

1 Introduction
3-Phenylpropylamine (3-PPA) has been extensively used in many different ways for scientific researches. It has been used to synthesize two-dimensional lithium beryllofluoro-layered compounds and vanadium oxide nanotubes [1–3]. Furthermore, it has been intercalated in the PbI2 films by exposing the thin films to amine vapours in a sealed container [4]. It is also a structural element of many pharmaceutical drugs such as tolterodine and 3-phenoxy-3-phenylpropanamine [5,6]. Complexes of this molecule have been used as an internal standard for various types of studies [7,8].
The GIAO/DFT (gauge including atomic orbitals/density functional theory) approach is extensively used for the calculations of chemical shifts for different types of compounds [9–18]. DFT calculations are hardly expensive and they enable accurate calculations on systems such as large organic molecules [17]. At present with this methodology, a variety of spin–spin coupling constants can be calculated with good accuracy in polyatomic systems [19,20]. NMR spectroscopy has an enormous potential for investigating conformations and configurations in organic compounds. After the discovery of NMR, such potential rapidly increased to cover various kinds of compounds, including biological, inorganic and organometallic compounds [21]. 1D and 2D hetero and homonuclear NMR methods enable one to get full assignment and structural information of organic compounds [22–24].
In this study, we report 1H, 13C, DEPT, COSY, NOESY and HETCOR NMR spectra, the magnitude of 1–3J(C, H) coupling constants and solvent effects on chemical shifts and coupling constants of 3-PPA. 1H, 13C NMR chemical shifts, 1–3J(C, H) coupling constants of 3-PPA have also been calculated by using the Hartree–Fock (HF), Becke–Lee–Yang–Parr (BLYP) and Becke-3–Lee–Yang–Parr (B3LYP) density functional methods with 6-311++G(d,p) basis set. Furthermore, bond lengths, bond and torsion angles of the title molecule have been determined with HF, BLYP and B3LYP at 6-311++G(d,p) level.
2 Computational details
The molecular structure of 3-PPA (in vacuum) in the ground state was optimized by HF, BLYP and B3LYP with the 6-311++G(d, p) basis set. For the NMR calculations, the molecular structure of 3-PPA was first fully optimized at HF/6-311++G(d,p), BLYP/6-311++G(d,p), B3LYP/6-311++G(d,p) levels in CDCl3 (ɛ = 4.90), CD2Cl2 (ɛ = 8.93), DMSO-d6 (ɛ = 46.70), MeOD (ɛ = 32.63) and CD3COCD3 (ɛ = 20.70) by using the IEFPCM (integral equation formalism polarizable continuum model) method [25,26]. By using the same methods and the basis set, it was seen that all the vibrational frequencies of 3-PPA were real. After optimization, 1H, 13C NMR chemical shifts (δH and δC) and 1–3J(C, H) coupling constants were calculated for all five solvents using the GIAO method [27] at the HF/6-311++G(d,p), BLYP/6-311++G(d,p) and B3LYP/6-311++G(d,p) level. Relative chemical shifts were then estimated by using the corresponding TMS shielding, calculated in advance at the same theoretical level as the reference. Although the absolute shielding calculations were performed for all nuclei, it was focused on 1H and 13C shifts since the experimental values referred to these nuclei. The molecular geometry was restricted and all the calculations were performed by using Gaussian 03 program package and GaussView program was used for molecular visualization [28,29].
3 NMR spectroscopy
NMR spectra were recorded in CDCl3, CD2Cl2, MeOD, DMSO-d6 and CD3COCD3 (0.3 mg ml−1) at room temperature on a Bruker AVANCE 500 (11.7 Tesla, 500.13 MHz for 1H, 125.76 MHz for 13C) using a 5 mm BBO probe. 1H and 13C chemical shifts (δ in ppm) were measured with an accuracy of 0.01 ppm and 0.02 ppm, respectively, and referred to TMS (1H, 13C, 0 ppm). In order to obtain the 1H NMR spectrum, the pulse sequence used a delay (D1) and acquisition time (AQ) of 1.0 s and 5.9 s, a spectral width of 5466.5 Hz, 64 K data points, 90° pulse (14.15 μs) and 16 scans. For 13C NMR spectrum, the pulse sequence used a delay (D1) and acquisition time (AQ) of 2.0 s and 1.6 s and a spectral width of 20161.3 Hz, 64 K data points, 90° pulse (8.3 μs) and 32 scans. DEPT spectra were obtained at θz = 45° where CH, CH2 and CH3 appear in the positive phase, θz = 90° where only CH appears in the positive phase and θz = 135° where CH, CH3 appear in the positive phase and CH2 appears in the negative phase. Two-dimensional COSY, NOESY and HETCOR techniques were measured using standard micro-programs provided by Bruker.
4 Results and discussion
1H and 13C NMR data of 3-PPA are reported in Table 1. As in Fig. 1 3-PPA molecule shows seven different carbon atoms, which is in agreement with the structure regarding the molecular symmetry. Due to that fact, in Fig. 2, seven carbon peaks are observed in 13C NMR spectrum of 3-PPA. The peak which appears around 143 ppm in the 13C NMR spectrum cannot be observed in DEPT 135, DEPT 90 and DEPT 45 spectra (Fig. 3). Therefore, it can be concluded that the peak belongs to the phenyl ring (C5) and does not contain any hydrogen atom.
Experimental and calculated 1H and 13C NMR chemical shifts (ppm) of 3-PPA.
| Nucleus | Exp. [38,39]a | Exp. | HF | BLYP | B3LYP | Exp. | HF | BLYP | B3LYP | Exp. | HF | BLYP | B3LYP |
| CDCl3 | DMSO-d6 | CD3COCD3 | |||||||||||
| C-5 | 142.08 | 143.18 | 153.40 | 152.67 | 153.53 | 143.82 | 154.01 | 153.38 | 154.38 | 143.57 | 153.92 | 153.36 | 154.22 |
| C-4, C-6 | 128.28 | 129.38 | 135.80 | 134.40 | 135.92 | 129.73 | 136.17 | 134.85 | 136.37 | 129.47 | 136.11 | 134.84 | 136.28 |
| C-1, C-3 | 128.25 | 129.34 | 137.10 | 133.78 | 135.55 | 129.65 | 137.41 | 134.18 | 136.01 | 129.26 | 137.36 | 134.19 | 135.93 |
| C-2 | 125.67 | 126.75 | 132.44 | 130.48 | 132.11 | 126.97 | 132.70 | 130.82 | 132.42 | 126.61 | 132.65 | 130.82 | 132.35 |
| C-9 | 41.78 | 42.80 | 42.23 | 52.55 | 49.69 | 42.73 | 42.30 | 52.67 | 49.72 | 51.30 | 42.29 | 52.69 | 49.68 |
| C-8 | 33.24b | 36.47 | 39.00 | 46.59 | 44.64 | 36.80 | 39.34 | 46.89 | 45.20 | 33.58 | 39.34 | 46.96 | 45.07 |
| C-7 | 35.43b | 34.27 | 34.50 | 42.13 | 40.31 | 34.12 | 34.42 | 42.04 | 40.42 | 30.17 | 34.44 | 42.11 | 40.22 |
| Phenyl, 5H | 7.34–7.11 | 7.26–7.10 | 7.85–7.82 | 7.59–7.50 | 7.73–7.64 | 7.26–7.11 | 7.97–7.94 | 7.72–7.61 | 7.85–7.74 | 7.28–7.11 | 7.94–7.92 | 7.70–7.50 | 7.83–7.72 |
| H20, H21 | 2.72 | 2.66 | 2.53 | 3.05 | 2.94 | 2.57 | 2.55 | 3.06 | 2.94 | 3.17 | 2.54 | 3.05 | 2.94 |
| H16, H17 | 2.65 | 2.60 | 2.42 | 2.57 | 2.56 | 2.53 | 2.45 | 2.61 | 2.59 | 2.66 | 2.44 | 2.60 | 2.59 |
| H18, H19 | 1.78 | 1.71 | 1.48 | 1.68 | 1.64 | 1.61 | 1.46 | 1.66 | 1.61 | 1.61 | 1.45 | 1.66 | 1.62 |
| H22, H23 | 1.19 | 1.06 | 0.70 | 1.16 | 1.05 | 1.43 | 0.93 | 1.41 | 1.29 | 3.34 | 0.89 | 1.37 | 1.24 |
| Nucleus | Exp. [38,39]a | Exp. | HF | BLYP | B3LYP | Exp. | HF | BLYP | B3LYP | ||||
| MeOD | CD2Cl2 | ||||||||||||
| C-5 | 142.08 | 142.24 | 154.01 | 153.44 | 154.25 | 142.81 | 153.69 | 153.03 | 153.90 | ||||
| C-4, C-6 | 128.28 | 128.31 | 136.13 | 134.88 | 136.36 | 128.66 | 135.96 | 134.67 | 136.11 | ||||
| C-1, C-3 | 128.25 | 128.29 | 137.47 | 134.22 | 135.96 | 128.55 | 137.25 | 134.00 | 135.76 | ||||
| C-2 | 125.67 | 125.72 | 132.73 | 130.84 | 132.42 | 125.91 | 132.56 | 130.69 | 132.26 | ||||
| C-9 | 41.78 | 41.07 | 42.34 | 52.65 | 49.72 | 42.07 | 42.26 | 52.65 | 49.72 | ||||
| C-8 | 33.24b | 34.70 | 39.35 | 47.05 | 45.08 | 35.97 | 39.19 | 46.78 | 44.84 | ||||
| C-7 | 35.43b | 31.16 | 34.51 | 42.07 | 40.23 | 33.50 | 34.47 | 42.15 | 40.26 | ||||
| Phenyl, 5H | 7.34–7.11 | 7.24–7.08 | 7.96–7.94 | 7.71–7.60 | 7.84–7.74 | 7.26–7.10 | 7.89–7.87 | 7.65–7.55 | 7.78–7.68 | ||||
| H20, H21 | 2.72 | 2.60 | 2.54 | 3.06 | 2.94 | 2.60 | 2.53 | 3.05 | 2.94 | ||||
| H16, H17 | 2.65 | 2.60 | 2.45 | 2.61 | 2.59 | 2.60 | 2.42 | 2.59 | 2.57 | ||||
| H18, H19 | 1.78 | 1.70 | 1.46 | 1.65 | 1.62 | 1.70 | 1.47 | 1.67 | 1.63 | ||||
| H22, H23 | 1.19 | - | 0.92 | 1.40 | 1.27 | 1.10 | 0.80 | 1.28 | 1.17 |

The optimized molecular structure of 3-PPA for methods used.

13C NMR spectra of 3-PPA, (a) CDCl3, (b) CD2Cl2, (c) DMSO-d6, (d) MeOD, (e) CD3COCD3.
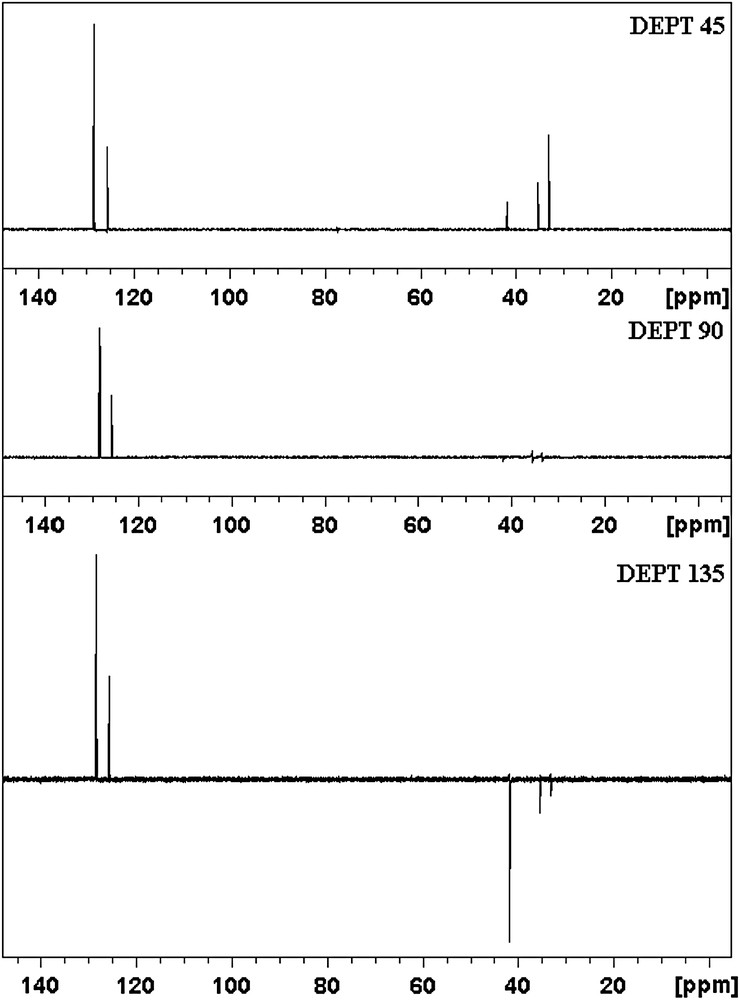
DEPT NMR spectra of 3-PPA.
1H NMR spectra of the title compounds are given in Fig. 4. Noticeable chemical shift differences were observed (Table 1) in varying the solvents. This difference is evident when we employed CDCl3 which is poor in forming hydrogen bonds with electronegative atoms when compared to DMSO-d6 and CD3COCD3 [30]. In this study, CD3COCD3 has the largest effect on proton chemical shifts due to strong hydrogen bonding. Unlike CDCl3, CD2Cl2, MeOD and DMSO-d6, CD3COCD3 also interacts with protons of C9. This interaction causes the up field frequency region shift of the corresponding H20,21 protons and C9 carbon of the 3-PPA. Contacts of type C–H⋯M where M is an electronegative atom are known to led, for some cases, an increase in the related 1J(C,H) coupling constants [31–36]. However, the magnitude of one bond coupling constants is almost the same for all solvents used in this study (Table 2). While working with MeOD, the peak coming from amine protons vanishes due to possible exchange between deuterium of MeOD and protons of NH2. This solvent dependence of the chemical shifts is being reported for the first time in 3-PPA.

1H NMR spectra of 3-PPA, (a) CDCl3, (b) CD2Cl2, (c) DMSO-d6, (d) MeOD, (e) CD3COCD3.
Experimental and calculated 1–3J(C, H) coupling constants (Hz) of 3-PPA.
| Nucleus | Exp. | BLYP | B3LYP | Exp. | BLYP | B3LYP | Exp. | BLYP | B3LYP | Exp. | BLYP | B3LYP | Exp. | BLYP | B3LYP |
| CDCl3 | DMSO-d6 | CD3COCD3 | MeOD | CD2Cl2 | |||||||||||
| 1J(C7-H16,17) | 125.78 | 121.15 | 120.06 | 125.85 | 121.49 | 120.41 | 127.51 | 121.43 | 120.34 | 125.86 | 121.49 | 120.39 | 126.47 | 121.29 | 120.28 |
| 1J(C8-H18,19) | 126.32 | 121.33 | 122.06 | 126.61 | 123.01 | 121.83 | 126.17 | 123.08 | 121.86 | 126.88 | 123.05 | 121.82 | 126.90 | 123.17 | 121.95 |
| 1J(C9-H20,21) | 133.49 | 125.89 | 124.59 | 132.11 | 126.27 | 124.99 | 132.07 | 126.18 | 124.88 | 134.57 | 126.22 | 124.88 | 133.58 | 126.05 | 124.56 |
| 1J(C1,3-H11,13) | 158.98 | 151.02 | 149.35 | 158.95 | 151.62 | 149.96 | 158.67 | 151.51 | 149.86 | 158.83 | 151.59 | 149.94 | 159.96 | 151.28 | 149.66 |
| 1J(C4,6-H14,15) | 156.20 | 149.12 | 147.54 | 157.34 | 149.90 | 148.29 | 156.38 | 149.76 | 148.17 | 156.14 | 149.89 | 148.34 | 155.75 | 149.48 | 148.88 |
| 1J(C2-H12) | 160.58 | 152.16 | 151.48 | 160.60 | 152.76 | 151.10 | 160.46 | 152.65 | 150.97 | 159.95 | 152.73 | 151.04 | 160.44 | 152.43 | 151.75 |
| 2J(C8-H16,17) | 4.26 | 3.19 | 4.03 | 3.91 | 3.24 | 4.10 | 4.25 | 3.23 | 4.06 | 3.42 | 3.24 | 4.05 | 3.92 | 3.22 | 4.10 |
| 2J(C8-H20,21) | 1.64 | 1.74 | 2.65 | 1.91 | 1.73 | 2.65 | - | 1.73 | 2.67 | 1.98 | 1.71 | 2.66 | 2.35 | 1.74 | 2.60 |
| 2J(C9-H18,19-H22,23) | 4.09 | 2.62 | 3.35 | 3.94 | 2.70 | 3.44 | 4.29 | 2.68 | 4.13 | 4.54 | 2.69 | 3.45 | 4.14 | 2.65 | 3.39 |
| 2J(C6,4-H11,13) | 1.67 | 2.94 | 1.62 | 1.28 | 3.05 | 1.71 | 2.19 | 3.03 | 1.71 | 1.90 | 3.04 | 1.72 | 2.58 | 2.99 | 1.67 |
| 3J(C7-H14,15) | 3.92 | 4.25 | 4.28 | 4.02 | 4.23 | 4.30 | 4.26 | 4.23 | 4.28 | 3.92 | 4.23 | 4.27 | 3.96 | 4.24 | 4.28 |
| 3J(C1,3-H13,11) | 7.27 | 6.86 | 7.02 | 7.12 | 6.85 | 7.01 | 8.43 | 6.85 | 7.01 | 8.36 | 6.85 | 7.01 | 8.15 | 6.86 | 7.01 |
| 3J(C4,6-H12) | 6.19 | 6.76 | 6.92 | 7.12 | 6.75 | 6.92 | 6.05 | 6.75 | 6.91 | 6.34 | 6.75 | 6.92 | 6.75 | 6.51 | 6.92 |
| 3J(C2-H14,15) | 7.37 | 6.48 | 6.70 | 7.38 | 6.48 | 6.67 | 7.66 | 6.48 | 6.81 | 7.34 | 6.48 | 6.68 | 7.29 | 6.48 | 6.71 |
All the coupling constants were derived from a proton coupled 13C NMR spectrum of the title molecule and the results are presented in Table 2. For nuclear shieldings, the Hartree–Fock approximation is a useful one, applicable to most systems of interest in NMR experiments, providing results of reasonable quality [18]. However, in this study, the results of the Hartree–Fock method for spin–spin coupling constants showed large deviations from experimental observations. Therefore, only BLYP and B3LYP results are presented in Table 2. Due to the electronegative nitrogen atom, N10, 1J(C9, H20–21) is larger than 1J(C8, H18–19) and 1J(C7, H16–17) in the propylamine chain.
From 2D COSY NMR spectrum (Fig. 5), all the connections between the hydrogen atoms on propylamine chain and the phenyl ring are clearly observed. The two-dimensional nuclear overhauser enhancement NMR (NOESY) experiment is capable of revealing spatial relationships among proximal protons [37]. In Fig. 5, the NOESY cross-peak for the title molecule indicates that the spatial distance between H14,15 and H16,17 protons is less than 5 Å. The calculated spatial distances for H15–H16 and H14–H17 have been found 2.523 Å and 2.435 Å, respectively. The correlations between C1,3–H11,13 (or C4,6–H14,15), C2–H12, C9–H20,21, C8–H18,19 and C7–H16,17 are clearly observed in the HETCOR spectrum (Fig. 5). It is also clear that there is no H atom bonded to C5 which supports the DEPT results, as expected. Some NMR properties of 3-PPA are reported in Refs. [38,39]. Unlike the study reported in Ref. [38], in the present study we have found that the C7 carbon atom appears at the lowest field frequency region instead of C8. This fact was also supported by HETCOR results.
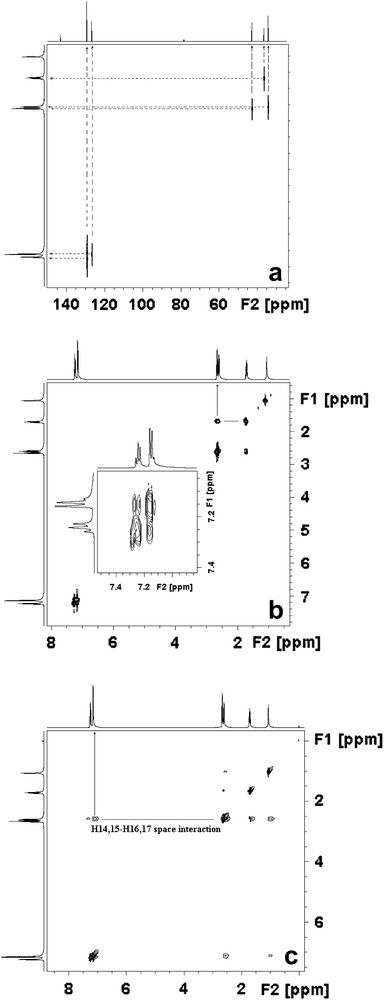
HETCOR (a), COSY (b) and NOESY (c) NMR spectra of 3-PPA (in CDCl3).
5 Geometrical structures
The optimized geometric parameters (bond lengths, bond angles and dihedral angles) by ab initio Hartree–Fock and density functional theory are listed in Table 3 in accordance with the atom numbering scheme presented in Fig. 1. The molecular structure of 3-phenylpropylamine, phenyl radical and 1-propylamine has not been reported by any diffraction technique. In order to make a comparison, experimental bond lengths for benzene and gauche ethylamine are r(C–H) = 1.084 Å, r(C–C) = 1.397 Å [40] and r(C–H) = 1.107 Å, r(C–C) = 1.524 Å, r(C–N) = 1.475 Å, r(N–H) = 1.052 Å, respectively [41]. 3-PPA shows C1 symmetry where the propylamine plane, which has trans-gauche (TG) orientation [42] is almost perpendicular to the phenyl plane. This result can be seen from dihedral angle D(8,7,5,4) of 3-PPA given in Table 3. We also observed that bond angles of phenyl A(4,5,6) and A(3,4,5) were shortened following conjugation with n-propylamine (Table 3). Huang et al. [43] and Nicolaides et al. [44] reported geometric parameters of phenyl radical (neutral, anion, cation). Bond distances and bond angles of phenyl have been given in the literature by Radziszewski et al. [45]. Structural parameters of n-propylamine have been investigated by Durig et al. [42]. Our data is consistent with those previously reported data and experimental data of benzene [40] and ethylamine [41].
The optimized geometry parameters of 3-PPA (in vacuum) calculated by the HF, BLYP and B3LYP methods using Gaussian 6-311++G(d,p) basis set.
| Coordinatea | Calculated values (HF) | Calculated values (BLYP) | Calculated values (B3LYP) | Coordinatea | Calculated values (HF) | Calculated values (BLYP) | Calculated values (B3LYP) | ||||||||||||
| 3PPA | Ph [40] | PA [39] | 3PPA | Ph [40] | PA | 3PPA | Ph [42] | PA | 3PPA | Ph [40] | PA [39] | 3PPA | Ph [40] | PA | 3PPA | Ph [40] | PA | ||
| R(1,2) | 1.386 | - | - | 1.404 | - | - | 1.394 | - | - | A(12,2,3) | 120.3 | 119.9 | - | 120.3 | 119.6 | - | 120.3 | 119.6 | - |
| R(2,3) | 1.385 | 1.401 | - | 1.404 | 1.407 | - | 1.394 | 1.397 | - | A(13,3,4) | 119.8 | 119.9 | - | 119.8 | 119.6 | - | 119.8 | 119.7 | - |
| R(3,4) | 1.386 | 1.402 | - | 1.404 | 1.415 | - | 1.394 | 1.404 | - | A(14,4,5) | 119.6 | 121.7 | - | 119.4 | 122.4 | - | 119.4 | 123.3 | - |
| R(4,5) | 1.390 | 1.388 | - | 1.410 | 1.387 | - | 1.399 | 1.378 | - | A(16,7,8) | 109.2 | - | 110.6 | 108.9 | - | 111.5 | 109.0 | - | 111.5 |
| R(5,7) | 1.385 | - | - | 1.403 | - | - | 1.393 | - | - | A(17,7,8) | 109.5 | - | 110.8 | 109.2 | - | 111.3 | 109.3 | - | 111.2 |
| R(7,8) | 1.534 | - | 1.528 | 1.553 | - | 1.545 | 1.540 | - | 1.531 | A(18,8,7) | 109.2 | - | 110.7 | 109.0 | - | 109.4 | 109.1 | - | 109.5 |
| R(8,9) | 1.524 | - | 1.525 | 1.541 | - | 1.543 | 1.529 | - | 1.528 | A(19,8,7) | 110.0 | - | 110.8 | 110.0 | - | 110.3 | 110.1 | - | 110.4 |
| R(9,10) | 1.456 | - | 1.456 | 1.484 | - | 1.490 | 1.468 | - | 1.469 | A(20,9,8) | 109.3 | - | 109.3 | 109.4 | - | 109.3 | 109.3 | - | 109.2 |
| R(12,2) | 1.075 | 1.076 | - | 1.091 | 1.093 | - | 1.084 | 1.086 | - | A(21,9,8) | 109.3 | - | 108.9 | 109.1 | - | 109.0 | 109.1 | - | 109.0 |
| R(13,3) | 1.076 | 1.076 | - | 1.091 | 1.094 | - | 1.085 | 1.087 | - | A(22,10,9) | 111.4 | - | 113.3 | 110.6 | - | 110.7 | 111.2 | - | 110.8 |
| R(14,4) | 1.077 | 1.075 | - | 1.092 | 1.094 | - | 1.086 | 1.086 | - | A(23,10,9) | 111.2 | - | 113.9 | 110.4 | - | 110.3 | 110.8 | - | 111.2 |
| R(16,7) | 1.087 | - | 1.086 | 1.102 | - | 1.102 | 1.096 | - | 1.095 | D(4,3,2,1) | −0.1 | - | - | −0.1 | - | - | −0.1 | - | - |
| R(17,7) | 1.087 | - | 1.087 | 1.103 | - | 1.103 | 1.096 | - | 1.098 | D(5,4,3,2) | −0.1 | 0.0 | - | −0.1 | 0.0 | - | −0.1 | 0.0 | - |
| R(18,8) | 1.086 | - | 1.085 | 1.101 | - | 1.100 | 1.094 | - | 1.095 | D(7,5,4,3) | −178.6 | - | - | −178.5 | - | - | −178.4 | - | - |
| R(19,8) | 1.089 | - | 1.089 | 1.104 | - | 1.104 | 1.097 | - | 1.098 | D(8,7,5,4) | 92.1 | - | - | 95.3 | - | - | 94.9 | - | - |
| R(20,9) | 1.093 | - | 1.093 | 1.110 | - | 1.113 | 1.103 | - | 1.103 | D(9,8,7,5) | −179.7 | - | −180.1 | −179.5 | - | −179.7 | −179.4 | - | −179.8 |
| R(21,9) | 1.087 | - | 1.086 | 1.102 | - | 1.102 | 1.096 | - | 1.096 | D(10,9,8,7) | −178.7 | - | 181.7 | −178.0 | - | −178.4 | −178.2 | - | −178.6 |
| R(22,10) | 1.000 | - | 1.002 | 1.024 | - | 1.021 | 1.016 | - | 1.016 | D(13,4,3,2) | 179.8 | 180.0 | - | 179.7 | 180.0 | - | 179.7 | 180.0 | - |
| R(23,10) | 1.001 | - | 1.002 | 1.023 | - | 1.020 | 1.015 | - | 1.015 | D(14,5,4,3) | 179.7 | 180.0 | - | 179.7 | 180.0 | - | 179.7 | 180.0 | - |
| A(1,2,3) | 119.4 | 120.7 | - | 119.5 | 120.6 | - | 119.5 | 120.6 | - | D(16,7,5,4) | −146.1 | - | - | −143.0 | - | - | −143.4 | - | - |
| A(2,3,4) | 120.2 | 120.1 | - | 120.1 | 120.1 | - | 120.1 | 120.2 | - | D(16,7,8,9) | 58.5 | - | 60.1 | 58.5 | - | 60.5 | 58.6 | - | 60.4 |
| A(3,4,5) | 121.0 | 117.1 | - | 121.1 | 116.6 | - | 121.0 | 116.6 | - | D(17,7,5,4) | −30.0 | - | - | −26.7 | - | - | −27.1 | - | - |
| A(4,5,6) | 118.2 | 124.7 | - | 118.1 | 125.6 | - | 118.2 | 125.7 | - | D(17,7,8,9) | −57.8 | - | −59.8 | −57.4 | - | −59.6 | −57.4 | - | −59.7 |
| A(5,7,8) | 113.0 | - | - | 113.3 | - | - | 113.1 | - | - | D(19,8,7,5) | 58.6 | - | - | 58.7 | - | - | 58.7 | - | - |
| A(7,5,4) | 120.9 | - | - | 121.0 | - | - | 121.0 | - | - | D(20,9,8,7) | −60.1 | - | −60.5 | −60.0 | - | −60.3 | −59.9 | - | −60.3 |
| A(7,5,6) | 120.8 | - | - | 120.9 | - | - | 120.8 | - | - | D(21,9,8,7) | 56.5 | - | 56.6 | 56.4 | - | 56.0 | 56.3 | - | 55.9 |
| A(9,8,7) | 112.7 | - | 112.9 | 112.9 | - | 113.1 | 112.8 | - | 113.0 | D(22,10,9,8) | 176.3 | - | 167.8 | 175.8 | - | 176.1 | 176.0 | - | 175.8 |
| A(10,9,8) | 110.8 | - | 110.7 | 110.7 | - | 111.8 | 110.7 | - | 111.9 | D(23,10,9,8) | −63.9 | - | −64.0 | −66.3 | - | −66.2 | −64.9 | - | −65.3 |
a Coordinate descriptions are carried out by the numbers in Fig. 1. R, A, and D characters represent bond length (Å), angle (0) and dihedral angle (0) type of coordinates, respectively. Ph: Phenyl, PA: Propylamine.
6 Conclusion
The experimental and the theoretical investigations of the 3-PPA molecule have been performed successfully by using NMR and quantum chemical calculations. Application of the GIAO method yields 1H and 13C chemical shifts that are in reasonable agreement with experimental chemical shifts with the exception of N–H Hs due to a very strong hydrogen bonding effect between acetone-d and the title molecule. On the other hand, when compared to B3LYP and BLYP, the HF approach seems to lack a description of the perturbations responsible for spin–spin coupling constants. The correlation values for proton and carbon chemical shifts are found to be 0.9740, 0.9744, 0.9746 and 0.9973, 0.9969, 0.9974 for HF, BLYP and B3LYP with the 6-311++G(d,p) basis set, respectively.
Acknowledgement
This study is dedicated to the 50th anniversary of Anadolu University.