

1 Introduction to artificial photosynthesis
The field of artificial photosynthesis (AP) consists of the design and synthesis of organic or inorganic systems that show selected aspects of the natural photosynthetic process [1–9]. The essential photochemistry of these synthetic systems follows from that of their natural counterparts: absorption of sunlight, energy transfer, regulation of energy flow, protection from photodamage, conversion of excited states to redox potential, conversion of redox potential to proton motive force, but they also include catalysis and ultimately could incorporate self-repair and replication. Following the path marked by nature, the goal of AP is to assemble molecular systems into larger scale constructs capable of storing a fraction of the energy carried by sunlight in energy rich compounds. AP is a diverse field in its infancy and at present involves contributions from disciplines ranging from physics through chemistry and materials science to synthetic biology. AP research today is largely focused on the understanding and mimicry of the steps employed by natural photosynthesis to produce energy rich fuels. In nature, these processes are linked to photosynthetic membranes where antenna pigments convert solar photons to excited states, regulate the flow of collected excitation energy to reaction centers (RCs), and suppress the undesirable production of reactive oxygen species such as molecular singlet oxygen. The excited reaction centers undergo photoinduced electron transfer to produce charge-separated states that store electrochemical energy. The first steps at incorporating AP constructs into membranes in which the membrane serves as an integral component of the energy conversion system have been taken (vide infra).
One of the many approaches for the assembly of AP model compounds is to use synthetic chromophores that are related to the natural pigments, bacteriochlorophylls, chlorophylls and carotenoids, and to employ covalent bonds in place of the proteins that hold and organize the pigments in nature. In the pigment-protein complexes of natural photosynthetic membranes, the electronic interactions, for example, between carotenoids and chlorophylls, that give rise to energy and electron transfer, are in part a consequence of the proximity of their π electron systems, which is a function of the distance and geometry imposed on the pigments by the protein structure. In general, these may be thought of mainly as through-space interactions. In our model systems, the interchromophore distance and geometry are controlled by the linker bonds joining the chromophores, electron donors and electron accepting moieties. There are two different roles for the linker: when the electronic structure of the linkage participates in the coupling between donor and acceptor moieties, the interaction is referred to as through bonds and in cases where the linkage does not take part to a measureable extent, the coupling is referred to as through space. These two interactions are not mutually exclusive; AP models can be designed to include both through space and through bond electronic couplings. In this work, we review a selection of the AP constructs developed in our laboratories that mimic many of the photochemical steps that occur in the natural photosynthetic systems. Reviews of our work including AP reaction centers, proton pumps, water oxidation, and proton reduction have been published elsewhere [2,8–10].
2 Artificial antennas
Chlorophylls and bacteriochlorophylls are prevalent throughout the natural photosynthetic antennas but they only absorb light strongly in the blue and red regions of the solar spectrum. Carotenoids play a key role in absorbing the blue and green photons of the solar spectrum (400–550 nm region), and by their incorporation, plants, algae, and photosynthetic bacteria are able to collect more energy. Once the energy is harvested by the antennas, an efficient energy transport mechanism is needed in order to pass the energy from the carotenoids, chlorophylls and other pigments to the reaction centers, where the excitation energy is transformed into chemical potential in the form of a charge separated state [11,12]. In this section, we will describe a series of artificial antennas; some exhibit very low excitation energy transfer (EET) efficiency and in others the efficiency reaches 100%.
The first artificial antenna molecules synthesized by our research group were carotenoporphyrins [13,14]. Carotenoids with different conjugation lengths were used to tune the wavelength of absorption. These systems consisted of a porphyrin covalently attached to carotenoids with 6 or 10 carbon–carbon double bonds (db) conjugated with a phenyl ring (compounds C1 and C2 in Fig. 1). The absorption spectra of C1 and C2 are essentially the sum of the spectra of related individual carotenoids and porphyrins, which indicates that there is no strong interaction between the chromophores in the ground state. The ester linkage constrains the edges of the carotenoid and porphyrin π-orbitals to be close to one another and provides weak electronic coupling. By steady state fluorescence excitation spectroscopy, it was found that in C1, the efficiency of EET from the carotenoid to the porphyrin is 80%. However, this short carotenoid absorbs light basically in the same spectral region as the porphyrin and hence does not improve significantly the spectral range used. The longer carotenoid employed in C2 collects light from an under-utilized region of the solar spectrum, but essentially none of the light absorbed by this carotenoid led to fluorescence emission from the porphyrin in C2, indicating that EET was not efficient. This result suggests that a greater degree of electronic interaction between the energy donor and acceptor moieties is required.
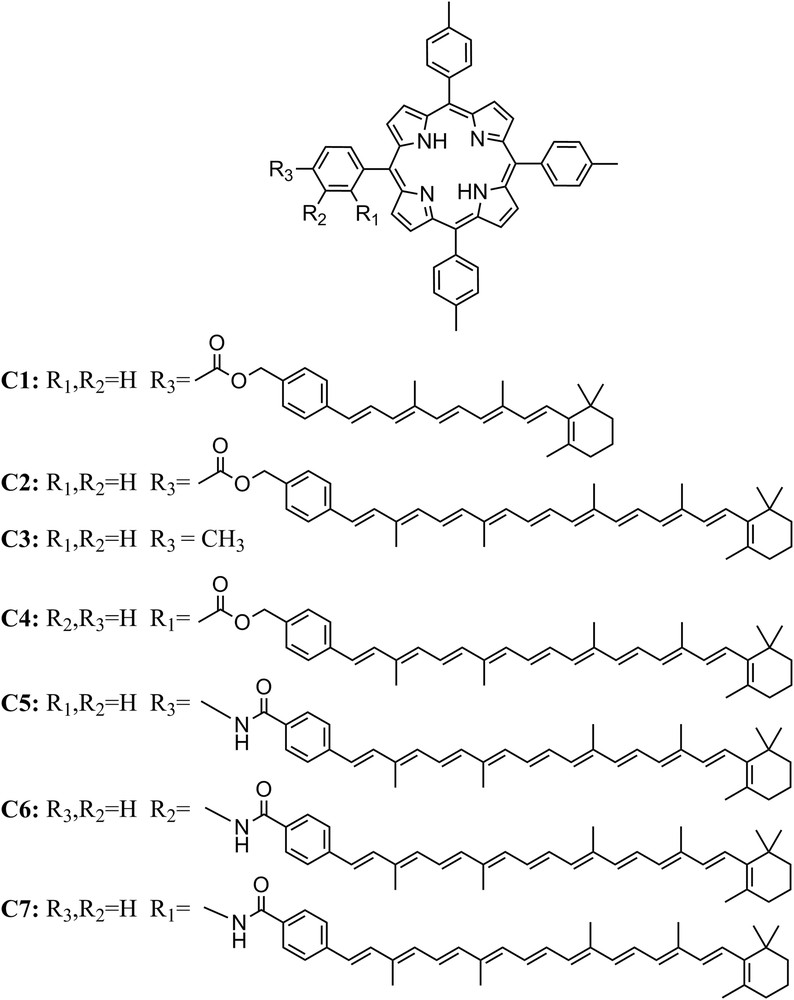
Molecular structures of carotenoporphyrin dyads and meso-tetra(p-tolyl)porphyrin (TTP).
A constitutional isomer of C2, dyad C4 (Fig. 1), wherein the carotenoid is attached to an ortho position of the meso aryl ring of the porphyrin was synthesized in order to achieve a closer interaction of the two chromophores, while maintaining the same ester linkage. NMR studies of C4 demonstrated that as a result of the ortho attachment of the carotenoid to the porphyrin meso aryl group, the carotenoid folds back across the porphyrin so that the π-systems are essentially in van der Waals contact [15]. The separation of the two π-stacked systems was determined by molecular mechanics calculations (∼4 Å) and 25% EET efficiency was determined by fluorescence excitation measurements. Dyads, C2 and C4, illustrate the use of through bond and through space coupling that can be accessed in model compounds. The electronic coupling provided by the linkage, as reflected by the number of bonds and the hybridization of the atoms, is essentially the same in both cases, and it was proven insufficient to mediate singlet energy transfer from the carotenoid to the porphyrin and fast triplet–triplet (T–T) energy transfer in C2. Nonetheless, when dyad C4 adopts a folded conformation, it acquires through space interactions between the π systems, resulting in improved singlet–singlet (S–S) energy transfer. This clearly demonstrates that through space, electronic interactions can be mimicked in covalently linked dyads when they adopt a conformation that provides the necessary orbital overlap.
Later, it was found that increased electronic interactions between the carotenoid and cyclic tetrapyrrole moieties can also be achieved by involving the linkage itself [16]. The role of the potential involvement of the linkage in the electronic interaction of carotenoids and cyclic tetrapyrroles was studied for carotenoporphyrin dyads C5–C7. These dyads consist of identical carotenoid and porphyrin moieties which are joined by a relatively rigid amide group, with the only difference being the point of attachment of the carotenoid to the porphyrin meso aryl group. The singlet–singlet energy transfer quantum yields were 0.17, 0.10 and 0.13 for C7, C6 and C5, respectively. A lower energy transfer quantum yield, and therefore a slower singlet energy transfer rate, was observed for the meta isomer, C6, than for para isomer, C5, despite the fact that molecular mechanics calculations showed that the separation of the two chromophores is smaller for C6. These results are consistent with energy transfer mediated by the linkage bonds rather than by a through-space mechanism. Qualitatively, this superexchange, or through-bond, hypothesis was evaluated using simple Hückel molecular orbital theory assuming that the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the linker were involved in the interaction. These results showed that for both the HOMO and LUMO the coefficient of the wavefunction is greater at the ortho and para positions than that at the meta position.
In general, the unusual characteristics of the carotenoid excited states impose strict constraints on the electronic coupling and thermodynamics necessary for the energy and electron transfer processes between them and cyclic tetrapyrroles which are crucial to photosynthesis and AP [11,12,17]. Even though the carotenoid S2 state lifetime is extremely short, we were able to achieve efficient EET in dyad C8 (Fig. 2) by careful attention to electronic coupling and thermodynamic parameters [18]. The S2 lifetime of a model for the carotenoid in dyad C8, a phenylacetamido-substituted carotenoid with 10 db, was found to be 150 fs as measured by fluorescence upconversion spectroscopy. In the dyad, the carotenoid S2 has a considerably shorter lifetime, ∼40 fs. Hence, if this quenching is attributed fully to EET from the carotenoid S2 state to the purpurin moiety, the rate constant for EET in this system (∼2 × 10−13 s−1) is of the order of that observed in natural light-harvesting antennas and the quantum yield of this process is 0.73.
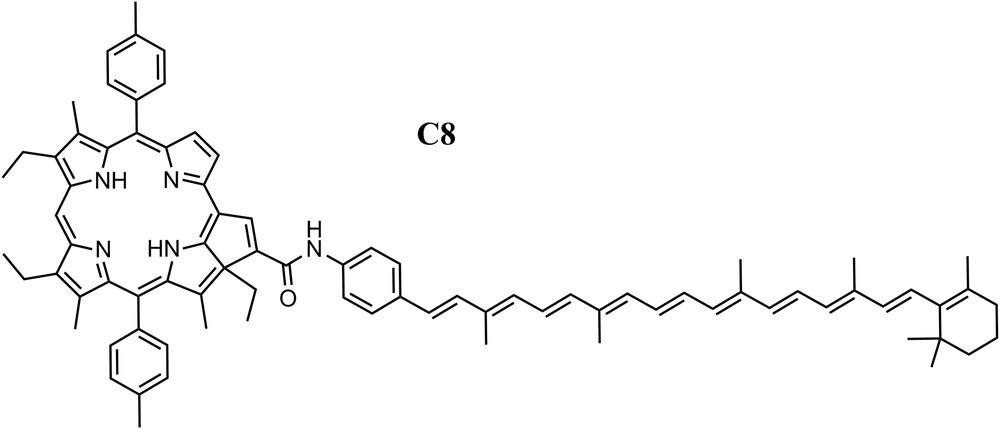
Molecular structure of dyad C8, a 10 double bond polyene linked through a phenylamide group to the fused cyclopentadiene ring of a purpurin.
Triad C9 (Fig. 3) was designed to examine the energy transfer from carotenoids to cyclic tetrapyrroles with different relative orientations [19]. Triad C9 is composed of two 9 db carotenoids linked on opposite sides to a silicon phthalocyanine (Pc) through ester linkages at the center of the macrocycle. The carotenoids extend axially at angles of approximately 90° to the plane of the cyclic tetrapyrrole. It was found that efficient EET was achieved by using either the S2 or the S1 states of the carotenoids. The lifetimes of these excited states were measured in a model carotenoid and in triad C9 to determine in detail how the carotenoid S1 and S2 excited states are involved in the flow of energy to Pc. By fluorescence upconversion spectroscopy, it was proven that the S2 state of the carotenoid was quenched from 82 fs in a model carotenoid to 53 fs in C9, resulting in an EET efficiency of 35% from the S2 state. The contribution of S1 was investigated by transient absorption measurements, which found a difference in the S1 state lifetime of a carotenoid in triad C9 (2.6 ps) and the carotenoid model (24.4 ps). Hence, taking into account that the quantum yield of formation of the S1 state was only 65%, the quantum yield of energy transfer from S1 is ∼58%. Thus, the overall quantum yield of EET from carotenoid to Pc from both the S2 (35%) and S1 (58%) states is 93%, in excellent agreement with the value estimated from steady state fluorescence excitation measurements [19]. Extending the carotenoid conjugation by one double bond from 9 to 10 in triad C10 (Fig. 3) reduced the overall EET efficiency from ∼90% to 30%. This change is a result of the S1 level in the longer carotenoid moving below that of the lowest singlet state of Pc, making energy transfer from the S1 state of carotenoid to the Pc thermodynamically unfavorable. Thus, in triad C10, EET to Pc occurs only from the carotenoid S2 state. Transient absorption studies on triads C9 and C10 also provided evidence for the formation of a state designated S* from S2 in both of the triads, but not in the isolated carotenoid models [20]. The efficient antenna function of the 9 db carotenoid in C9 is associated not only with the S1 and S2 states; all three states, S2, S1, and S*, contribute singlet electronic energy to Pc. This illustrates the importance of multiple pathways for the EET process when using carotenoids in the construction of highly efficient artificial photosynthetic antennas.
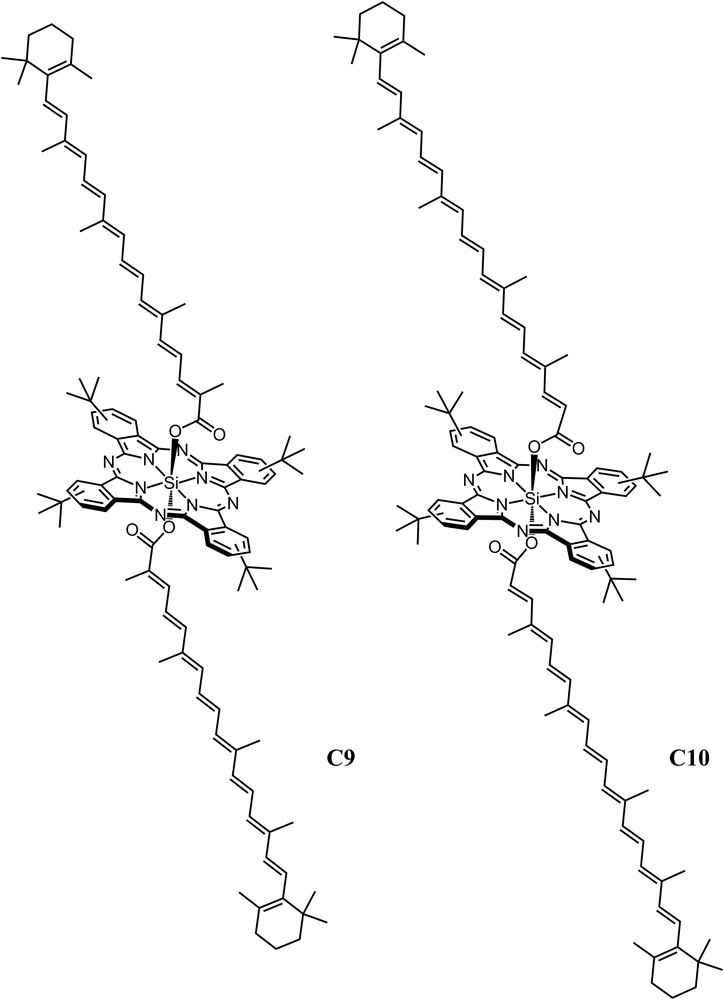
Axially substituted silicon phthalocyanine triads with polyenes containing 9 double bonds, C9, and 10 double bonds, C10.
Similar dependence of the EET from carotenoids to Pc on the carotenoid length was observed in another set of dyads. In contrast to the case above where the carotenoids were axially linked to the Pc, in dyads C11, C12 and C13, where the carotenoids have 9, 10, and 11 double bonds, respectively, the carotenoids are attached to the periphery of the macrocycle of a zinc Pc via an amide linkage [21]. In all these systems, a pathway of energy deactivation within the carotenoid manifold in which the S* state acts as an intermediate state in the S2 to S1 internal conversion on a sub-picosecond time scale was detected. In dyad C11, similar to the case with triad C9, all carotenoid singlet excited states, S2, S* and S1, contribute to singlet–singlet energy transfer to Pc, making the process very efficient (>90%), while for dyads C12 and C13, the S1 energy transfer channel is precluded by thermodynamic considerations and only S2 was capable of transferring energy to Pc (Fig. 4).
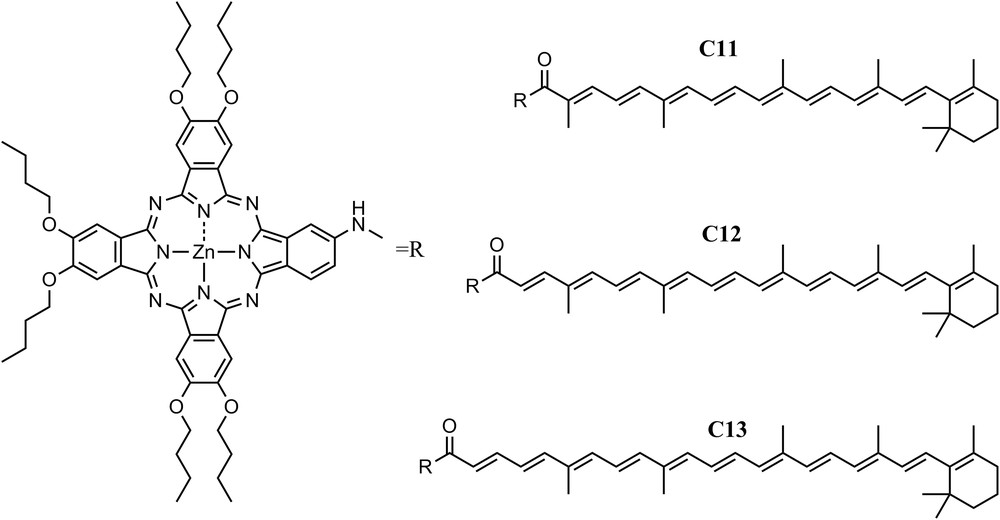
Three carotenophthalocyanine dyads where the two chromophores are connected via an amide linkage. Dyad C11 has a polyene with 9 double bonds, C12 with 10 double bonds, and C13 with 11 double bonds.
To improve the ability to gather sunlight and following nature's strategy, antenna systems consisting of chromophore arrays have been designed. A recently reported synthetic method has been employed to prepare arrays of free base and zinc porphyrins [22]. In the arrays, the porphyrins are arranged around a central benzene ring (C14). The presence of only one meso-aryl substituent on each porphyrin allows strong electronic interactions between the porphyrin macrocycles. In similar arrays containing two or six porphyrins, a variety of evidence indicates that the porphyrins exist as twist-stacked dimers reminiscent of the special pairs of bacteriochlorophylls found in some photosynthetic bacteria. These dimers feature van der Waals contact between the macrocycles and demonstrate excitonic splitting due to π–π interactions. The excitonic effect splits and blue-shifts the Soret absorptions, and slightly broadens the Q-band absorptions and shifts them to longer wavelengths. The interactions also lower the first oxidation potentials by ca. 100 mV; the arrays show evidence for delocalization of the radical cation over both porphyrins in the dimer. The arrays demonstrate singlet–singlet energy transfer among the chromophores. Arrays of this type will be good models for some aspects of the interactions of photosynthetic pigments, including those of reaction center special pairs and possibly quantum coherence effects [23] (Fig. 5).
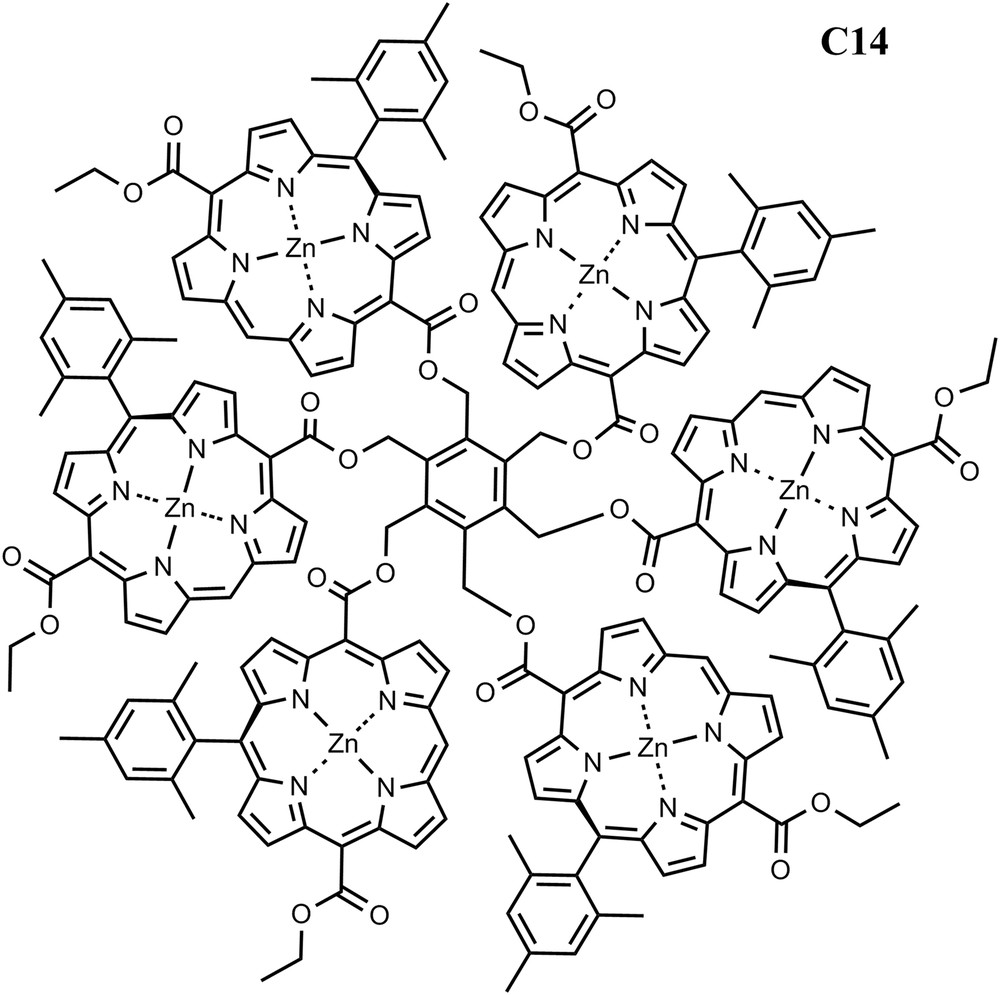
Structure of porphyrin array C14.
3 Artificial reaction centers
3.1 Molecular reaction centers
The initial approaches to mimicking reaction centers for AP in the late 1970s consisted of linking two molecular analogs of components involved in the natural photosynthetic reaction center with covalent bonds (e.g., porphyrins, carotenoids, chlorophylls, pheophytins, phthalocyanines and quinones) [24,25]. In this context, quinones are compounds with suitable characteristics for becoming part of AP constructs because they are ubiquitous electron acceptors in nature. Quinones display a series of useful properties: they have appropriate redox potentials to accept electrons from the primary and secondary donors of artificial reaction centers, undergo stepwise conversion into stable reduction products (hydroquinones) via semiquinones, form hydrogen bonds, undergo protonation/deprotonation reactions with ease, and are relatively small mobile molecules that can shuttle redox equivalents within membranes [26]. For these reasons, early work in the AP field was focused on building porphyrin–quinone (P–Q) reaction center dyads; upon porphyrin excitation, these compounds undergo electron transfer to form a P+–Q− state [27,28]. Although this charge separated state can be produced efficiently under optimal conditions, fast (sub-nanosecond) charge recombination precludes facile coupling of its photo-generated redox potential to slower catalytic steps necessary for fuel synthesis in AP systems. In the 1980s, the Gust, Moore and Moore group (GMM) designed and synthesized a series of carotenoid–porphyrin (Car–P) dyads [29,30], some of which were shown to undergo hole transfer from the oxidized porphyrin (P+) to the carotenoid to yield the Car+–P species [31].
Knowing that Car+–P forms spontaneously from Car–P+ and that P+–Q− forms spontaneously from 1P–Q (where 1P is the first excited single state of P), we reasoned that Car+–P–Q− could be produced spontaneously from Car–1P–Q by photoinduced electron transfer to form Car–P+–Q− followed by hole (h+) transfer to the carotenoid. In such a molecular triad, the final charge-separated state would be formed by a stepwise process in which each step could be fast and efficient, and this final state could have a long lifetime due to reduced electronic coupling between the radical ions. Indeed, early results from studies of bacterial reaction centers suggested that in the natural system charge separation over the thickness of a bilayer lipid membrane (ca. 3 to 4 nm) was accomplished by short range, sequential electron transfer steps which spatially separate positive and negative charges across a substantial distance and gave rise to long lived transmembrane charge separation [32].
Synthesis and subsequent study of a Car–P–Q triad (C15) [33,34] marked a major improvement in stabilizing charge separation in an artificial reaction center and established a platform for engineering improved artificial reaction centers as well as studying other process characteristics of photosynthesis. This triad reaction center (Fig. 6) consisted of a ditolylporphyrin covalently bonded via amide linkages to a quinone electron acceptor and a carotenoid secondary electron donor. Transient spectroscopic studies of C15 showed that illumination with visible light (600 nm) generates the porphyrin first excited singlet state, Car–1P–Q; this species then decays to the first charge-separated state, Car–P+–Q−. Competing with recombination, a second electron transfer from the carotenoid to the porphyrin radical cation produces the final charge-separated state Car+–P–Q−. Monitoring the transient absorbance of the oxidized carotenoid moiety (in the 970 nm region) showed that the final charge separated state had a lifetime of 170 ns and was produced with a quantum yield of 0.04 in dichloromethane; the lifetime of the final charge separated state increased to 2.5 μs with a quantum yield of 0.25 in electrolyte-saturated solvents [33]. Triad C15, model compounds C16, and C17 and subsequent carotenoporphyrin-acceptor triads characteristically adopt a linear conformation in solution without folding of the appended groups back over the plane of the porphyrin [33,35]. The final state retains 1.1 eV of the 1.9 eV of the porphyrin first excited singlet state [7]. The improved stability of the final charge-separated state of this construct relative to those of the preceding dyads resides in the greater spatial separation, and therefore electronic decoupling, of the separated charges and in the endergonic steps necessary to form either the Car–P+–Q− or Car+–P−–Q intermediates that could be involved in the overall energetically favorable charge recombination to the ground state of the triad. The concepts demonstrated by C15, C16, and C17 combined with Marcus theory [36–39] are universal and have been extended to more complex multiple-component systems establishing a platform for engineering efficient artificial reaction centers [8,9].

Molecular structure of triad C15 and model compounds C16 and C17.
The introduction of a fullerene electron-accepting unit in place of a quinone advanced the performance of the artificial reaction centers [40]. Although it is not observed in any known biological system, the use of a fullerene acceptor moiety offers definite advantages in imitating the photochemical processes observed in natural photosynthesis. Compared to quinones, fullerenes perform remarkably well in reaction center constructs due to their small solvent and internal reorganization energies for electron transfer and relative insensitivity of the radical anion stabilization to the solvent dielectric constant [40–42]. In comparison with triad complexes employing quinones, those with fullerene acceptor moieties in general show more rapid photoinduced charge separation and slower charge recombination, can perform charge separation in a variety of solvents and even at low temperature in glasses [40,43], and can recombine to triplet excited states rather than the ground state [40,44]. Such behavior is characteristic of the natural system, making fullerenes an ideal example where compounds unknown to biology can substantially aid in the development and performance of the overall biomimetic artificial systems [45].
The first iteration of the C–P–C60 based triad, C18, featured a β-alkyl substituted porphyrin and formed the final Car+–P–C60− state with a yield of 0.14 in 2-methyltetrahydrofuran [40]. Spectroscopic studies showed that the state decays with a lifetime of 170 ns. Progressive molecular engineering of the complex by introduction of small changes in the linkage leads to compound C19 which undergoes charge separation with quantum yields up to 0.88 and has a lifetime of up to 1 μs, depending upon the conditions. The various electron transfer rate constants are relatively insensitive to solvent and temperature, and the triad functions in media ranging from fluid solutions at ambient temperatures to a rigid organic glass at 8 K. Unlike C15, in most solvents recombination of Car+–P–C60− yields the carotenoid triplet state 3C–P–C60 rather than the ground state [43]. The introduction of a ditolylporphyrin into C20 [46], and then dimesitylporphyrin into C21 [44] steadily improved the performance of the reaction centers, producing complexes capable of obtaining quantum yields of the final charge-separated state near unity (Fig. 7).
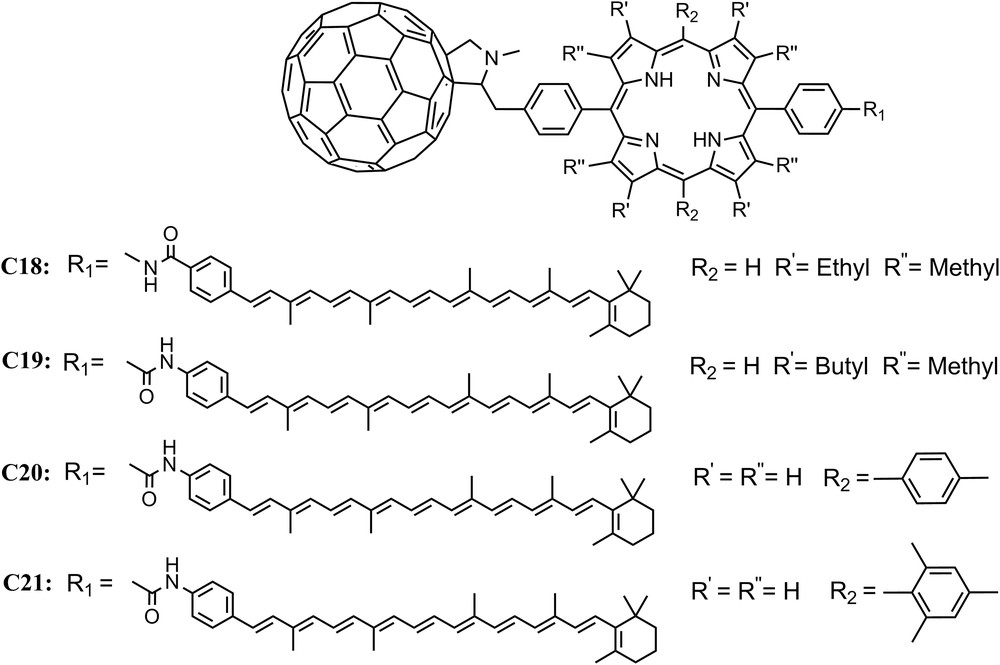
Molecular structures of triads C18, C19, C20 and C21.
The extension of the triad design led to the creation of tetrad and pentad complexes which demonstrated the feasibility of carrying out multiple electron transfer steps across larger complexes resulting in greater spatial separation of the charges and longer lifetimes for the charge-separated state [47–49]. In order to increase the distance between the final electron donor and acceptor, a series of tetrachromophoric molecules C22, C23 and C24 consisting of a porphyrin (P) covalently linked to both a carotenoid polyene (Car) and a rigid diquinone moiety (QA–QB) to give Car–P–QA–QB were prepared. Upon excitation, compound C24 produces a long-lived (1.9 μs) charge-separated state Car+–P–QA–QB−, with a quantum yield of 0.11. Comparison of these results with those for related C22 and C23 clearly demonstrates that although the tetrad is a considerably more complex system, the additional electron-transfer step led to an increased quantum yield of long-lived, high-energy charge-separated states [49].
As just described, covalently linked carotenoid–porphyrin–quinone (C–P–Q) triads and carotenoid–porphyrin–diquinone tetrad molecules mimic the multistep electron-transfer strategy to yield long-lived charge-separated states upon excitation. However, electron, singlet energy, and triplet energy transfer between tetrapyrroles are also key facets of natural photosynthetic charge separation. The triads and tetrads mentioned above do not model such processes, although a number of dyad and triad molecules containing covalently linked tetrapyrroles indeed demonstrate electron and energy transfer phenomena. An example is C25, which consists of two covalently linked porphyrins (PA–PB), one bearing a carotenoid polyene and the other a naphthoquinone electron acceptor. Upon photoexcitation, transient spectroscopic measurements led to the conclusion that C25 ultimately yields a Car+–PA–PB–Q− charge separated state with a quantum yield of ca. 0.25 and a lifetime of 2.9 μs. The tetrad also demonstrates singlet and triplet energy transfer behavior, which mimics photosynthetic antenna function by chlorophylls and carotenoids and carotenoid photoprotection from singlet oxygen damage [47] (Fig. 8).
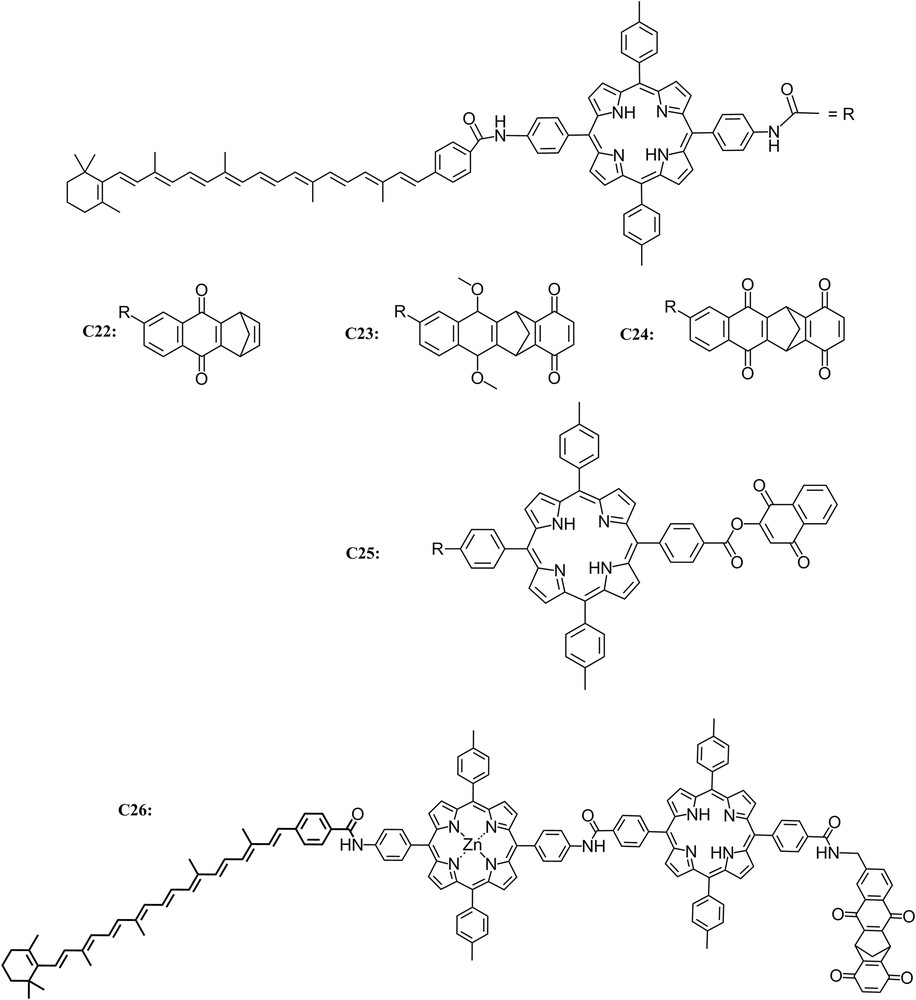
Molecular structure of triads C22, C23 and C24, tetrad C25 and pentad C26.
Using the knowledge gained with the previous triads and tetrads, a synthetic five-part molecular system was prepared, C26, which uses a multistep electron transfer strategy similar to that of photosynthetic organisms to capture light energy and convert it to chemical potential in the form of long-lived charge separation. It consists of two covalently linked porphyrin moieties, one containing a zinc ion (PZn) and the other present as the free base (P). The metalated porphyrin bears a carotenoid polyene (Car) and the other a diquinone species (QA–QB). Excitation of the free-base porphyrin in a chloroform solution of the pentad yields an initial charge-separated state, Car–PZn–P+–QA−–QB, with a quantum yield of 0.85. Subsequent electron transfer steps lead to a final charge-separated state, Car+–PZn–P–QA–QB−, which is formed with an overall quantum yield of 0.83 and has a lifetime of 55 μs. The artificial photosynthetic system retains ca. 53% of the initial excitation energy (1.9 eV) in the long-lived, charge-separated state [48].
3.2 Charge separation across a phospholipid membrane
In natural photosynthesis, photoinduced charge transfer across the thylakoid membrane is coupled to proton translocation across the bilayer lipid membrane, thus establishing a proton gradient. Dissipation of the proton chemical potential back across the membrane via the transmembrane ATP-synthase enzyme drives the production of ATP. Designing constructs that mimic this process may offer insight into schemes and development of artificial systems for solar energy conversion. When a planar phospholipid bilayer membrane impregnated with triad C27, which consisted of a photochemically active porphyrin, an electron donor (the carotenoid moiety) and an electron acceptor moiety (the quinone), was illuminated with visible light, a steady-state photocurrent was observed in an external circuit bridging the phospholipid bilayer membrane when suitable electron donor and acceptor species were present in the aqueous phases. Such an effect was not observed when the same experiment was performed with compounds C28 and C29. Those results could be understood taking into consideration that triad C27 undergoes charge separation across the planar phospholipid membrane following two intramolecular steps, the first induced by visible light excitation, while C28 and C29 (reference compounds) do not. Thus the planar lipid membrane containing C27 mimics key features of the photodriven transmembrane electron transfer process characteristics of photosynthetic organisms [50] (Fig. 9).
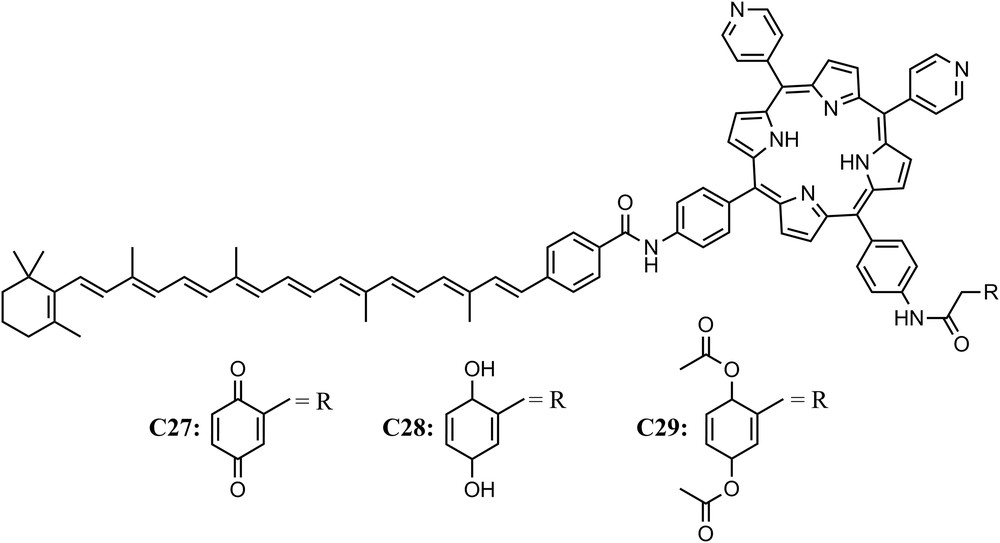
Molecular structure of triads C27 and reference compounds (C28 and C29).
These constructs were later used in more complex systems to study related photosynthetic processes like photoinduced proton and calcium ion gradient generation across lipid membranes and the use of the proton gradient to drive the enzymatic formation of ATP [51,52]. Inserting a similar C–P–Q type reaction center into the bilayer of a liposome set the stage for a system capable of pumping protons across the lipid layer [51]. Due to the overall amphiphilic nature of the C–P–Q triad used, the asymmetric insertion of the reaction center into the liposome with the carotenoid moiety toward the interior and the more polar quinone to the exterior was possible. The excitation of the triad resulted in a potential gradient within the phospholipid membrane; oxidizing potential inside the liposome, where the carotenoid was located, and a reducing potential toward the liposome periphery, where the quinone was located. A freely diffusing quinone electron/proton carrier within the membrane of the liposome with a midpoint potential between that of the oxidized carotenoid and reduced quinone moieties of the reaction center shuttles protons across the membrane, resulting in the acidification of the interior of the liposome. In that manner, a light-induced proton gradient was generated across the bilayer. In order to harness this proton motive force, a CF0F1-ATP synthase was incorporated into the liposome along with the reaction center and redox mediator components [52]. With proton translocation driven by the photocycle described above, dissipation of the pH gradient coupled to the catalytic conversion of ADP and Pi to ATP was carried out by the CF0F1-ATP synthase and resulted in the net conversion of incident light energy into that of a high-energy chemical species. Quantitative analysis of the system reveals that in low light the absorption of 14 photons results in the production of one ATP molecule and, with illumination by 633 nm light, roughly 4% of the absorbed energy was conserved in the form of a chemical bond. In a subsequent study, the same C–P–Q reaction center was used in conjunction with a quinone-like molecule which had the ability to bind Ca2+ in a redox state dependent way [53]. This system was capable of pumping Ca2+ ions against a concentration gradient by the asymmetric arrangement of the C–P–Q reaction center in liposomes. Although the quantum yield was only 1%, a significant potential difference was measured across the membrane, extending Mitchell's mechanism of accumulating membrane potential using a redox loop to drive the translocation of divalent cations in addition to protons.
3.3 Hybrid reaction center
3.3.1 Electron injection into a metal oxide semiconductor
AP engineering needs to go further than natural photosynthesis in order to ensure a sufficient energy production to satisfy human needs; hence it is necessary to introduce compounds/materials which are not used in natural photosynthesis. The study of electron and energy transfer processes in simple and well-defined dye semiconductor colloidal nanoparticles (SNPs) is important not only for direct photocatalytic applications but also for understanding the key processes that control the efficiency of devices such as dye sensitized solar cells and photoelectrochemical cells. Due to their simplicity, dye-SNP systems allow detailed studies of the parameters that control photosensitization such as dye binding (and its correlation with the dye structure) and the relative energies of the states involved in the photoinduced electron transfer process. In order to study the photoinjection of an electron into the conduction band of TiO2, a series of perylene derivatives with different redox potentials was synthesized. These systems were studied in reverse micelles when working in water and with naked TiO2 nanoparticles (NPs) with organic groups on their surfaces when working in organic solvents. Using particle encapsulation methods involving reverse micelles, dye-TiO2 NP systems were obtained where the size was controlled to an average radius of ∼4 nm. Using this strategy simple dye-TiO2 systems consisting of perylene derivatives and TiO2 NP were encapsulated inside AOT (anionic detergent) reverse micelles (perylene-TiO2@micelles) [54]. The binding strategy chosen for the perylene-TiO2@micelle systems involves the opening of a cyclic anhydride group by hydrolysis to generate two carboxylic acid groups that can attach to the semiconductor surface. For the series of analogous perylene derivatives C30, C31 and C32 in Fig. 10 it was shown that the dye to TiO2 binding efficiency depended among other factors on the dye molecular structure. In particular, C30 and C31 showed efficient binding whereas C32 did not bind, presumably due to steric effects induced by the bulky tert-butyl groups. It was also observed that the micelle structure plays a role in the binding process; fluorescence anisotropy measurements indicated that C30 and C31 were preferentially solubilized in the micellar structure to create a relatively large local concentration that presumably favored the attachment of the dye to the TiO2 surface.

Structures of perylene derivatives (C30, C31 and C32) used in the hybrid systems attached to TiO2 through carboxylate groups. The diagram displays the first oxidation potential versus SCE for the ground state and the photoexcited state of C30, C31 and C32 and the TiO2 CB and VB.
Simpler model systems constructed with the same perylene dyes but attached to “naked” TiO2 SNPs (dye-TiO2) suspended in an organic solvent were prepared and studied [55]. In these systems, the SNPs were synthesized following a method that produces surfactant-free nanoparticles with a narrow size distribution (4 ± 1 nm) [56] which are readily suspended in an organic solvent such as tetrahydrofuran. Since the need for micelle encapsulation with this approach is eliminated, it produces simpler colloidal systems, which are closely related to the structures encountered in functioning dye-semiconductor devices. The “naked” systems showed similar results to the encapsulated systems with respect to dye binding and sensitization. However, the distinct solvent environment sensed in each case (THF and micelle-water-pool for the “naked” and encapsulated systems, respectively) creates a considerably different energetic situation. In particular, the CB edge in the “naked” system is more negative than that of the micelle encapsulated system [55]. Therefore, the driving force for photoinduced electron transfer (PeT) in “naked” systems is smaller than that in the micelle system and the rates for photoinduced charge separation (kPeT) follow the relationship kPeT(naked) < kPeT(micelle). Therefore, these systems illustrate that factors affecting the position of the semiconductor ECB also need to be carefully considered.
3.3.2 Hole injection into a p-type metal oxide semiconductor
Excited-state hole injection into the valence band of Cu5Ta11O30 nanoparticles was investigated through sensitization with zinc porphyrin dyes using simulated solar irradiance. The dye C33 and its analog in which the pyridine groups are methylated (C34) were attached to nanoparticles of Cu5Ta11O30 with average sizes of ∼10–15 nm. Those dyes were found to have excited state reduction potentials appropriate for p-type dye sensitization of the nanoparticles. The dye-sensitized NP-Cu5Ta11O30 exhibited fluorescence quenching consistent with electron transfer from the NP-Cu5Ta11O30 to the dye, and forward and recombination rates were obtained by transient absorption measurements. Hole injection times of 8 ps and <100 fs were observed for C33 and C34, respectively [57] (Fig. 11).
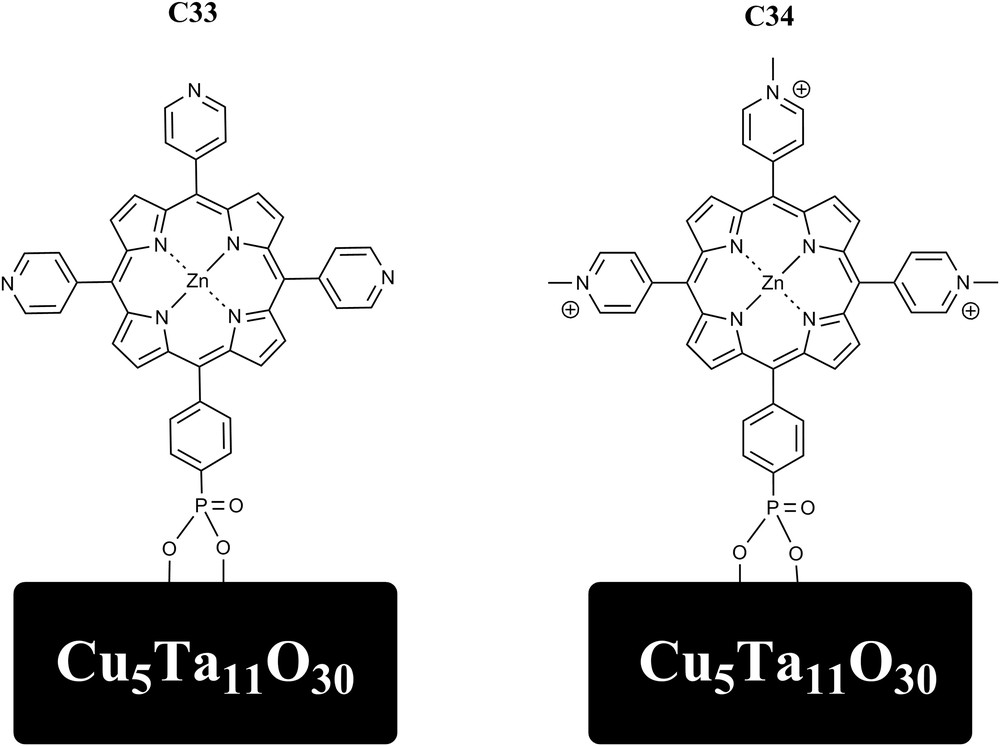
Molecular Structures of C33 and C34 attached to Cu5Ta11O30 nanoparticles through phosphonate groups.
3.4 Proton coupled electron transfer (PCET) in photosynthetic artificial models
Even though charge separation in simple dye-SNPs has been extensively demonstrated, for photocatalytic applications such as water oxidation these systems require careful control of the dark redox driven processes and the thermodynamics and kinetics of the photoinduced redox potential generation. In fact, most catalytic centers for water oxidation operate at relatively slow rates as compared to the recombination rates of the redox species produced after initial photoinduced charge separation. The recombination rate of these species can be lengthened by altering the thermodynamics (e.g., increasing their potential energy) to push the recombination reaction further into the Marcus inverted region. Nevertheless, in some cases this strategy may not be sufficient to achieve a high yield of catalyst oxidation. In order to address these issues, the GMM group developed a series of model dye-SNP systems based on the bio-inspired PCET strategy. The approach was tested in a molecular triad C35 [58] and was also used in several hybrid systems. These hybrid constructs consist of molecular dyads composed of Bi–PhOH moieties covalently attached to a PF10 porphyrin which bears a carboxylic acid as an anchoring group to attach to SNPs (see Fig. 12, systems C36, C37 and C38). For all these dye-SNP constructs, the primary photoinduced electron donor is the high-potential porphyrin (PF10), while the SNP acts as primary electron acceptor (replacing the organic β-tetracyanoporphyrin moiety of triad C35), and either the benzimidazole–phenol (Bi–PhOH) or the phenol–benzimidazole (PhOH–Bi) acts as a secondary electron donor. It was shown that in all these systems the phenolic hydrogen is capable of forming a strong hydrogen bond with the lone pair electrons of an imidazole nitrogen. Upon oxidation of the phenol by PF10+, the phenolic proton is transferred to the benzimidazole group by a PCET reaction to produce a neutral phenoxyl radical (E = 1.06 V vs SCE in the case of C36) that is thermodynamically capable of water oxidation (E0 = +0.58 V vs SCE, pH 7). As shown in Fig. 12 the dye-SNP system C36 consists of a benzimidazole–phenol (PhOH–Bi) moiety covalently bound to PF10 and the primary electron acceptor is a TiO2 nanoparticle. The final charge separated state of this compound involves a phenoxyl radical and injected electrons into the TiO2 nanoparticle, as was demonstrated by X-band and D-band EPR spectroscopy at low temperatures [59]. It was shown that upon illumination at 4.2 K only 48% of the holes localized on PhOH–Bi and 52% resided on PF10. However, when the experiments were performed at 80 K, 95% of the holes were localized on the PhOH–Bi moiety. This temperature dependence of the charge shift is attributed to restricted nuclear motion at low temperatures. The C37 system is similar in construction to that of C36, but it bears a benzimidazole–phenol group (Bi–PhOH) in which the phenol moiety (instead of the benzimidazole) is covalently attached to PF10 [60]. In C37 a stronger internal hydrogen bond than that in C36 is observed. For construct C37, the detection of the phenoxyl radicals was also performed by high-field EPR. Those studies showed that a higher energy state formed at 13 K and a relaxed state were observed after increasing the temperature up to 100 K and then reducing it back to 13 K for the measurement. After performing DFT calculations, it was suggested that solvent molecules around the initial site O–H⋯N undergo a reorganization to solvate the newly formed O⋯H–N site. This reorganization is likely not possible at 13 K, and the system is frozen in a higher-energy state with a gx value of 2.0056. However, when the system is warmed up to 100 K, the solvent molecules around the new O⋯H–N site reorganize. This solvent reorganization and some minor structural rearrangements around the oxidized benzimidazole–phenol result in a relaxed final structure with a gx value of 2.0061. Most likely the PCET process in the case of C36 and C37 occurs by a concerted mechanism [58]. On the other hand, the thermal relaxation observed by high-field EPR of C37 is not a general phenomenon. As mentioned before, construct C36 with a weaker intramolecular hydrogen bond and the benzimidazole group as part of the linkage between the porphyrin and phenol moieties does not show such relaxation [59]. Thus, the simplicity of these dye-SNP systems and the ability of C37 to reproduce the EPR thermal relaxation observed in the TyrD–His189–P680 portion of the PSII electron transport chain helped to show that the strong intramolecular hydrogen bond in the Bi–PhOH group of C37 is a key structural component responsible for its biomimetic behavior [60].

Structures of molecular triad C35 and dye-SNP systems C36, C37 and C38. TiO2 and SnO2 indicate the composition of the semiconductor nanoparticles to which the dye was attached through a carboxylate group.
System C38 uses small (2.5 nm) SnO2 nanoparticles instead of TiO2 as SNPs. The SnO2 acts as the primary electron acceptor and again PF10 is the photoinduced electron donor and a Bi–PhOH group in which the phenol moiety is covalently attached to PF10 acts as a secondary electron donor [61]. Because the TiO2 band gap and PhO absorb in the same spectral region (400 nm), it is difficult to measure the PhO lifetime. Hence, we changed the semiconductor nanoparticle. In construct C38, the generation of the final charge separated state BiH+–PhO–PF10–SnO2(e−) was proven using time-resolved transient absorption measurements in aqueous solution at pH 7 at room temperature. Global analysis of the kinetics at different wavelengths showed that a 93 μs component is associated with the decay of BiH+–PhO–PF10–SnO2(e−). This determination is based on the lack of PF10 ground state bleaching and the observation of PhO and SnO2(e−) induced absorption around 400 nm and 600–1000 nm, respectively. This long lived charged separated species (BiH+–PhO–PF10–SnO2(e−)) demonstrates the key role played by the Bi–PhOH in stabilizing the charge separated state even when semiconductors with high electron mobility, such as SnO2, are involved.
4 Artificial antenna-reaction centers
EET between (bacterio)chlorophyll molecules within antennas and funneling of the energy to reaction centers is ubiquitous in light harvesting complexes of natural photosynthetic systems. In collaboration with J. Lindsey, we constructed artificial antenna molecules consisting of four covalently linked zinc tetraarylporphyrins, e.g., tetrad C39, and linked them to free base porphyrin-fullerene artificial reaction centers to give hexads C40 and C41 [45,62]. It was shown that the excitation of any peripheral zinc porphyrin moiety is followed by EET to the central zinc porphyrin with a time constant of ∼50 ps. The excitation energy is then transferred to the free base porphyrin of C40 in 240 ps, and subsequently electron transfer to the fullerene with a time constant of 3 ps takes place. The final charge-separated state has a lifetime of ∼1 ns and is generated with a quantum yield of 0.69 based on light absorbed by the zinc porphyrin antenna (Fig. 13).
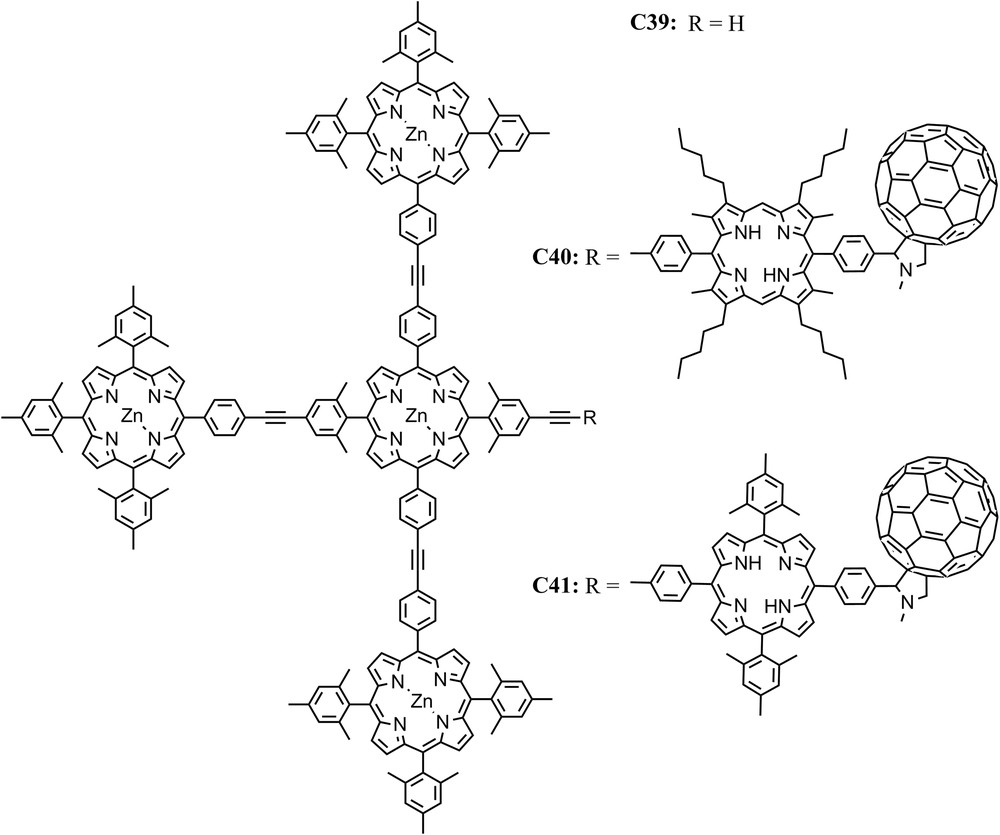
Structures of a tetrad antenna C39 and two different hexad antenna-reaction center models, one with an octa-alkyl-meso-diphenylporphyrin-C60 reaction center, C40, and another with a meso-tetra-aryl porphyrin-C60 reaction center, C41.
We note that the EET rate between the zinc porphyrin in the antenna of C40 and the free base porphyrin was relatively slow (240 ps)−1 compared with EET among the zinc porphyrins (50 ps)−1. In order to explain this behavior, a careful investigation of the frontier molecular orbitals of the porphyrins involved in the linkage was performed. It shows that the symmetry of the HOMOs of porphyrins is either a2u, which has lobes at the meso positions, or a1u which has nodes at the meso positions. Actually, there are examples of meso-linked arrays of porphyrins with a2u HOMOs which undergo rapid energy transfer, whereas similar arrays of porphyrins with a1u HOMOs undergo much slower energy transfer [63–65]. The free base porphyrin of C40 with the presence of the eight β-alkyl substituents results in the HOMO showing a1u symmetry and therefore a diminished electronic coupling with the linker. Furthermore, in molecules with β-alkyl substituents, steric effects result in an increased dihedral angle between the planes of the porphyrin ring and the meso-aryl ring. These factors certainly reduce the electronic coupling and thereby reduce the through-bond mediated contributions to EET. Increasing the rate of this process by addressing the factors that control the symmetry of the HOMO and steric effects, and thereby the electronic coupling that mediates EET, is an example of using theory to guide design to optimize function. This strategy resulted in an increase in the quantum yield of the final charge separated state from 69% in C40 to 90% in C41 [45].
Following these and related principles, it was possible to design antennas consisting of different multiple chromophores; with these systems efficient light harvesting throughout the visible spectrum was achieved. Functional mimics of a photosynthetic antenna-reaction center complex comprising five bis-(phenylethynyl)anthracene antenna moieties and a porphyrin-fullerene dyad organized by a central hexaphenylbenzene core have been prepared, C42, and studied spectroscopically. The molecules successfully integrate singlet–singlet energy transfer and photoinduced electron transfer. Energy transfer from the five antennas to the porphyrin occurs on the picosecond time scale with a quantum yield of 1.0. Comparisons with model compounds and theory suggest that the Förster mechanism plays a major role in the extremely rapid energy transfer, which occurs at rates comparable to those seen in some photosynthetic antenna systems. However, a through-bond, electron exchange mechanism also contributes. Once the excitation energy populates the porphyrin first excited singlet state it donates an electron to the attached fullerene to yield a P+–C60− charge-separated state, which has a lifetime of several nanoseconds. The quantum yield of charge separation based on light absorbed by the antenna chromophores is 80% for the free base molecule and 96% for the zinc analog [66] (Fig. 14).

Structures of antenna-reaction center C42.
In order to ensure efficient utilization of the solar spectrum, photosynthetic organisms use a variety of antenna chromophores to absorb light and transfer excitation to a reaction center, where photoinduced charge separation occurs. A synthetic molecular heptad that features two bis(phenylethynyl)anthracene and two borondipyrromethene antennas linked to a hexaphenylbenzene core that also bears two zinc porphyrins is an example of an artificial antenna with absorption that extends throughout the visible spectrum. A fullerene electron acceptor self-assembles to both porphyrins via dative bonds (C43). Excitation energy is transferred very efficiently from all four antennas to the porphyrins, which are the excitation energy sink. Singlet–singlet energy transfer occurs both directly and by a stepwise funnel-like pathway wherein excitation moves down a thermodynamic gradient. Finally, the porphyrin excited state donates an electron to the fullerene with a time constant of 3 ps to generate a charge separated state with a lifetime of 230 ps. The overall quantum yield is close to unity. In the absence of the fullerene, the porphyrin excited singlet state donates an electron to a borondipyrromethene on a slower time scale. This molecule demonstrates that by incorporating antennas, it is possible for a molecular system to harvest efficiently light throughout the visible from ultraviolet wavelengths out to ∼650 nm [67] (Fig. 15).

Structure of tetrad antenna-reaction center C43.
5 Conclusion
The fundamental photochemical and photophysical interactions among chromophores that are used in photosynthesis for light gathering and photoinduced charge separation can be realized and studied in simpler, covalently linked molecular systems and in hybrid systems consisting of dye pigments interacting with semiconductor nanoparticles. The study of these simpler systems contributes to the understanding of energy conversion in the much more complex natural systems. In addition, the principles exemplified in the simpler synthetic constructs can be used to engineer photosynthesis-like functions into artificial photosynthetic solar energy conversion schemes.
Acknowledgments
This work was supported by the Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Energy Biosciences, Department of Energy under contract DE-FG02-03ER15393 and the Center for Bio-Inspired Solar Fuel Production, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001016.