

1 Introduction
Epoxides are versatile and valuable intermediates in the synthesis of several biologically active compounds and they play an important role in industry. The ease of preparation of these compounds and their facile ring-opening reactions have contributed to make epoxides an attractive way of functionalising olefins. Some chemical routes to produce epoxides use reagents such as organic peroxyacids [1], sodium hypochlorite [2] or hydrogen peroxide [3] as a source of oxygen. The use of these reagents in the presence of a variety of catalysts to synthesize epoxides has also been reviewed recently [4].
Electrochemical methodology has been investigated as a promising alternative route to epoxides, motivated by the inherent advantages of ease of operation and limited environmental impact [5]. Moreover, indirect electrochemical reactions in comparison with direct electrochemical reactions present, in general, important advantages such as: low consumption of electricity; improvement of yield; an increase of reaction rate; reaction of electrochemically inactive substrates, by decrease of activation energy [6]. The high industrial demand for epoxides has led to a need for a “greener” route for their production. It was reported [5a] that electrogenerated hydrogen peroxide in ionic liquids, under a carbon dioxide-saturated environment and in the presence of a catalytic amount of a manganese salt, was used in the epoxidation of lipophilic alkenes. Allylic alcohols were successfully electroepoxidized by means of hydrogen peroxide generated in situ in the presence of titanium silicalite-1 [5b]. Electrosynthesis of epoxides and dibromides from olefinic carboxylic acids was achieved [7] by indirect oxidation (bromination) through a Br−/Br+ mediator leading to the corresponding 1,2-dibromide. The epoxidation was obtained by a multi-step process involving the cathodic reduction of dioxygen to hydrogen peroxide and also the action of a vanadium catalyst. Electroepoxidation can also be achieved by bromide ion mediation [8] at room temperature. The reaction proceeds through an indirect electrochemical reaction avoiding certain specific disadvantages of direct electrolysis, for example, in passivation of the electrode surface or slow heterogeneous electron transfer. Tori et al. [9] reported that the electrooxidation of halide salts is useful for the generation of reactive species of halogen atoms under mild conditions. In fact, it has been shown that the Br− ion discharged from NaBr can be a good mediator for the epoxidation of dimethyl 4-cyclohexene-1,2-dicarboxylate and acyclic alkenes [9] and n-hexene [10] in water–organic solvent media.
Herein, using the methodology reported in the literature [9], we describe the electrochemical epoxidation of styrene, (E)-1,2-diphenylethene (trans-stilbene), 1-[(E)-prop-1-enyl]benzene (trans-β-methylstyrene), 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene (limonene), 1-methyl-4-(propan-2-ylidene)cyclohex-1-ene (terpinolene), (E)-3,7-dimethylocta-2,6-dien-1-ol (geraniol), 1-isopropyl-4-methylcyclohexa-1,3-diene (α-terpinene), 1-isopropyl-4-methylcyclohexa-1,4-diene (γ-terpinene) and 2-(4-methylcyclohex-3-enyl)propan-2-ol (α-terpineol), and using sodium bromide as mediator in acetonitrile–water mixtures at constant-current electrolyses. The yields obtained are compared with those published for the epoxidation of the same alkenes using different methodologies.
2 Results and discussion
2.1 Electrochemistry of Br2/Br− system
The cyclic voltammetric study of Br2/Br− system was carried out in MeCN:H2O (1:4) containing NaClO4 (0.1 mol dm−3) as supporting electrolyte at a platinum electrode. Under these conditions the Br− ion presents an oxidation peak at 0.99 V and the corresponding reduction peak at 0.91 V versus Ag/AgCl due to the one-electron process described in the following equation:
| (1) |
2.2 Electroepoxidation of synthetic alkenes: styrene, (E)-1,2-diphenylethene and 1-[(E)-prop-1-enyl]benzene
The study was initiated by carrying out constant-current electrolysis of 20 mM styrene (1) in the presence of 20 mM NaBr as mediator using a current density of 33 mA cm−2 and Pt electrodes in MeCN:H2O (4:1). It was observed, based on 1H NMR data [12], that 2-phenyloxirane (4) was obtained in 27% yield (Table 1, entry 1). In order to improve the yield of epoxide we studied the influence of the concentration of NaBr and found that an increase from 20 mM to 30 mM led to an increase in the yield of 4 to 57% (Table 1, entry 2). The influence of the current density was also explored and an increase from 33 to 66 mA cm−2 was found to produce the epoxide, as the major product, in a yield of 79% (Table 1, entry 3), together with bromohydrin 3 (10%) and dibromo derivative 6 (11%) as by-products (Fig. 1). These products were identified on the basis of 1H NMR data. In previous studies [13] it was reported that styrene oxide was obtained in yields between 30 and 100% using constant-potential electrolyses and Mn–Salen catalysts.
Coulometric data and product yields for catalytic oxidation of synthetic alkenes (20 mM) in the presence of sodium bromide (20 mM) in MeCN:H2O (4:1) at platinum electrodes.
| Entry | Substrate | Current densitya (mA cm−2) | F/molb | Conversion (%) | Epoxide yield (ηm/%) | Current efficiency (ηc/%) | Product |
| 1 | 1 | 33 | 2 | 100 | 27 | 21 | 4 |
| 2 | 1c | 33 | 2 | 100 | 57 | 74 | 4 |
| 3 | 1 | 66 | 2 | 100 | 79 | 38 | 4 |
| 4 | 8 | 33d | 5 | 66 | 23 | 47 | 9 |
| 5 | 8 | 33d | 8 | 84 | 24 | 50 | 9 |
| 6 | 8 | 3.3d | 4 | 82 | 38 | 67 | 9 |
| 7 | 11 | 33 | 5 | 100 | 30 | 29 | 12 |
a Platinum anode (A = 1.1 × 1.1 cm2).
b Number of faradays per molecule of the starting material.
c [NaBr] = 30 mM.
d Platinum anode (A = 2.5 × 2.5 cm2); ηm = [obtained mass of epoxide (g)/theoretical mass of epoxide (g)] × 100; ηc = [moles of product obtained by a charge c/theoretical moles of product obtained by a charge c] × 100; conversion (%) = [(initial moles of substrate − moles of substrate at the end of reaction)/initial moles of substrate] × 100.
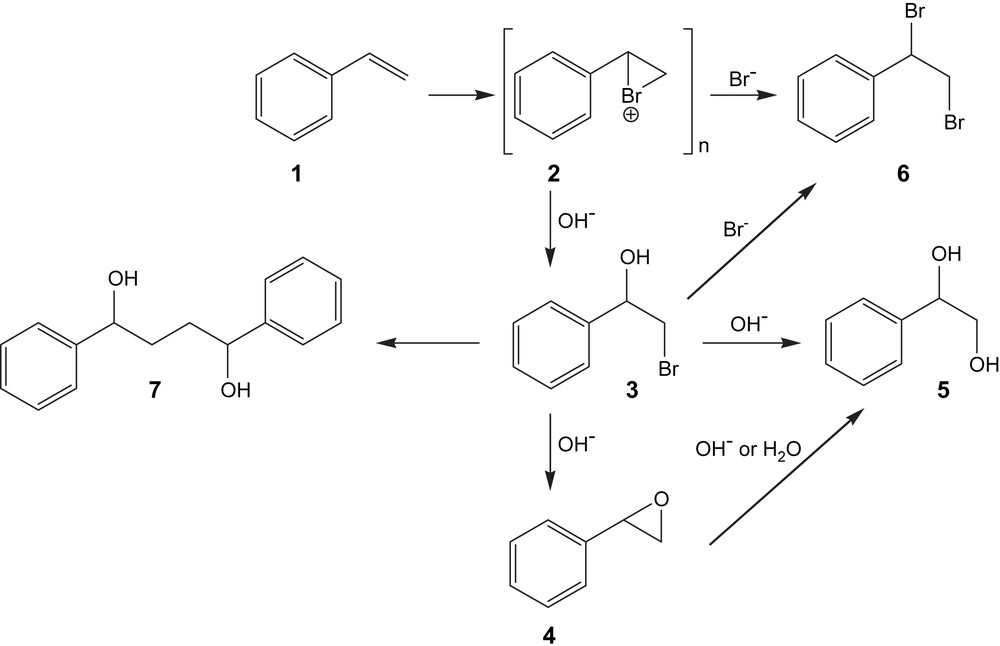
The proposed mechanism for styrene electroepoxidation mediated by NaBr and for the identified by-products.
Electroepoxidation of (E)-1,2-diphenylethene (8) was carried out using the same experimental conditions and a current density of 33 mA cm−2 and led to the formation of 2,3-diphenyloxirane (9) in 23% yield (Table 1, entry 4). The main by-product identified was the corresponding dibromide 10 (16%) (Fig. 2). Using the same current density but increasing the charge passed, the degree of conversion increased but the epoxide yield did not (Table 1, entry 5). Subsequently, the influence of the current density was studied and it was found that using a current density of 3.3 mA cm−2, the electroepoxidation of (E)-1,2-diphenylethene (8) led to the formation of 2,3-diphenyloxirane (9) in 38% yield (Table 1, entry 6). An epoxide yield between 70% and 93% was obtained with cobalt catalysts or potassium peroxomonosulfate [14].
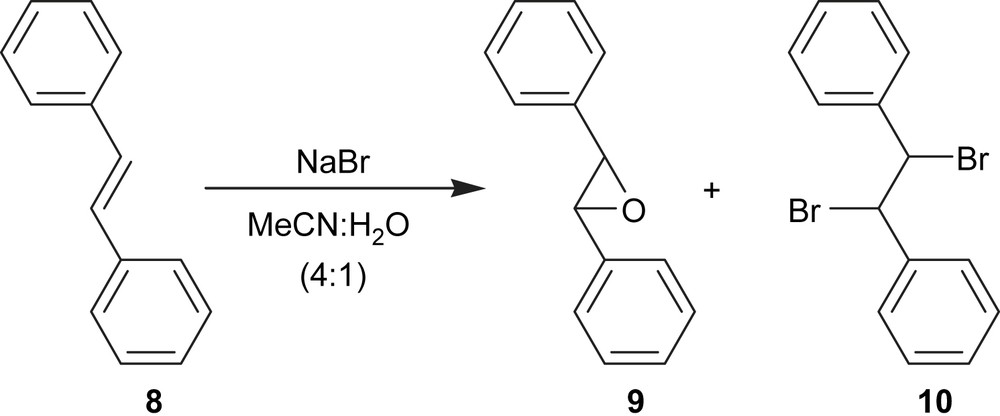
Electroepoxidation of (E)-1,2-diphenylethene.
The electroepoxidation of 1-[(E)-prop-1-enyl]benzene (11) was performed using the same experimental conditions and a current density of 33 mA cm−2. Under these conditions a conversion of 100% of 1-[(E)-prop-1-enyl]benzene was obtained (11) but 2-methyl-3-phenyloxirane (12) was produced with 30% yield and a current efficiency of 29% (Fig. 3) (Table 1, entry 7). Epoxide (12) was obtained in 5–54% yield when Ni(II) complexes were used as catalysts [15].
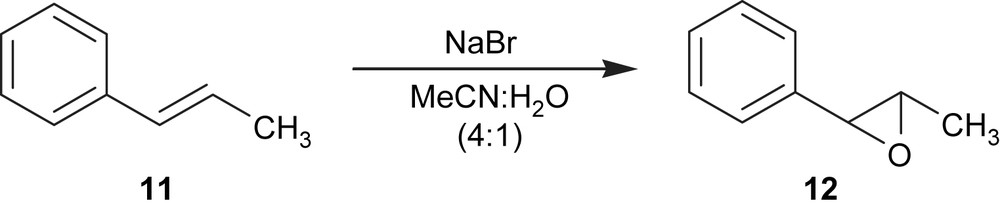
Electroepoxidation of 1-[(E)-prop-1-enyl]benzene.
In all the epoxidations carried out the main by-products obtained were the corresponding bromohydrin (4–23%) and dibromide (11–20%) derivatives. In agreement with Alkire and Köhler [10] the bromohydrin precedes the formation of epoxide while dibromide formation prevents the formation of epoxide.
2.3 Electroepoxidation of monoterpenes: 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene, 1-methyl-4-(propan-2-ylidene)cyclohex-1-ene, (E)-3,7-dimethylocta-2,6-dien-1-ol, 1-isopropyl-4-methylcyclohexa-1,3-diene, 1-isopropyl-4-methylcyclohexa-1,4-diene, and 2-(4-methylcyclohex-3-enyl)propan-2-ol
The terpene epoxides are important building blocks for natural product synthesis and as starting materials in the synthesis of flavours, fragrances and therapeutically active substances [16]. The electrochemical epoxidation was performed by slightly modifying the experimental conditions (see conditions in Table 2 caption) used for the electroepoxidation of synthetic alkenes. The results obtained are presented in Table 2.
Coulometric data and product yields for catalytic oxidation of natural terpenes (40 mM) in the presence of sodium bromide (40 mM) in MeCN:H2O (4:1) at a current density of 3.3 mA cm−2 at platinum electrodes.a
| Entry | Substrate | F/molb | Conversion (%) | Epoxide yield (ηm/%) | Current efficiency (ηc/%) | Product |
| 1 | 13 | 2c,d | 83 | 25 | 25 | 14 |
| 2 | 13 | 3c | 83 | 37 | 41 | 14 |
| 3 | 13 | 2 | 93 | 34 | 37 | 14 |
| 4 | 16 | 2 | 86 | 40 | 45 | 17 |
| 5 | 18 | 3 | 53 | 43 | 65 | 19 |
| 6 | 18 | 5 | 73 | 24 | 23 | 20 |
| 7 | 21 | 2 | 88 | 32 | 27 | 22 |
| 8 | 23 | 2 | 82 | 41 | 52 | 24 |
| 9 | 25 | 2 | 83 | 56 | 61 | 26 |
a Platinum anode (A = 2.5 × 2.5 cm2).
b Number of faradays per molecule of the starting material.
c Platinum anode (A = 1.1 × 1.1 cm2).
d Current density equal to 33 mA cm−2; ηm = [obtained mass of epoxide (g)/theoretical mass of epoxide (g)] × 100; ηc = [moles of product obtained by a charge c/theoretical moles of product obtained by a charge c] × 100; conversion (%) = [(initial moles of substrate − moles of substrate at the end of reaction)/initial moles of substrate] × 100.
2.3.1 1-Methyl-4-(prop-1-en-2-yl)cyclohex-1-ene
The constant-current electrolysis of 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene (13) (20 mM) in MeCN:H2O (4:1) as substrate in the presence of NaBr (40 mM), with a current density of 33 mA cm−2 using Pt electrodes (1.1 × 1.1 cm2), after 5 F/mol, produced an unexpected compound (15) in 25% yield (Fig. 4). The structure proposed for compound 15 was derived from the NMR (1H and 13C) analysis and is in agreement with the literature [17]. This compound was obtained when the concentration of 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene was half of the concentration of NaBr and the current density was 33 mA cm−2. Maintaining this current density but increasing the 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene concentration to 40 mM, only compound 14 was obtained in 25% yield (Table 2, entry 1). The 1H NMR spectrum of this compound was in good agreement with that already published [12].
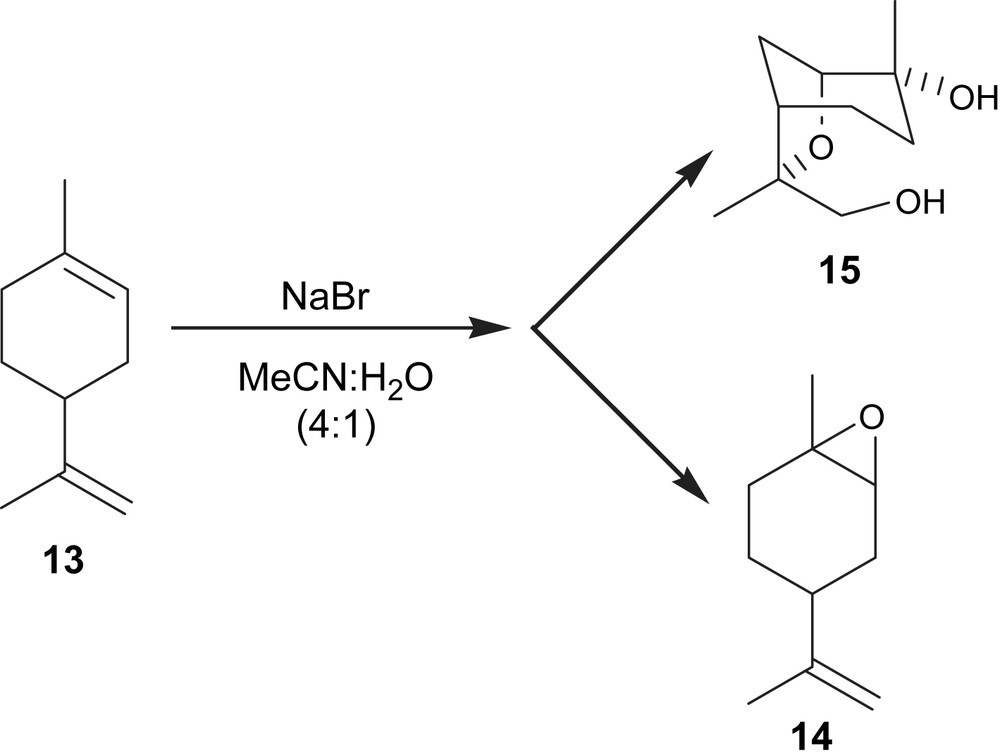
Electroepoxidation of 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene.
In order to study the influence of the current density, the electroepoxidation of 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene was performed using a current density of 3.3 mA cm−2. It was observed that a decrease in the current density reduced the rate of electrolysis and 11 h were necessary to achieve 3 F/mol (Table 2, entry 2). The Pt electrode area was increased to 2.5 × 2.5 cm2 and although the reaction time diminished (2.5 h) and the degree of conversion (93%) was higher, an epoxide yield of only 34% was obtained (Table 2, entry 3). The analysis of these results confirms that the epoxide was obtained in relatively low yields. It was reported [18] that the electroepoxidation of 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene catalysed by Mn(salen), Ni(salen) or heteropolytungstates complexes afforded the expected epoxide (14) in 96% yield in optimised experimental conditions. The analysis of 1H NMR spectral data of compound 14 revealed the existence of two diastereomers in the ratio of 3:2.
2.3.2 1-Methyl-4-(propan-2-ylidene)cyclohex-1-ene
The electroepoxidation of 1-methyl-4-(propan-2-ylidene)cyclohex-1-ene (16) was also mediated by NaBr in MeCN:H2O (4:1) (Fig. 5) using the optimised conditions applied in 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene epoxidation. The 2,2,6-trimethyl-1-oxaspiro[2.5]oct-5-ene (17) was obtained in 40% yield with a current efficiency of 45% for a conversion of 86% (Table 2, entry 4). The yield obtained is similar to that reported (36%) when alumina was used as a heterogeneous catalyst and hydrogen peroxide as the oxidising agent [16].
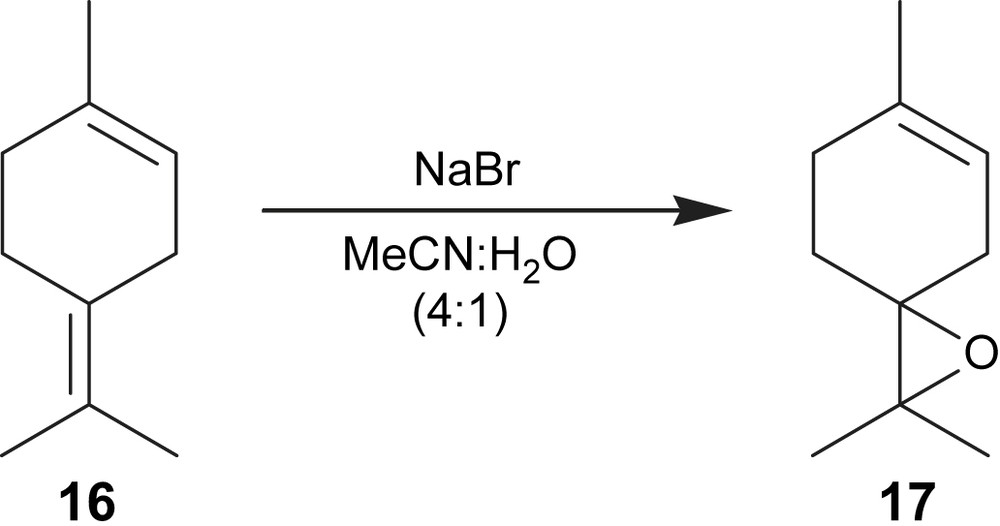
Electroepoxidation of 1-methyl-4-(propan-2-ylidene)cyclohex-1-ene.
2.3.3 (E)-3,7-Dimethylocta-2,6-dien-1-ol
The electroepoxidation of (E)-3,7-dimethylocta-2,6-dien-1-ol (18) mediated by NaBr led to the formation of (E)-3-methyl-5-(3,3-dimethyloxiran-2-yl)pent-2-en-1-ol (19) and {3-methyl-3-[2-(3,3-dimethyloxiran-2-yl)ethyl]oxiran-2-yl}methanol (20) as the main products (Fig. 6). (E)-3-Methyl-5-(3,3-dimethyloxiran-2-yl)pent-2-en-1-ol (19) was obtained in 43% yield and with a current efficiency of 65% (Table 2, entry 5). The influence of the charge passed during the reaction was studied in order to improve the yield of 19 but it was observed that by increasing the charge passed (5 F/mol) the yield of (E)-3-methyl-5-(3,3-dimethyloxiran-2-yl)pent-2-en-1-ol decreased and that of {3-methyl-3-[2-(3,3-dimethyloxiran-2-yl)ethyl]oxiran-2-yl}methanol (20) increased (Table 2, entry 6). Comparing these results with those published [19] it can be seen that the yield of (E)-3-methyl-5-(3,3-dimethyloxiran-2-yl)pent-2-en-1-ol (19) is lower than that obtained (67%) with titanosilicates and hydrogen peroxide. However, using the electrochemical approach it is possible to obtain the diepoxide.
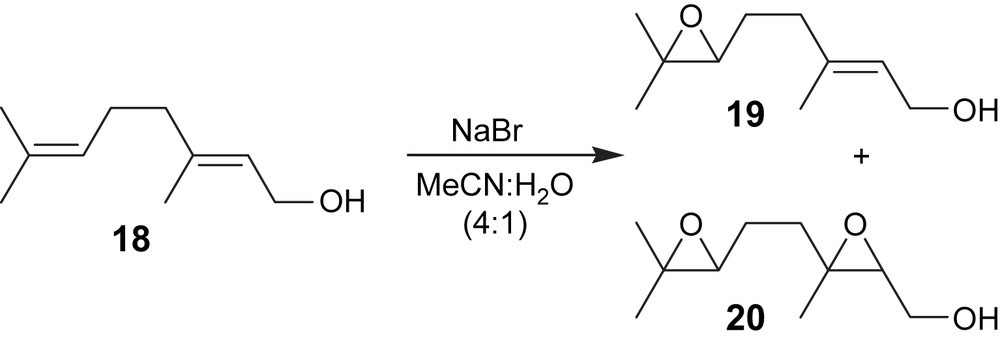
Electroepoxidation of (E)-3,7-dimethylocta-2,6-dien-1-ol.
The analysis of 13C NMR data of compound 20 reveals a mixture of two diastereomers with a diastereomeric ratio of 3:1 which is in agreement with the published data [20].
2.3.4 Other natural monoterpenes
We have also performed the electroepoxidation of 1-isopropyl-4-methylcyclohexa-1,3-diene (21) (Fig. 7) and compound 22 was obtained in 32% yield (Table 2, entry 7). A migration of the double bond, via an SN2′ type mechanism was observed. Due to the high reactivity of 1-isopropyl-4-methylcyclohexa-1,3-diene, Uguina et al. [16] also obtained a larger amount of secondary products resulting in a low selectivity in the formation of the epoxide.
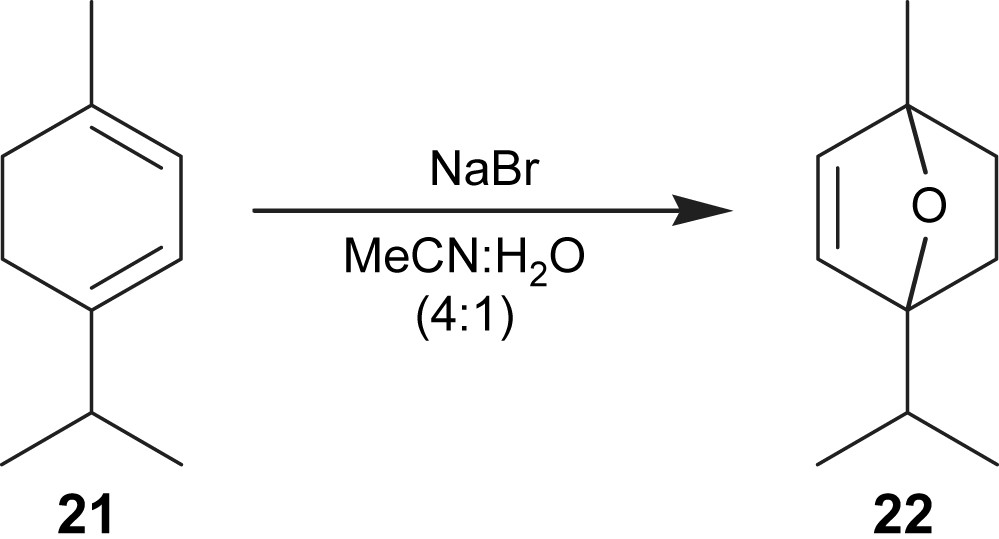
Electroepoxidation of 1-isopropyl-4-methylcyclohexa-1,3-diene.
In the 1-isopropyl-4-methylcyclohexa-1,4-diene (23) epoxidation (Fig. 8) the epoxide (24) was obtained in a yield of 41% with a conversion of 82% (Table 2, entry 8). Sakaguchi et al. [21] have reported that the epoxide 24 could be obtained in a yield of 78%, with aqueous hydrogen peroxide and peroxotungstophosphate as catalyst under biphasic conditions using chloroform as the solvent.
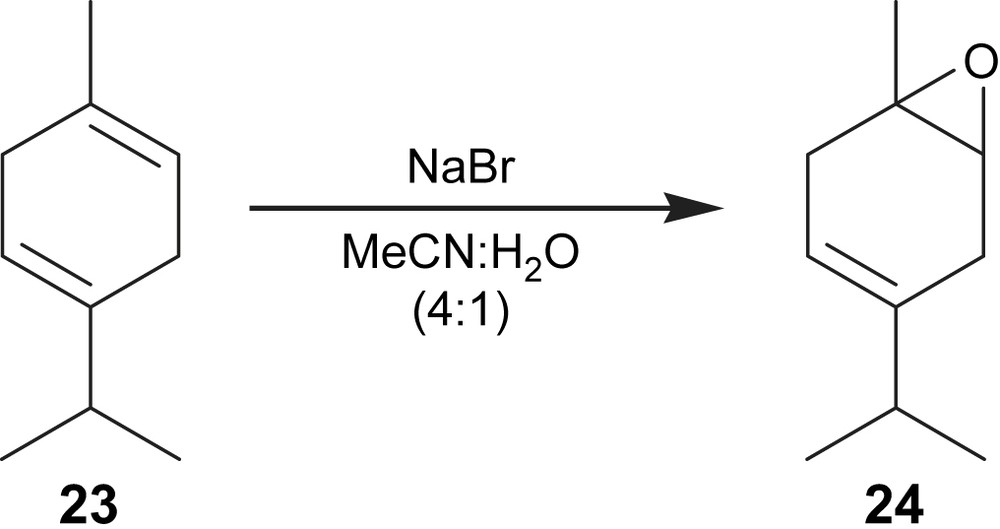
Electroepoxidation of 1-isopropyl-4-methylcyclohexa-1,4-diene.
In the case of 2-(4-methylcyclohex-3-enyl)propan-2-ol (25), the conversion of the substrate was 83% and the epoxide 26 (Fig. 9) yield was 56% with a current efficiency of 61% (Table 2, entry 9). Chan et al. [20b] have reported yields of epoxide 26 in the range 62–86% using a beta-cyclodextrin-substituted dioxirane as catalyst.
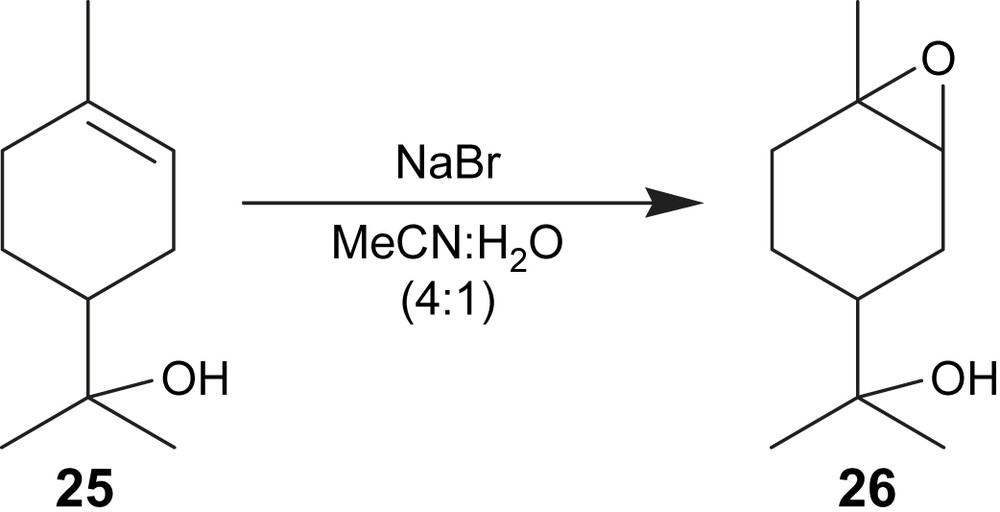
Electroepoxidation of 2-(4-methylcyclohex-3-enyl)propan-2-ol.
2.4 Turn-over of NaBr mediator solution
The turn-over number of the NaBr aqueous solution to mediate a new electroepoxidation was evaluated using 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene as substrate. After removal of the oxidation products (Table 2, entry 3), the NaBr solution was used for a 2nd run. This process was repeated for four runs. The results obtained are presented in Table 3. From an analysis of the data reported in Table 3, it is clear that the epoxide yield obtained decreased substantially from the 3rd to the 4th run. From the 1st to the 2nd run there is also a small decrease in the epoxide yield. In each cycle of the mediator there are bromine losses, mainly by the formation of brominated by-products. As the NaBr concentration diminishes the epoxide yield also decreases. We considered that the yield obtained in the initial experiment (Table 2, entry 3), (34%) is close to the 39% obtained in the first run (Table 3, entry 1). We have concluded that the same NaBr solution can be used at least two times further after the initial electrolysis without significant loss of activity of NaBr.
1-Methyl-4-(prop-1-en-2-yl)-7-oxa-bicyclo[4.1.0]heptane (14) yields obtained re-using the solution of NaBr of the epoxidation of 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene (Table 2, entry 3) with current density of 3.3 mA cm−2 at platinum anode (2.5 × 2.5 cm2). [1-Methyl-4-(prop-1-en-2-yl)cyclohex-1-ene] = 40 mM.
| Run | 1-Methyl-4-(prop-1-en-2-yl)-7-oxa-bicyclo[4.1.0]heptane (%) | |
| ηm | ηc | |
| 1 | 39 | 34 |
| 2 | 35 | 31 |
| 3 | 29 | 21 |
| 4 | 20 | 12 |
Taking into consideration our results for the electrochemical epoxidation in the presence of NaBr and based on the mechanism proposed by Alkire and Köhler [10], we propose the following mechanism.
The initial steps are anodic oxidation of bromide and cathodic reduction of water:
| (2) |
| (3) |
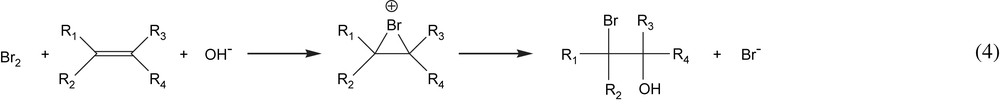
The halohydrin then consumes another OH− ion and is converted to the epoxide:
3 Conclusion
The epoxidation of synthetic alkenes and natural terpenes was achieved through an indirect electrochemical reaction, at constant current, mediated by NaBr in MeCN:H2O (4:1) at room temperature in an undivided cell under stirring using platinum electrodes (2.5 × 2.5 cm2 or 1.1 × 1.1 cm2). By adjusting the experimental conditions it was possible to obtain the desired epoxides, although other oxidation products can be formed.
Optimised experimental conditions were established with styrene as substrate. The electroepoxidation of all other substrates was carried out using these conditions with minor modifications.
There are several reasons that may explain the relatively low epoxide yields obtained. Epoxides are known to present high reactivity leading them to form other compounds, particularly, diols. In the present study, diols were detected by 13C NMR as by-products in all reactions but were not quantified. A further reason for low yields might be a result of the electrophilic addition reaction of bromine to double bonds. This reaction explains the formation of mono- and dibromide by-products. The low yield of monoterpene epoxides might be explained by the high volatility of these compounds.
However, although the yields are modest, this method may provide an alternative to classical chemical methods, avoiding the use of oxidants such as NaClO. The method used in this study is simple, economic and carried out at room temperature.
4 Experimental part
4.1 Reagents
Alkenes and monoterpenes were used as-received (Sigma–Aldrich, >98%) and all solvents were purified prior to use.
4.2 Constant-current electrolysis
Electrolyses were carried out in a single compartment cell with electrodes separated by 5 mm, under agitation at constant current using a DC power supply (Good Will Instruments GSP-3030D). Electrolyte volumes were 25 cm3 or 50 cm2, using 1.1 × 1.1 cm2 or 2.5 × 2.5 cm2 Pt electrodes, respectively. The electrolysis was monitored by TLC and terminated when the substrate was completely converted or the number of by-products increased with time without further consumption of the substrate. After the electrolysis, sodium metabisulfite (1%) was added to the reaction mixture, which was extracted with ethyl ether or chloroform (3 × 15 mL) and washed with water. The combined organic extracts were dried (Na2SO4) and concentrated. The crude residue was submitted to flash chromatography on silica gel 60 M (230–400 mesh).
1H and 13C NMR data were recorded on a Bruker AC 250P Spectrometer in CDCl3; chemical shifts were measured, in parts per million, versus the residual peak of the solvent. The infrared spectra were recorded on a Unicam Mattson 5000 FT spectrophotometer, using capillary films supported on NaCl windows or on a Bomem MB-series FT-IR/DiffusIR spectrophotometer, using Nujol mulls on NaCl windows.
The compounds were identified by means of 1H NMR (250 MHz, CDCl3) and 13C NMR (63 MHz) spectrometry, δ ppm.
4.2.1 2-Phenyloxirane (4)
1H NMR (CDCl3, 250 MHz): 7.40–7.27 (5H, m, ArH); 3.83 (1H, dd, J1 = 3.9; J2 = 2.5 Hz, H-2); 3.12 (1H, dd, J1 = 5.4; J2 = 3.9 Hz, H-1); 2.77 (1H, dd, J1 = 5.4; J2 = 2.5 Hz, H-1).
4.2.2 2,3-Diphenyloxirane (9)
1H NMR (CDCl3, 250 MHz): 7.56–7.36 (10H, m, ArH); 3.99 (2H, s, H-1 e H-2). 13C NMR (CDCl3, 63 MHz): 137.3 (C-1′ and C-1″); 128.7 (C-3′, C-5′, C-3″ and C-5″); 128.5 (C-4′ and C-4″); 125.7 (C-2′, C-6′, C-2″ and C-6″); 63.0 (C-1 and C-2).
4.2.3 2-Methyl-3-phenyloxirane (12)
1H NMR (CDCl3, 250 MHz): 7.41–7.21 (5H, m, ArH); 5.04 (1H, d, J = 10.0 Hz, H-2); 4.60 (1H, dq, J1 = 10.0; J2 = 6.5 Hz, H-1); 2.05 (3H, d, J = 6.5 Hz, H-1′). 13C NMR (CDCl3, 63 MHz): 140.7 (C-1″); 128.9 (C-4″); 128.8 (C-3″ and C-5″); 127.8 (C-2″ and C-6″); 59.3 (C-2); 51.3 (C-1); 26.0 (C-1′). νmax (cm−1): 2923, 2852, 1560, 1260 and 840 (C–O–C), 1215, 1094, 820.
4.2.4 1-Methyl-4-(prop-1-en-2-yl)-7-oxa-bicyclo[4.1.0]heptane (14)
1H NMR (CDCl3, 250 MHz): 4.65 (2H, br s, H-10); 3.05 (0.4H, t, J = 2.4 Hz, H-2); 2.99 (0.6H, d, J = 5.4 Hz, H-2); 2.18–1.97 (1H, m, H-4); 1.87–1.81 (2H, m, 1H-6 and 1H-3); 1.74–1.50 (2H, m, 1H-5 and 1H-3); 1.68 (1.2H, s, H-9); 1.66 (1.8H, s, H-9); 1.41–1.20 (2H, m, 1H-5 and 1H-6), 1.31 (1.8H, s, H-7); 1.29 (1.2H, s, H-7).
4.2.5 7-(Hydroxymethyl)-4,7-dimethyl-6-oxa-bicyclo[3.2.1]octan-4-ol (15)
1H NMR (CDCl3, 250 MHz): 3.87 (1H, d, J = 11.1 Hz, CH2–OH part A of an AB system, H-8); 3.86 (1H, d, J = 4.8 Hz, H-5); 3.64 (1H, d, J = 11.1 Hz, CH2–OH part B of an AB system, H-8); 2.05–1.98 (1H, m, H-1); 1.86–1.61 (6H, m, H-2, H-3 and H-10); 1.29 (3H, s, H-9); 1.22 (3H, s, H-11). 13C NMR (CDCl3, 63 MHz): 84.8 (C-5); 84.3 (C-7); 72.5 (C-4); 65.8 (CH2–OH); 40.1 (C-1); 35.6 (C-3 or C-8); 33.8 (C-8 or C-3); 24.5 (C-9 or C-11); 24.4 (C-9 or C-11); 24.2 (C-2).
4.2.6 2,2,6-Trimethyl-1-oxaspiro[2.5]oct-5-ene (17)
1H NMR (CDCl3, 250 MHz): 5.30 (1H, dq, J1 = 3.1; J2 = 1.6 Hz, H-2); 2.33–1.96 (4H), 1.74–1.67 (2H), 1.65 (3H, m, H-7); 1.31 and 1.28 (6H, 2s, H-10 and H-9). 13C NMR (CDCl3, 63 MHz): 134.4 (C-1); 119.2 (C-2); 64.4 (C-4); 62.4 (C-8); 30.7 (C-6); 28.9 (C-3); 27.0 (C-5); 23.4 (C-7); 20.9 (C-9); 20.5 (C-10). νmax (cm−1): 3018 (CCH), 2925, 2915, 1635, 1221 and 852 (C–O–C), 756.
4.2.7 (E)-3-Methyl-5-(3,3-dimethyloxiran-2-yl)pent-2-en-1-ol (19)
1H NMR (CDCl3, 250 MHz): 5.39 (1H, t, J = 6.8 Hz, H-2), 4.08 (2H, broad d, J = 6.8 Hz, H-1); 2.67 (1H, t, J = 6.2 Hz, H-6); 2.17 (1H, broad s, OH); 2.13–2.05 (2H, m, H-4); 1.63 (3H, broad s, H-10); 1.60–1.55 (2H, m, H-5); 1.25 and 1.21 (6H, 2s, H-8 and H-9). 13C NMR (CDCl3, 63 MHz): 137.7 (C-3); 124.2 (C-2); 64.1 (C-6); 58.8 (C-1); 58.5 (C-7); 36.2 (C-4); 27.0 (C-5); 24.7 (C-8); 18.6 (C-9); 16.1 (C-10).
4.2.8 {3-Methyl-3-[2-(3,3-dimethyloxiran-2-yl)ethyl]oxiran-2-yl}methanol (20)
1H NMR (CDCl3, 250 MHz): 3.80–3.68 (2H, m, H-1); 2.99 (1H, dd, J1 = 8.3; J2 = 3.8 Hz, H-2); 2.78–2.64 (2H, m, H-6); 1.93 (1H, s, OH); 1.88–1.47 (4H, m, H-4 and H-5); 1.33 (0.75H, H-8); 1.32 (2.25H, H-8); 1.29 and 1.27 (6H, 2s, H-10 and H-9). 13C NMR (CDCl3, 63 MHz): 64.5 and 63.9 (C-6); 62.9 and 62.5 (C-2); 61.4 and 61.0 (C-1); 60.9 and 60.8 (C-7); 59.0 and 58.7 (C-3); 36.3 and 35.2 (C-4); 24.9 and 24.6 (C-5); 25.0 (C-8); 18.9 and 18.8 (C-9); 17.1 and 16.5 (C-10).
4.2.9 1-Isopropyl-4-methyl-7-oxa-bicyclo[2.2.1]hept-2-ene (22)
1H NMR (CDCl3, 250 MHz): 5.72 (1H, dd, J1 = 10.0; J2 = 1.4 Hz, H-2); 5.61(1H, dd, J1 = 10.0; J2 = 1.5 Hz, H-3); 1.31 (3H, s, H-1″); 0.96 (3H, d, J = 6.8 Hz, H-2′); 0.89 (3H, d, J = 6.9 Hz, H-3′). 13C NMR (CDCl3, 63 MHz): 135.5 (C-3); 133.6 (C-2); 71.7 (C-4); 67.4 (C-1); 37.5 (C-1′); 33.6 (C-5); 29.7 (C-1″); 27.1 (C-6); 17.7 (C-2″ or C-3); 16.5 (C-3′ or C-2′).
4.2.10 4-Isopropyl-1-methyl-7-oxa-bicyclo[4.1.0]hept-3-ene (24)
1H NMR (CDCl3, 250 MHz): 5.15 (1H, dd, J1 = 3.4; J2 = 1.9 Hz, H-5); 3.10 (1H, br s, H-2); 2.13 (1H, sept, J = 6.8 Hz, H-8); 1.35 (3H, s, H-7); 0.97 (3H, d, J = 6.8 Hz, H-9); 0.96 (3H, d, J = 6.8 Hz, H-10). 13C NMR (CDCl3, 63 MHz): 137.7 (C-4); 113.9 (C-5); 60.4 (C-2); 56.6 (C-1); 34.6 (C-8); 30.6 (C-6); 27.2 (C-3); 23.0 (C-7); 21.4 (C-9 or C-10); 21.1 (C-10 or C-9). νmax (cm−1): 2948, 2879, 1640, 1224 and 845 (C–O–C), 750.
4.2.11 2-(6-Methyl-7-oxa-bicyclo[4.1.0]heptan-3-yl)propan-2-ol (26)
1H NMR (CDCl3, 250 MHz): 2.92 (1H, d, J = 5.3 Hz, H-2); 2.14–1.42 (6H, m, H-3, H-6 and H-5); 1.70–1.51 (1H, m, H-4); 1.23, 1.05 and 1.03 (9H, 3s, H-7, H-10 and H-9). 13C NMR (CDCl3, 63 MHz): 72.4 (C-8); 59.3 (C-2); 57.8 (C-1); 44.1 (C-4); 30.8 (C-6); 27.3 (C-7); 26.1 (C-9 or C-10); 25.9 (C-3); 24.4 (C-10 or C-9).
Acknowledgements
One of the authors (M.I.) gratefully acknowledges a GRICES PhD scholarship and INABE (Angola) for financial support.