

1 Introduction
Metal–thiolate bonds have been found in many sites of proteins where they play important biochemical functions [1–3]. For example, nature utilizes iron- and nickel-ligated thiolates to promote superoxide reductase [4] and superoxide dismutase [5,6] functions, respectively. Besides its fundamental role in concentration modulation [7], structural [8] and redox [7,9] functions, metal-bound thiolate also reveals their nucleophilic character.
Zinc-bound thiolate has been shown to promote alkyl group transfer in several zinc enzymes [10]. This reactivity is reproduced by many zinc biomimetic complexes [11,12] as well as by iron- [13], cadmium- and mercury–thiolate complexes [14], as well as by other metal–heteroatom intermediates [15]. The influence of the nature of the metal cation on the nucleophilicity of p-toluenethiolate has been studied both experimentally and by DFT computation, revealing that nickel- and zinc–thiolate complexes are more reactive than iron- and cobalt–thiolate complexes [16]. In the case of zinc complexes, both the composition of the ligand set [17–20], in terms of donating capability and steric hindrance, and the presence of hydrogen bonding toward the reactive thiolate [21–24] have been shown to modulate the reactivity against electrophiles.
Prediction of the nucleophilicity remains a challenging task [25]. Attempts to theoretically define a general nucleophilicity index [26,27], based for example on atomic charges, energy of the highest occupied molecular orbital, force constants or, in the context of conceptual density functional theory [28], chemical hardness and Fukui function, have been shown to be conclusive only for a limited variety of compounds. In the case of zinc-bound thiolates, several studies provide the rate constants of their reaction against electrophile but it is difficult from these data to have a general view of their relative nucleophilicities as the experimental conditions are not the same [19,21,23,24,29]. Such a determination would however be of great significance in order to evaluate the relative reactivity of enzymes and to determine which thiolate is reactive in zinc active sites including more than one cysteinate.
In a previous study [30], we have shown that the computed Gibbs free energy of the SN2 activation barrier can explain the relative reactivity of zinc–thiolate complexes against methyl iodide. In this study, we have determined the SN2 activation barrier for a variety of zinc complexes. In order to conserve as much as possible along the reaction path the tetrahedral arrangement around zinc observed in biomimetic complexes and enzymatic active sites and to avoid unexpected structural deformation [31], these complexes include a relatively rigid tripodal core. This will allow us to compare the donating capability of tripodal ligands and to determine a good correlation between the zinc-bound thiolate nucleophilicity and the thiolate gas phase basicity.
2 Computational methods
Calculations were performed with Gaussian 03 [32]. Geometry optimisations were conducted using the B3LYP method at the 6-31G(d,p) level for the B, N, C, O, S, H atoms. The contracted Wachters basis [14s9p5d1f/9s5p3d1f] was used to describe the zinc atom [33]. The CRENBL relativistic effective core potential and associated valence basis set were employed to model the iodine atom [34]. This basis set is referred to as BS1. Each stationary point has been characterized with frequency analysis and shows the correct number of negative eigenvalues (0 for a local minimum and one for a transition state).
Energies were calculated for the stationary points at the B3LYP level using an extended basis set labelled BS2. It consists in the 6-311+G(2d,2p) for B, N, C, O, S, H, the extended Wachters basis [15s11p6d2f/10s7p4d2f] for Zn and the Aug-cc-pVTZ-PP basis set and pseudo-potential for I [35]. We have demonstrated previously that this level of calculation gives reliable geometries and relative energies on zinc complexes [30,36].
The relative gas phase Gibbs free energy was deduced from the equation:
| ΔGgas = ΔEelec + ΔZPE + ΔET − TΔS |
All calculations have been made in the gas phase, thus permitting to know the intrinsic properties and reactivities of the complexes. Furthermore, we have shown previously [30] that inclusion of solvation effect does not modify the relative reactivity of zinc–thiolate complexes. The gas phase nucleophilicity trends can thus be extrapolated to solvated biomimetic complexes or enzymatic active sites.
3 Results and discussion
A series of 16 zinc biomimetic complexes depicted in Scheme 1 has been examined in this work.
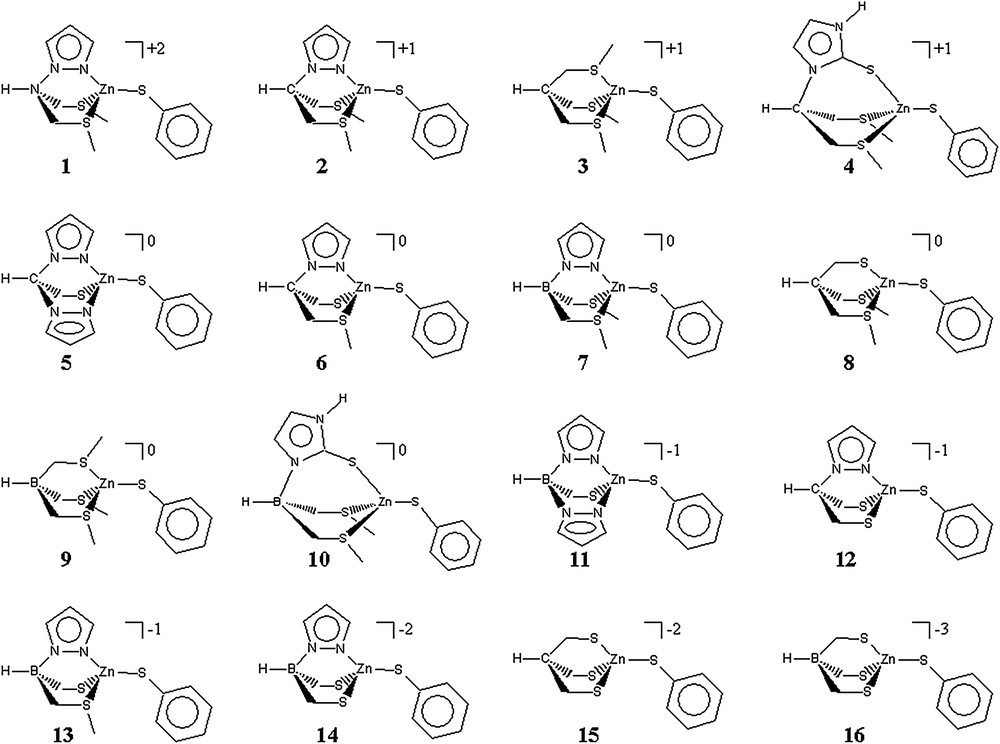
Thiophenolate zinc complexes studied herein.
They are derived from experimental complexes bearing a tripodal ligand and one monodentate phenylthiolate ligand. The tripod ligands possess a mixture of pyrazole-type nitrogen, and of thioimidazole-, thioether- or alkylthiolate-type sulfur as the donors for zinc. This allows to obtain N2S2, NS3 or S4 donor sets which model respectively the His2Cys2, HisCys3 and Cys4 coordination observed in many active sites of zinc enzymes [37–39]. Furthermore, the tripodal core is either a borate, a methylene or an ammonium moiety, thus permitting to modulate the global charge of the complexes. Consequently, our series of zinc–thiolate complexes goes from the dicationic [ZnNS3]+2 1 to the trianionic [ZnS4]−3 16 species.
Optimisation of the geometry of these complexes led to a tetrahedral coordination around zinc in all cases except 16 for which the phenylthiolate ligand dissociates from the metal, leading to a tricoordinated zinc complex. The high electronic density around zinc in 16 explains this incapability to keep four Zn–S bonds. Compared to 15 for which a ZnS4 core could be obtained, this also means that the negative charge of the borate group in 16 influences the zinc atom even in the absence of conjugated arms. We can thus hypothesize that the presence of a negatively charged group in the neighbourhood of a [Zn(Cys)4]−2 enzyme active site facilitates the sulfur–zinc bond dissociation. Reciprocally, the shorter Zn–thiolate bond in 1 (2.182 Å) compared to 2 (2.204 Å) shows that a cationic site close to the coordination sphere tightens the Zn–S bonds.
3.1 Reactivity of zinc-bound phenylthiolate
For complexes 1–15, we have determined the Gibbs free energy barrier of their SN2 reaction with methyl iodide (Scheme 2 and Fig. 1).
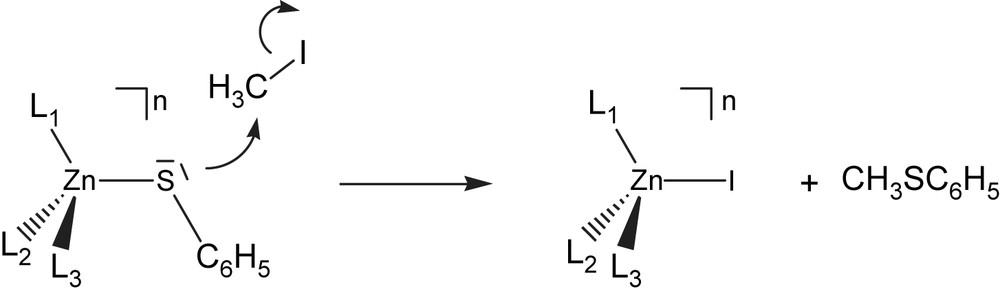
SN2 alkylation of zinc-bound phenylthiolate with MeI.
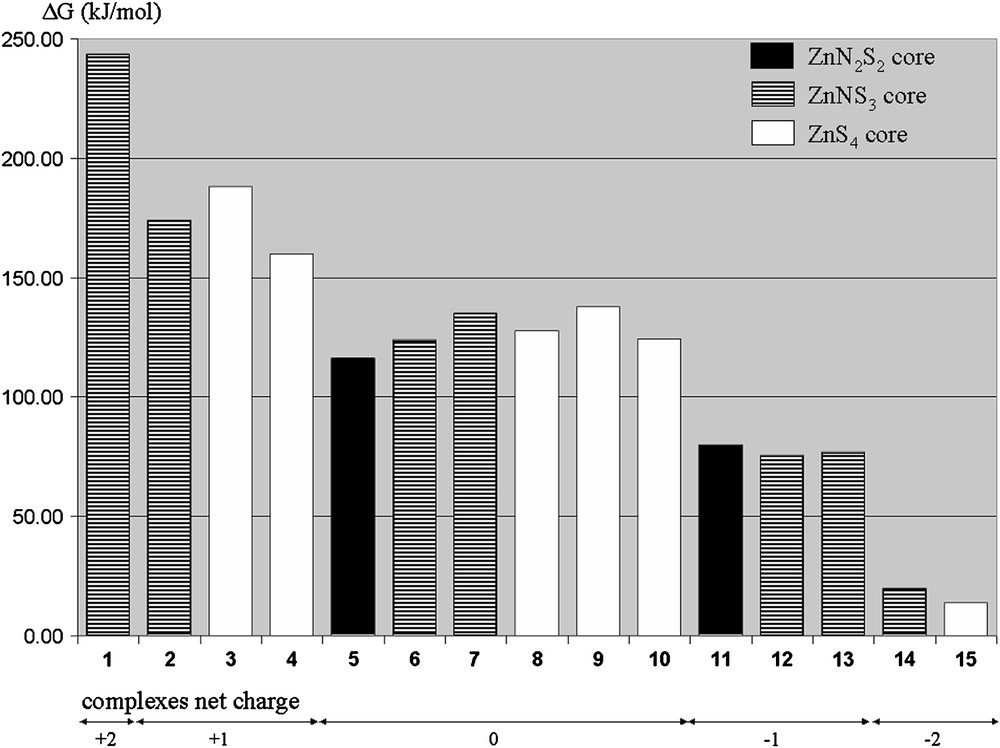
Gibbs free energy barrier (ΔG) for the reaction between MeI and complexes 1–15, calculated at the B3LYP/BS2//B3LYP/BS1 level (in kJ/mol).
The least and the more reactive complexes are 1 and 15 with a barrier of 243 and 15 kJ/mol, respectively. The big difference between these extreme values illustrates the importance of the coordination sphere around zinc on the intrinsic nucleophilicity of the thiolate. Fig. 1 shows however that the nature of only the atom donor set for zinc does not determine the nucleophilicity of the complex. Indeed, a ZnN2S2 core may be either more or less reactive than ZnN3S or ZnS4 cores, and the same holds for the same comparison between ZnN3S and ZnS4 cores. This shows that the conclusion obtained in previous studies [17,19] that ZnS4 complexes are more reactive than ZnNS3 and ZnN2S2 complexes cannot be generalized to a broader series. It should be noticed that a counter-example of the previous rule has already been mentioned [19].
Fig. 1 shows that the nucleophilicity depends on the complex net charge. Indeed dianionic, monoanionic, neutral, monocationic and dicationic complexes have their Gibbs free energy barrier in the range 0–50, 50–100, 100–150, 150–200 and 200–250 kJ/mol, respectively. This is in agreement with the decreased reactivity order measured from [ZnS4]−2 to [ZnNS3]−1 and [ZnN2S2]0 complexes [17,29].
As for the complete set of complexes, the relative reactivity between complexes bearing the same charge does not depend on the nature of the atom donors for zinc. This is illustrated for example by the fact that 2, a [ZnNS3]+1 complex, is respectively more and less reactive than 3 and 4, which are both [ZnS4]+1 complexes. On the contrary, the nucleophilicity of the zinc-bound phenylthiolate is influenced by the nature of the tripod arms. Substituting a pyrazolyl arm by a thioether arm, as from 2 to 3 or from 5 to 6, reduces indeed the reactivity of the phenylthiolate. By comparing the activation barrier for all complexes, it is thus possible to determine a scale of the ligands which induces the larger nucleophilicity (Fig. 2).
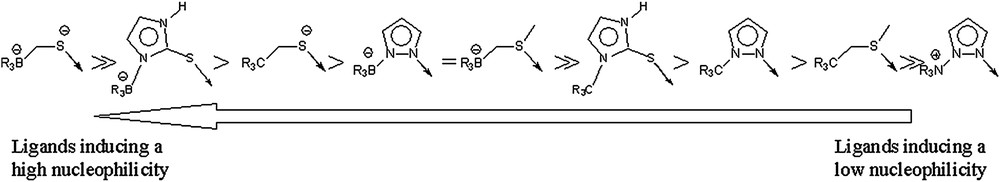
Scale of ligands influencing the nucleophilicity of zinc-bound thiolate.
This scale is in agreement with experimental results indicating that substituting a pyrazolyl nitrogen ligand by a thioimidazole sulfur ligand increases the reactivity [19]. It also shows that thioimidazole is intermediate between a thioether and a thiolate group in terms of their capability of thiolate enrichment [40,41]. On the contrary we may notice that a thioimidazole borate is a more donating ligand than an alkylthiolate.
3.2 Reactivity of zinc-bound alkylthiolate
Some of the tripodal ligands possess an alkylthiolate arm which can be alkylated, as already observed experimentally [23]. For complexes 5, 6, 11, 12 and 13, we have calculated the transition state corresponding to the SN2 reaction between the zinc-bound alkylthiolate and methyl iodide. The values of the Gibbs free energy barrier for this reaction are given in Table 1 in parallel to the values obtained for the same reaction on the phenylthiolate.
Gibbs free energy barrier (ΔG) for the reaction between MeI and zinc-bound alkyl- or arylthiolate, calculated at the B3LYP/BS2//B3LYP/BS1 level (in kJ/mol).
| Complex | ΔG for the arylthiolate | ΔG for the alkylthiolate |
| 5 | 116 | 124 |
| 6 | 124 | 135 |
| 6′ | 137 | 141 |
| 11 | 80 | 52 |
| 12 | 76 | 64 |
| 13 | 77 | 46 |
| 13′ | 92 | 53 |
These results indicate that the alkylthiolate nucleophilicity in a given complex stands in the same range as the one observed for the phenylthiolate in the same complex. Furthermore, the alkylthiolate reactivity may be either higher (in 11–13) or smaller (in 5 and 6) than the reactivity of the phenylthiolate. The order of reactivity of these zinc-bound thiolates is thus not determined by their substituents but by the nature of the ligand set around zinc. In our series, it seems that the presence of a second negative charge in the tripod ligand, as in 11–13, induces a higher reactivity of the alkylthiolate arm.
3.3 Influence of hydrogen bonding
We and others have shown in previous studies [21–24,30] that substituting the phenylthiolate by the o-NHC(O)H–phenylthiolate, inducing an H bond with the sulfur atom of the arylthiolate, reduces the reactivity of the corresponding zinc-bound arylthiolate. The same aryl substitution on complexes 6 and 13 leads to two new complexes, noted 6′ and 13′, respectively, for which it is possible to evaluate the influence of the H bond toward the arylthiolate on the nucleophilicity of both the aryl- and the alkylthiolate.
As expected (Table 1), moving from 6 to 6′ or from 13 to 13′ induces an increase by 13–15 kJ/mol of the Gibbs free energy barrier for the reaction on the arylthiolate. More surprisingly, this also leads to a decrease of reactivity of the alkylthiolate as the Gibbs free energy barrier increases by 6–7 kJ/mol. The optimized geometry of complexes 13 and 13′ (or 6 and 6′) (Fig. 3) gives an explanation of this trend.

Optimized structures of 13 and 13′ at the B3LYP/BS1 level. Bond length in Å. Yellow: sulfur, blue: nitrogen, gray: carbon, white: hydrogen, red: oxygen, pink: boron, light blue: zinc.
The H bond reduces the Lewis basicity of the o-NHC(O)H–phenylthiolate compared to the bare phenylthiolate. Consequently, the Zn–S(aryl) bond is slightly longer in 13′ than in 13. Due to the d10 electronic structure of zinc(II), we observe a “push–pull” effect between the ligand set around the metal [31]. Indeed, the lengthening of the Zn–S(aryl) bond induces a shortening of all other Zn-ligand bonds. Thus, the alkylthiolate of 13′ is slightly more tightly coordinated to zinc than the same alkylthiolate of 13. In other words, the charge transfer between the alkylthiolate and Zn2+ is larger in 13′ than in 13, thus explaining the lower nucleophilicity of the alkylthiolate of 13′ compared to the same thiolate in 13.
These results indicate that hydrogen bond has not only a local effect on the reactivity of the thiolate group toward which it is directed, but also a global effect on the complex reactivity.
3.4 Nucleophilicity index
Having in hand the calculated reactivity of a large panel of zinc-bound thiolates, is it possible to find a zinc-bound thiolate nucleophilicity scale based on the structure of the complex? It has been postulated that the Zn–S bond lengths [19] or the HOMO energy of the complex [18] could correlate with the complex reactivity.
Fig. 4A shows the relationship obtained between the energy barrier and the Zn–S(phenylthiolate) bond length for complexes 1–15. As expected, an increase of the metal–sulfur bond length corresponds approximately to an increase of the reactivity of the phenylthiolate. The linear correlation remains however modest. Extension of this relationship to all zinc–thiolate sites, thus including alkylthiolate and o-NHC(O)H–phenylthiolate (Fig. 4B) shows a clear disruption of the previous trend, as observed by others [20]. This means that the reactivity of various zinc-bound thiolates cannot be deduced from the zinc–thiolate bond length.
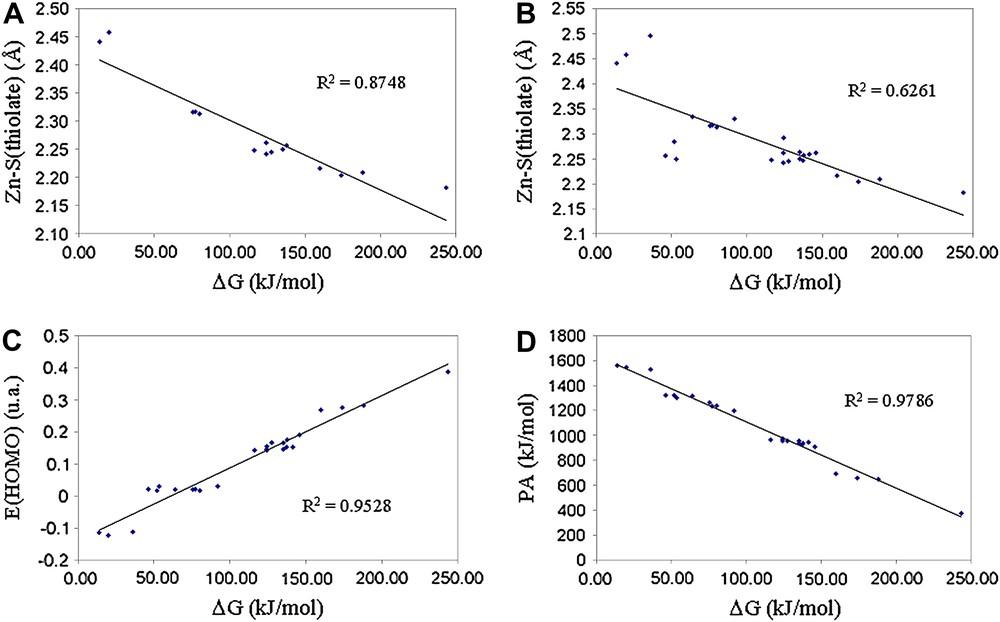
Plot of calculated Gibbs free energy barrier (ΔG) versus (A) Zn–S bond length for phenylthiolate, (B) Zn–S bond length for all thiolates, (C) energy of the HOMO for all complexes and (D) proton affinity for all thiolates.
On the contrary, the energy of the HOMO of all the complexes studied correlates reasonably with the calculated energy barrier (Fig. 4C) and may thus be used to compare the reactivity of different zinc–thiolate complexes, as already observed for other Zn–S complexes [18,20]. This however does not allow to determine the most reactive site of a zinc complex if several thiolate ligands are bound to the metal.
In order to be able to predict the most reactive thiolate of a complex, we also examined the proton affinity of the zinc-bound sulfur atoms. It is well known that basicity and nucleophilicity are related [18,42]. However, due to the differences between these concepts, they are often not directly proportional [43]. Fig. 4D shows a good correlation between the proton affinity and the activation barrier of all the zinc–thiolate moieties studied here. This shows that the zinc-bound thiolate basicity can be used as a pertinent nucleophilicity index in order to predict the relative reactivity of these coordinated thiolates.
4 Conclusion
Based on the calculated reactivity of 15 zinc–thiolate complexes, we have shown that the dominant factor inducing the nucleophilicity is the charge of the ligand set and the nature of the chemical groups bound to zinc. Thus the N/S ratio of atom donor set is not a sufficient parameter to determine the reactivity if the N and S ligands are not exclusively histidine and cysteinate side chains, respectively. The substituent of the thiolate does not give a clear indication of its reactivity, whereas the presence of H bond reduces the nucleophilicity of all reactive sites of a complex. The good correlation between basicity and nucleophilicity of a large series of zinc-bound thiolates gives confidence to the pertinence of the proton affinity as a nucleophilicity index. Work is in progress to use this index in order to predict the relative reactivity of enzymatic zinc active sites and to determine which cysteinate would be the most reactive in these sites.
Acknowledgment
We are grateful to the Centre National de la Recherche Scientifique (CNRS) and Ecole Polytechnique for financial support. We also thank the Institut du Développement et des Ressources en Informatique Scientifique (IDRIS, grant number 0543) and the Centre Informatique National de l'Enseignement Supérieur (CINES, grant number dcm2335) for computational resources.