

1 Introduction
Although known since 1957, cationic η6-(arene)tricarbonylmanganese complexes [1–4] have received little attention in comparison with the isoelectronic neutral η6-(arene)tricarbonylchromium derivatives [5–8], which have been widely used in organic synthesis. The main feature of cationic manganese complexes is their high electrophilic properties which allowed addition reactions of nucleophiles such as, inter alia, hydrides [9–14], to the coordinated arenes, leading to the formation of stable η5-(cyclohexadienyl)Mn(CO)3 complexes. The high solubility and the ‘non-charged’ nature of these last complexes gave them some interesting properties that were used in different fields. At first, they have been proved to be useful for the purification of η6-(halogenoarene)Mn+(CO)3 complexes, which are difficult to obtain in a pure form after coordination of the corresponding arenes to the Mn(CO)3 entity. Indeed, it has been well reported that complexation of bromobenzene, for example, suffered rather extensive loss of bromine and yielded a mixture of cationic bromobenzene and benzene complexes 1 and 2 (Fig. 1) [15, 16]. Thus, reaction of this mixture of complexes 1 and 2 with an hydride source afforded a mixture of neutral η5-(bromocyclohexadienyl) and η5-(cyclohexadienyl)Mn(CO)3 3 and 4 that could easily be separated by silica gel chromatography column. Hydride abstraction by CPh3+ PF6– from complex 3 gave the pure bromobenzene complex 1 (Fig. 1). Another source of difficulty to obtain η6-(halogenoarene) in a pure state comes from the problem due to their reactivity with polar solvents [16] and attempts to recrystallise the crude material from acetone–ethanol are frustrated by the reaction of the halogenoarene complex with EtOH, giving the ethoxybenzene complex. Only a relatively few studies about the reactivity of the neutral η5-Mn complexes were developed and involved attack at the terminus of the π-system leading to the formation of cyclohexadienes [4, 17] after an oxidative process. As far as we are concerned, we have used the anionic chromium complex 6 as a hydride donor towards cationic benzene Mn complex 2, thus demonstrating not only the easy hydride addition to Mn complexes, but also the reversibility of hydride addition in the case of η6-(benzene)Cr(CO)3 complex. Indeed, hydride H– was trapped with cationic η6-(benzene)Mn+(CO)3 2 to give the neutral η5-Mn complex 4 and the η6-Cr complex 5 (Fig. 2) [18]. We also described the first cine and tele nucleophilic substitutions of η5-(halogenocyclohexadienyl)Mn(CO)3 complexes after treatment with hydrides and a proton source [19]. Finally, very recently, we showed that η5-Mn complexes [19–23] could represent key building blocks for the synthesis of a new generation of electron-withdrawing group substituted η6-(arene)Mn+(CO)3 complexes, via palladium-catalyzed reactions (Fig. 3) [24, 25]. Indeed, Pd-catalyzed reactions of complex 7 in the presence of AsPh3 efficiently afforded the corresponding η5-Mn derivatives 8 substituted by carbon, oxygen, nitrogen, sulphur and phosphorous nucleophiles or η5 complexes 9 under CO atmosphere. Removal of the exo-hydrogen with CPh3BF4 yielded the η6-complexes 10 or 11. In the course of these studies, using differently substituted η5-(halogenocyclohexadienyl)Mn(CO)3 complexes as starting material, we prepared the two regioisomer chloro complexes 13 and 14. In this paper, we describe their structures in solution by means of NMR spectroscopy and in the solid state by means of X-ray analysis.
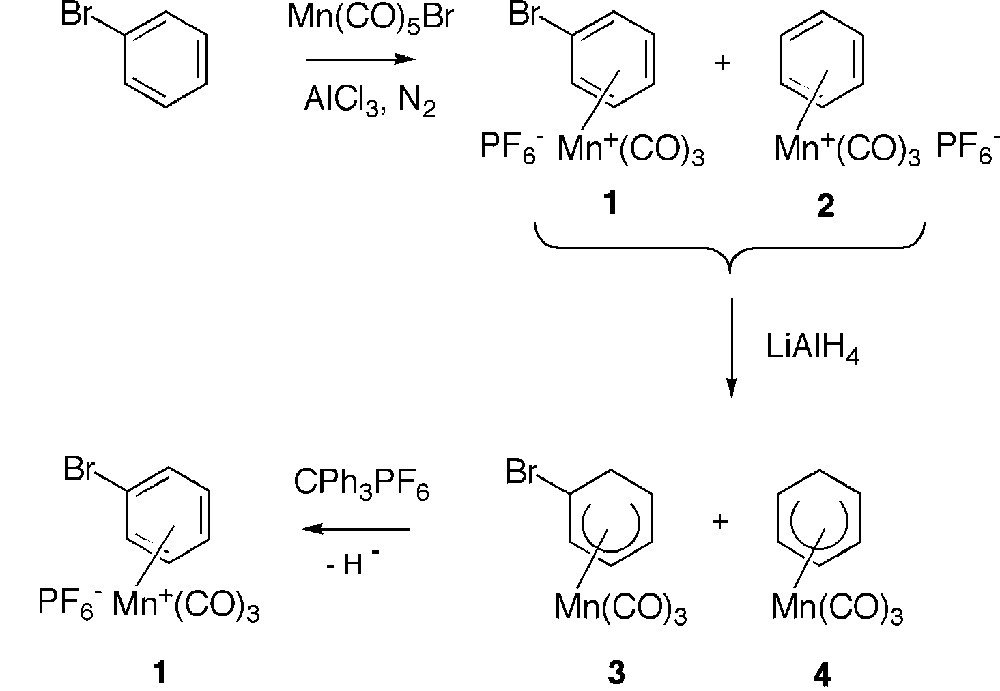
Complexation of bromobenzene to Mn(CO)3 entity.
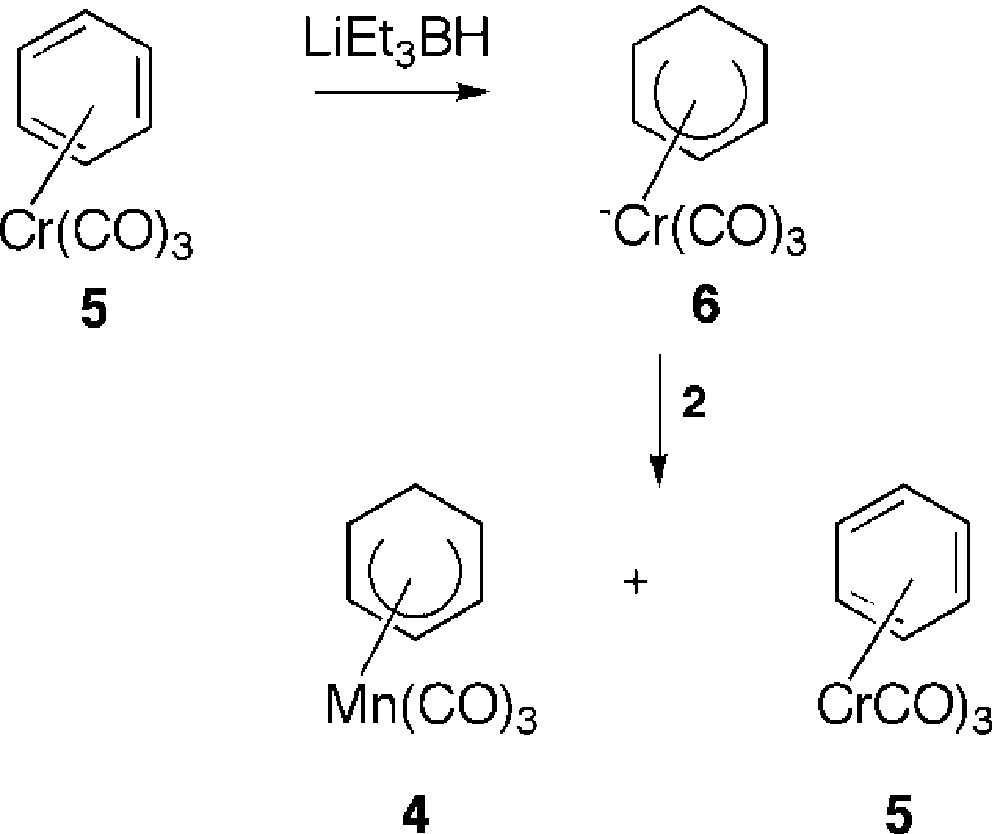
Easy hydride addition to Mn complexes.
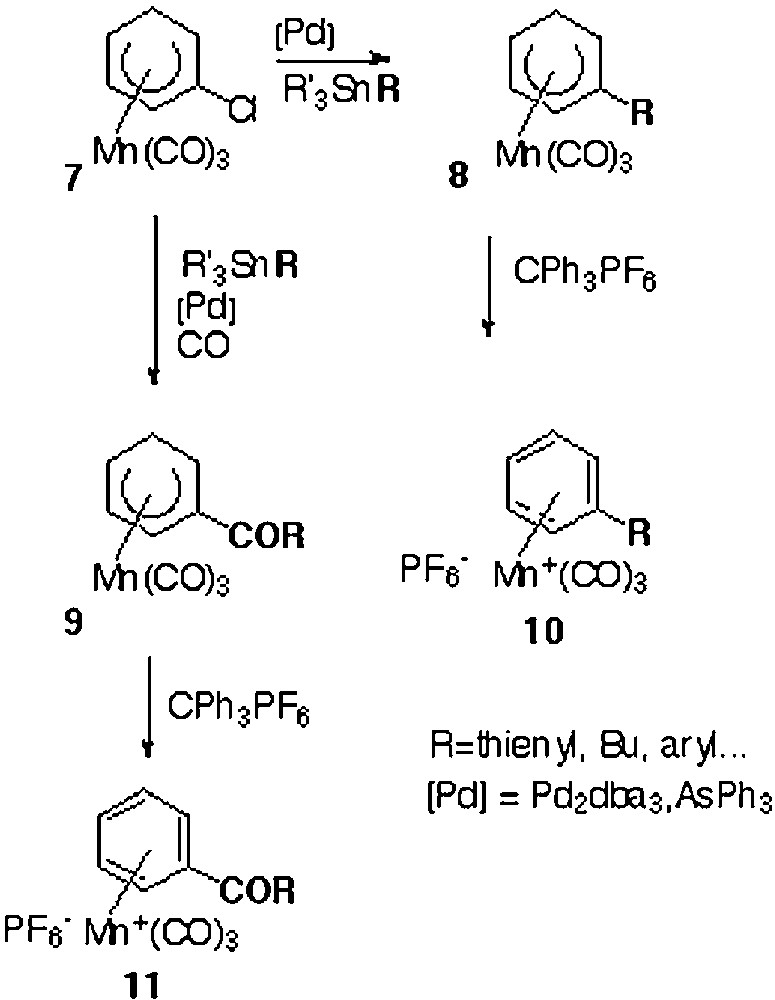
Palladium-catalyzed reactions applied to the syntheses of new Mn complexes.
2 Results and discussion
Addition of LiAlH4 to p-chlorotoluene tricarbonylmanganese complex 12 gave a 65:35 mixture of 1-chloro and 4-chloro complexes 13 and 14 in 90% yield [19, 26] Other hydride sources have been described in the literature, for example: LiEt3BH, LiB(i-PrO)3H [9], KBH4, KBEt3H, KB(i-PrO)3H, LiB[CH(CH3)C2H5]3H [10], Bu4NBH4 [11], NaBH4 [14]. This confirms that the major isomer 13 corresponds to an addition of the hydride ortho to the chloride group in harmony with the inductive effect predominating over the mesomeric effect for chloride atom (Fig. 4).
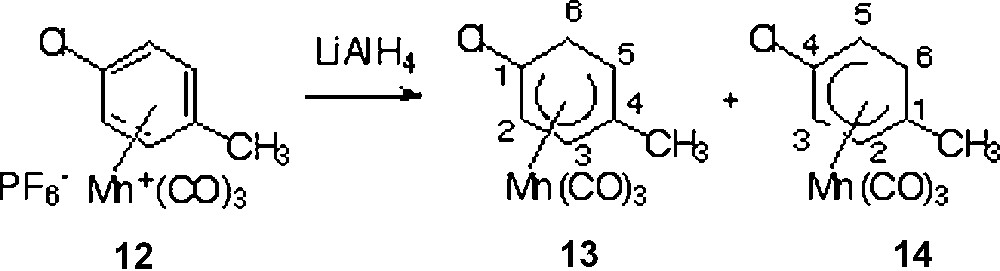
Synthesis of complexes 13 and 14.
2.1 1H NMR study
The chemical shifts of protons of para-chlorotoluene 15 are given in Table 1, entry 1, as well as those of the Mn and Cr corresponding complexes 12 and 16 (entries 2 and 5). The shielding of each proton Δδ1 = δ – δ′ (difference in chemical shift between H2,6 of complex 15 and the same protons in 12) and Δδ2 = δ – δ" (difference in chemical shift between H2,6 of complex 15 and the same protons in 16) is calculated in entries 3 and 6 and the difference of chemical shifts δH3 – δH2 is reported in column 5. These data are in a good agreement with other 1H NMR data comparing arene Cr and arene Mn complexes [27]. The chemical shifts of the cationic η6-Mn complexes resonated at higher frequencies than the corresponding neutral η6-Cr complexes. The H2 and H6 protons ortho to the chloride atom resonated in each case at the highest frequency (Table 1, entries 1, 2, 5). The difference of chemical shifts δH3 – δH2 is amplified in the case of Cr and Mn complexes (0.22 and 0.23 ppm) (entries 2 and 5) with respect to the free arene: 0.09 ppm in Me2CO d6 for example (column 5). The shielding effects due to the complexation Δδ1 and Δδ2 are smaller for the H2 and H6 protons (0.10 and 1.36 ppm) than for the H3 and H5 protons (0.18 and 1.50 ppm) (Table 1, entries 3 and 6). We did not try to predict the regioselectivity of the addition of an anion to complex 12 because it is well precedented in the literature [28] that regioselectivities of nucleophilic additions are strongly dependent upon the substituent of the ring, the reaction medium and the nature of the nucleophile. It is pointed out that “the effect of substituent on the regioselectivity seems to be very complex” and that addition of nucleophiles can even proceed with inversion of regioselectivity by changing the nature of the solvent. Nevertheless, in our case, it was not surprising to observe a major addition of the hydride to the C2 carbon ortho to the chloride atom because there is a synergic effect of the methyl and the chloro groups. Indeed the methyl moiety activates the meta position and the chloro moiety activates the ortho position. Selected 1H NMR chemical shifts of η5-cyclohexadienyl complexes 13 and 14 are given Table 2 in C6D6 and in CDCl3 showing clearly an increasing sp2 character from H5 to H2 and to H3 protons. In complex 14, an unexpected high value, difficult to explain, was observed for the difference δ3 – δ2 in the range 1.3–1.7 ppm, whereas the H3 and H2 positions seem relatively similar for complex 13 (δ3 – δ2 in the range 0.10–0.52 ppm). A characteristic 1J gem of 12–14 Hz is observed for the H6 hydrogens: the H6exo hydrogen is coupled only with the H6endo hydrogen because the first one is almost perpendicular to the cyclohexadienyl moiety whereas H6endo is usually a doublet of doublet J = 5–6 and J = 12–14 Hz.
Selected 1H NMR data of complexes 12 and 16.
| Entry | Arene | H2,6a | H3,5a | δH3 – δ H2a |
| 1 | δ 7.26 b,c,f | 7.17 b, c, f | 0.09 | |
| δ 7.26 d,f | 7.17 d, f | 0.09 | ||
| 15 | ||||
| 2 | δ′ 7.16 b, c, f | 6.94 b, c, f 6.48 d, e | 0.22 | |
| 12 | ||||
| δ′ 6.66 d,e | 0.18 | |||
| 3 | Δ δ 1 b = δ – δ' | 0.10 | 0.18 | |
| 4 | Δ δ 1 d | 0.60 | 0.69 | |
| 5 | δ" 5.90 b, c, f | 5.67 b, c, f | 0.23 | |
| δ" 5.77 d,f | 5.54 d, f | 0.23 | ||
| 16 | ||||
| 6 | Δ δ 2 b = δ – δ" | 1.36 | 1.50 | |
| 7 | Δ δ2d | 1.49 | 1.63 |
Selected 1H NMR data of complexes 13 and 14.
| complexes | H2 | H3 | δ3 – δ2 | H5 | δ3 – δ5 | Hendo | H6exo |
| 4.48a | 4.58a | 0.10a | 2.29a | 2.28a | 2.73a | 2.07a | |
| 5.00b | 5.52b | 0.52b | 3.09b | 3.43b | 3.02b | 2.62b | |
| 3.58a | 5.30a | 1.72a | 2.60a | 2.69a | 2.60a | 2.03a | |
| 4.60b | 5.91b | 1.31b | 3.07b | 2.84b | 2.36b | 2.66b |
2.2 X-ray structures of complexes 13 and 14
Fig. 5 shows two Cameron views of complex 13, the first one corresponding to the projection of the tricarbonylmanganese tripod on the cyclohexadienyl plane. The dienyl carbon atoms C1, C2, C3, C4 and C5 define a nearly perfect plane (maximum deviation 0.06 Å). The cyclohexadienyl ring is folded about C1–C5 with a dihedral angle of 33.2°. The carbon–carbon bond distances in the cyclohexadienyl ring (except the 1.369 Å value for C1–C2 bond that shows a greater double-bond character) are not significantly different from each other and can be compared with the 1.391 and 1.417 Å extreme values described in the case of tricarbonyl 6-phenyl, η5-(1,2,3,4,5-cyclohexadieny)manganese complex [29] or the 1.41 Å average value of the tricarbonyl η5-(cyclohexadienyl)manganese complex [30]. In each case, the mean carbon–carbon bond length is less than the value of 1.43 Å for the carbon–carbon distance in π-cyclopentadienyl systems, indicating that carbon–carbon bond of complexed π-dienyl system are stronger than those in the corresponding π-cyclopentadienyl case. The central atom C3 of the dienyl system is only 2.137(5) Å from manganese ion, whereas the intermediate carbon atoms C2 and C4 and the terminal atoms C1 and C5 are 2.245(7) and 2.157(5) Å and 2.197(5) and 2.183(5) Å from the metal atom, respectively. The carbon–carbon distances in the bent back portion are normal single bond lengths 1.470(8) and 1.463(8) Å. The C10–O3 bond eclipses the saturated carbon C6 and the carbon-chloride bond is anti-eclipsed with respect to the projection of the Mn(CO)3 tripod (Fig. 5).
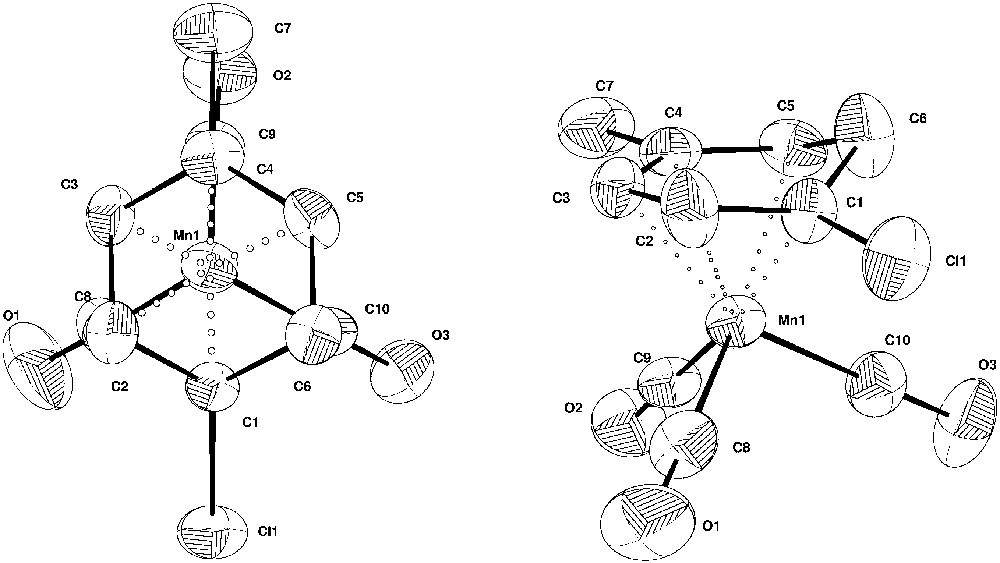
Cameron views of complex 13 (C7H8Cl)Mn(CO)3 with the thermal ellipsoids at the 50% probability level.
Fig. 6 shows two Cameron views of complex 14, the first one corresponding to the projection of the tricarbonylmanganese tripod on the cyclohexadienyl plane. The five unsaturated carbon atoms of the cyclohexadienyl ring are almost coplanar (maximum deviation 0.03 Å) while the remaining atom C6 lies in the C1 C6 C5 plane, which makes a 36.6° angle with the other plane. This value can be compared with the dihedral angle of 42.5° found by Churchill and Scholer [30] in the case of tricarbonyl-η5-(1,2,3,4,5-cyclohexadienyl)manganese complex, with the dihedral angle of 36.5(5)° found by Sweigart et al. for the tricarbonyl 6-phenyl, η5-(1,2,3,4,5-cyclohexadienyl)manganese complex [29], with the 42.2(3)° value found by Chung et al., for the tricarbonyl 1-N(CH2CH2O)3Si, 6-phenyl, η5-(1,2,3,4,5-cyclohexadienyl) manganese complex [28] and with the 37° value found recently with respect to the (exo-η5-C10H8)Mn(CO)3 complex [31]. However, it is fairly different from the dihedral angle of 29.6° in the case of the tricarbonyl 4-isobutyronitrile, η5-(3a,5,6,7,7a-indenyl) manganese complex 17 [32] and with the dihedral angle of 25.5° for complex 18 (Fig. 7) that we recently prepared [21]. C10–O3 bond eclipses the sp3 carbon C6. The carbon–carbon bond distances in the cyclohexadienyl ring are not significantly different from each other, indicating the delocalization of electrons in this portion of the ring and are also similar to those described in the literature [29, 30]. Nevertheless, the bond distances C1–C2 (1.379 Å) and C4–C5 (1.392 Å) are slightly shorter than the C2–C3 (1.402 Å) and C3–C4 (1.414 Å) bond lengths. The carbon–carbon lengths in the bent back portion are those of single bonds: 1.469(7) and 1.488(7). The metal–carbon distances in the cyclohexadienyl ring show a normal pattern in which the metal atom is closer to the C3 carbon atom and further from the substituted C1 carbon atom. Indeed, the central atom C3 is only 2.125(5) from the manganese ion, the intermediate carbon atoms C2 and C4 are 2.210(5) and 2.129(4) from the manganese and the terminal atoms C1 and C5 are 2.259(4) and 2.191(4) from the metal. The carbon–chloride bond is eclipsed by the Mn–C9–02 bond (Fig. 6).
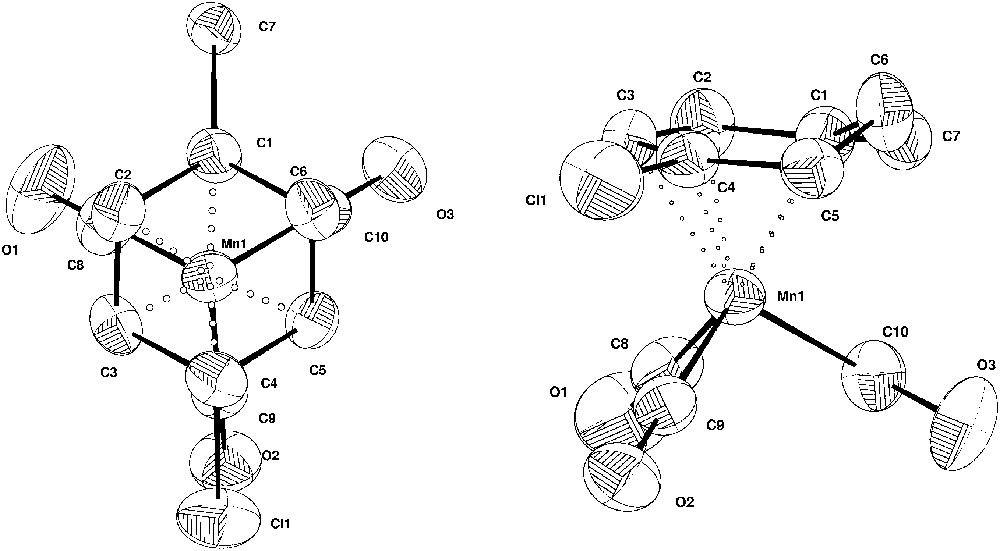
Cameron views of complex 14 (C7H8Cl)Mn(CO)3 with the thermal ellipsoids at the 50% probability level.

Two examples of η5-(cyclohexadienyl)Mn(CO)3 complexes 17 and 18, described in the literature.
3 Experimental section
3.1 Synthesis of complexes 13 and 14
(C7H8Cl)Mn(CO)3 were synthesized and fully characterized as previously described [16, 19].
Crystals of 13 and 14 were obtained by slow cooling of a pentane solution at 20 °C. A crystal (plate) of each of them of dimensions 0.1 × 0.38 × 0.56 mm and 0.16 × 0.5 × 0.7 mm, respectively, was mounted in a glass capillary under nitrogen.
3.2 Crystallography
Selected crystals were set up on an Enraf Nonius CAD4 diffractometer. Accurate unit cell dimensions and crystal orientation matrices together with their estimated standard deviations were obtained from least-squares refinements of the setting angles of 25 reflections. For both compounds, two standard reflections were monitored periodically; no intensity decay occurred during any of the data collections. Corrections were made for Lorentz and polarization effects Empirical absorption corrections were applied (CARSPECIAUX 121/P “symbol” Scan). Crystallographic data and other pertinent information are summarized in Table 3.
Crystal data of complexes 13 and 14.
| Formula | (C7 H8 Cl)Mn(CO)3 compound 13 | (C7 H8 Cl)Mn(CO)3 compound 14 |
| Chemical formula weight | 266.6 | 266.6 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/n | P21/n |
| a (Å) | 7.400(2) | 7.390(4) |
| b (Å) | 14.368(4) | 14.464(6) |
| c (Å) | 10.153(3) | 13.133(4) |
| β (°) | 98.23(2) | 98.57(4) |
| V (Å) 3 | 1068(4) | 1071(6) |
| Z | 4 | 4 |
| d calc (g cm – 3) | 1.658 | 1.653 |
| λ (Mo Kα) (Å) | 0.71069 | 0.71069 |
| μ (Mo Kα) (cm – 1) | 14.2 | 14.2 |
| Number of reflections for lattice parameters | 25 | 25 |
| Range for lattice parameters (°) | 14–15 | 14–15 |
| Temperature (K) | 295 | 295 |
| Collection method | ω 2θ | ω 2θ |
| Absorption corrected by | ψ scan | ψ scan |
| Correction (min/max) | 0.55/1 | 0.58/1 |
| Number of reflections collected | 2073 | 2069 |
| Number of uniques reflections | 1870 | 1872 |
| Number of reflections with I > 3 σ (I) | 1116 | 1352 |
| 2 θ range (°) | 3 to 50 | 3 to 50 |
| R = Σ(|Fo| – |Fc|)/Σ|Fo| | 0.0426 | 0.0436 |
| Rw = [Σ w (|Fo| – |Fc|)2/Σw |Fo|2]1/2 | 0.0505 | 0.0495 |
| S = [Σ w (|Fo| – |Fc|)2/( m – n)]1/2 | 1.13 | 1.18 |
| Number of parameters refined | 161 | 161 |
| Weighting scheme | w = w′ [1 – {Δ F /6 σ (F)}2]a,b | w = w′ [1 – {ΔF /6 σ (F)}2]a,b |
| ( Δ / σ ) | < 0.02 | 0.02 |
3.3 Structure solution and refinement
For the two compounds, the observed systematic absences are, h + l = 2 n + 1 for h0l and k = 2 n + 1 for 0k0, and the space group is then P21/n. Computations were performed by using PC version of CRYSTALS [33]. Scattering factors for all atoms were as incorporated in CRYSTALS.
Structures were solved by direct method [34]. All remaining non-hydrogen atoms were found by electron density map calculations. Their atomic coordinates were refined, and they were affected isotropic then anisotropic displacement factors.
At this stage, hydrogen atoms were located on a difference electron density map; their coordinates were refined with an overall isotropic displacement factor. Least-square refinement was carried out by minimising the function Σ w (|Fo| – |Fc|)2, where Fo and Fc are the observed and calculated structure factors. The criteria for a satisfactory complete analysis were the ratios of the rms shift to standard deviation being less than 0.02 and no significant features in the final difference map.
The molecular geometry and the atom numbering scheme of compounds 13 and 14 are shown in Figs. 5 and 6 produced by the program CAMERON [35] included in the CRYSTALS crystallographic program system.
Supplementary material
All crystallographic data have been sent in electronic format to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif files Nos. CCDC 200287 and 200288. They can be obtained by contacting the CCDC [Fax : (internat.) 44–1223/336–033 ; E-mail : deposit@ccdc.cam.ac.uk].
Acknowledgements
Financial support from CNRS and EU COST D14 Programm are greatly acknowledged.