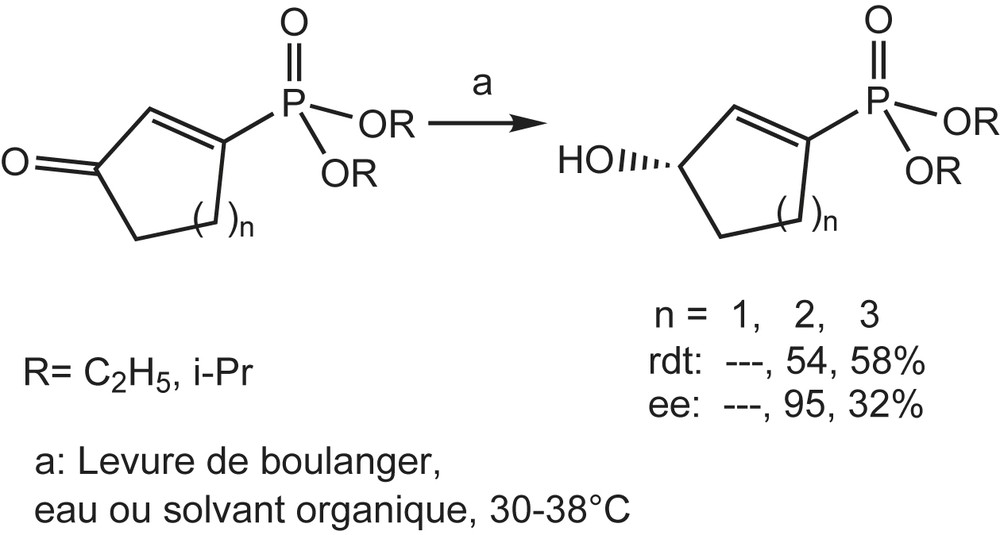
Cependant, lorsqu'il s'agit des systèmes à cinq chaînons, c'est le diéthyl (3-oxocyclopentyl)-phosphonate qui a été isolé avec un rendement de 45%. La modification des conditions de réduction n'a pas amélioré l'excès énantiomérique, sauf dans le cas des systèmes à six chaînons où il s'est avéré que le groupement alkyle du phosphoryle influence nettement et permet une optimisation des excès énantiomériques jusqu'à 95%.
Comme la levure du boulanger possède un grand potentiel catalytique [81,82] pour la bioconversion des diéthyl β,γ,δ-oxoalkylphosphonates et afin d'éviter la décomposition du substrat, les bio-réductions ont été effectuées dans des conditions anhydres [108], employant des cellules lyophilisées immobilisées sur la célite R630.
3.3.3 Préparation des dérivés d'oxazolidinone phosphonate
La combinaison de la partie aminophosphonate avec les benzoxazolones différemment substituées donne accès à une grande variété de structures susceptibles de montrer des propriétés biologiques et pharmacologiques intéressantes, puisque les oxazolidinones sont une classe importante d'agents antibactériens. Pour cette synthèse, le γ-cétophosphonate est couplé à l'amine aromatique dérivée du 2-amino-5-nitrophénol disponible et obtenue par une séquence en trois étapes (protection, cyclisation, et réduction) pour donner le produit d'amination réductrice (Schéma 128) ; étape clé pour l'accès aux acides γ-amino-phosphoniques. Afin d'améliorer leur activité biologique, ces nouveaux dérivés d'oxazolidinone phosphonate [132] sont sujets à des transformations au niveau des deux groupements amino, ainsi qu'en position α par rapport au groupe phosphonate.
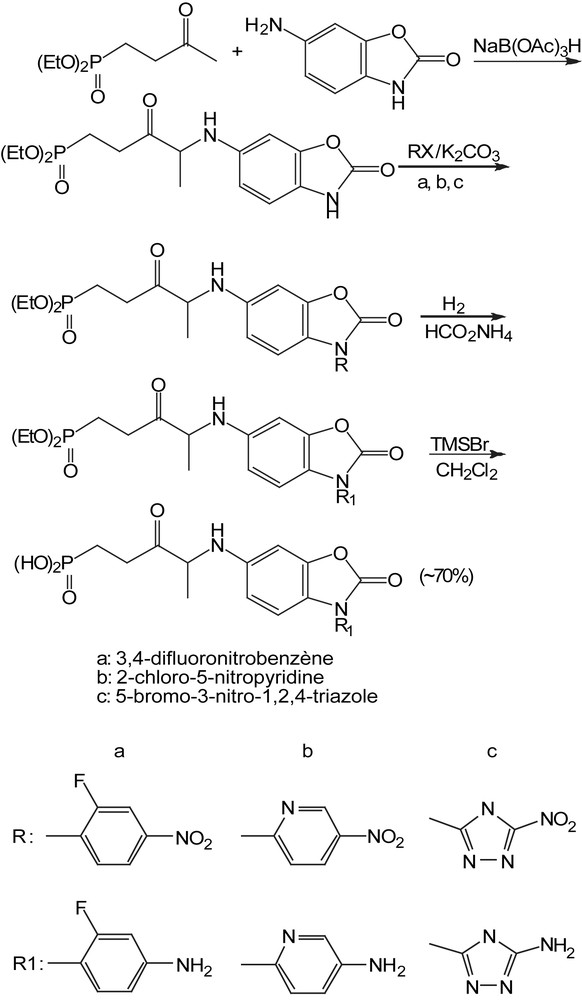
3.3.4 Préparation d'un phosphonate de vinyle
Pour illustrer l'utilité synthétique de la méthodologie décrite dans le (§2.3.8), les γ-formyl-phosphonates enrichis sont facilement convertis en phosphonates de vinyle β-substitués, sans perte d'énantiosélectivité (Schéma 129). La réduction du phosphonate de départ avec le NaBH4, suivie de la protection de l'alcool primaire avec le groupe TBDMS donne l'alcool protégé correspondant avec un rendement global élevé (84%). La réaction de HWE avec du formaldéhyde en solution aqueuse de NaOH à 50% conduit au phosphonate de vinyle β-substitué énantiomériquement enrichi avec un rendement élevé (81%) et avec conservation de l'excès énantiomérique (90%). Ce nouveau composé peut être impliqué dans une variété de transformations synthétiques telles que l'ozonolyse, cycloaddition, addition conjuguée, formation de méthylcétone [49].
4 Conclusion
Cette revue décrit le potentiel synthétique des oxophosphonates en couvrant les applications de 2000 à ce jour. Ils se sont avérés des intermédiaires très « versatiles » pour la synthèse des hydroxy-, amino-, diamino- et halogénophosphonates, dont l'intérêt dans les domaines médicinale, pharmaceutique et agrochimique est incontestable. Les différentes fonctionnalisations permettent aussi de préparer une large variété d'hétérocycles azotés phosphorés, de taille très variable (3 à 7 chaînons). Dans une première partie, les synthèses des β et α-oxophosphonates ont été présentées. Ensuite, la réactivité de ces derniers a été étudié, plus particulièrement les réactions de réductions, d'amination, de substitution, d'alkylation et de cycloaddition.