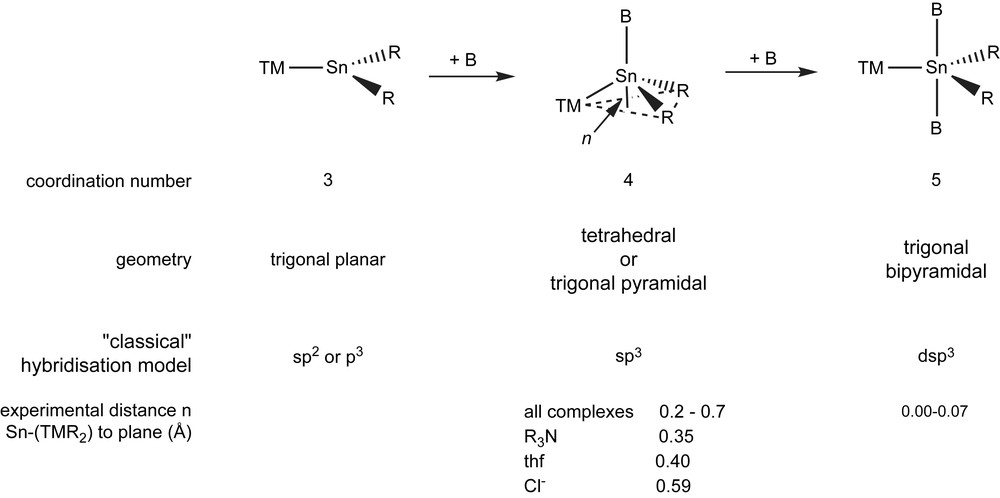
1 Stannylenes R2Sn as ligands
1.1 Electronic structure of stannylenes R2Sn
Stannylenes (“stannanediyls”) are divalent tin compounds which show electron deficiency with respect to the Xe electron configuration. The nomenclature had been used for the first time during the 1960s even if Sn(II) compounds are known for more than 150 years [1]. It logically fits into the column of low valent group-14 species like carbenes (“methylenes”), silylenes, germylenes and plumbylenes. As opposed to the lighter homologues, stannylenes have been characterised in solution and the solid state quite early. The tin species are electrochemically more stable (standard reduction potential Sn4+ → Sn2+: 0.15 V; Sn2+ → Sn0: −0.141 V [2]) than their lighter congeners. They can be isolated as monomers if the appropriate sterically or electronically stabilising substituents are chosen.
The electron distribution around the low valent tin atom is highly asymmetric as seen by the non-linear arrangement of the substituents. Two descriptions of the electronic ground state are usually discussed: sp2 hybridisation as found with carbene species (for the singlet state), with an idealised R–Sn–R bond angle of 120°. However, hybridisation is getting more and more unfavourable with heavy elements. Hence, an unhybridised model with an R–Sn–R angle of 90° view is preferred to describe the orbital distribution around Sn(II). In real structures, angles between 90° and 100° are generally found, favouring the unhybridised view with widening of the angle due to steric factors. The inclusion of (n − 1)d-orbitals has found not to be necessary to best describe the physico-chemical properties by various calculation methods (Fig. 1) [3].
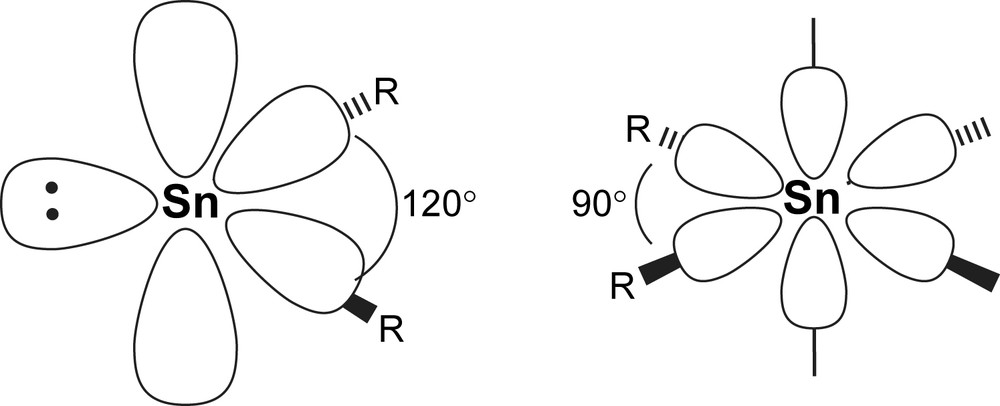
Alternative hybridisation models for Sn(II) compounds: sp2 hybridisation (left) with filled sp2 lobe versus unhybridised model (filled s-orbital not shown).
In the sp2-hybridised view, the lone pair of electrons is situated in one of the sp2 lobes and points away from the substituents. In the unhybridised model, the electron pair is distributed spherically around the tin atom. Common to both models is the empty p-orbital perpendicular to the plane formed by the R2Sn moiety.
It is the presence of the lone pair of electrons that makes these compounds potential ligands in transition metal (TM) complexes. Prominent valence isoelectronic ligands are e.g. carbenes and carbon monoxide, CO. They are at the same time isolobal [4] to phosphines, PR3. All those ligands are formally able to accept π-back bonding from symmetry adapted filled TM orbitals (Fig. 2).
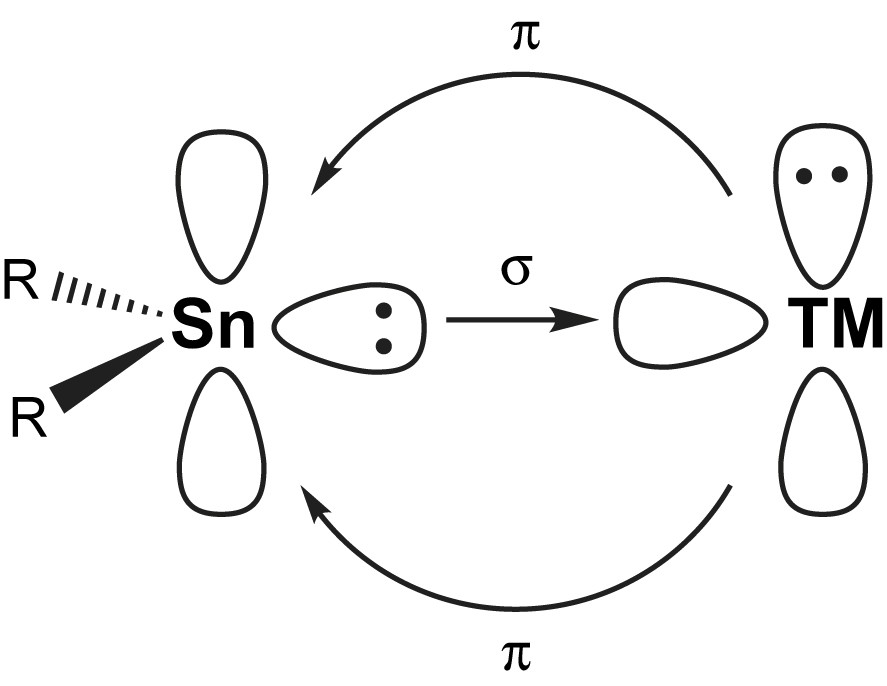
Simplified view of the bonding in R2Sn–TM complexes in analogy to carbenes and CO.
To reduce the lack of electron density, Sn(II) compounds easily accept Lewis bases perpendicular to the SnR2 plane. In absence of donor solvents, this leads to aggregation forming oligo- and polymers. Coordination of such aggregates to TMs leads to lower aggregation grades. However, the tendency away from intra- to intermolecular Lewis bases increases upon TM coordination. The majority of stannylene complexes form base adducts.
Stabilisation of divalent species by the help of TM complexes is in the focus of the present review. This feature-rich chemistry has been the subject of several reviews [5–9], many of which are mentioning TM coordination as one aspect of stannylene chemistry. Others focus on specific stannylene ligands or special aspects like synthesis and reactivity. The most comprehensive reviews have been presented by Holt et al. in 1989 [10] and especially by Petz in 1986 [11]. Both summarise synthetic, reactivity, crystallographic, infrared and Raman vibrational information of the known R2Sn–TM complexes. 119Sn NMR in the 1980s was not common as today so that tendencies in 119Sn NMR properties could be extracted only for a few selected examples. Most reviews on 119Sn NMR spectroscopy discuss the experimental requirements to record and process NMR spectra and discuss the chemical shift and coupling properties of Sn(IV) and Sn(II) compounds. However, NMR spectroscopy of TM stannylene complexes has not yet been in the focus of earlier accounts. The growing number of NMR and structural data gives us the possibility to start investigating spectroscopy–structure relationships.
The present review article aims at compiling 119Sn NMR data and molecular structural data of the complexes in which a stannylene interacts as terminal ligand with the transition metal based on the experimental data given in the literature for R2Sn–TM complexes up to 2007 (and partially beyond). This should increase our understanding of the TM–Sn bond, the influence of TM fragment coordination on the electronic structure of the stannylene and vice versa. With this review, which will be completed by a bibliographic update published elsewhere [12], we want to stimulate further theoretical and experimental investigations in this growing field.
1.2 Theoretical description of the TM–Sn(II) bond
The lone pair of electrons in stannylenes acts as a soft Lewis base towards e.g. (soft) transition metal complex fragments. Stannylene ligands may therefore be described in analogy to Fischer carbene (R2C) complexes [13]. The empty p-orbital may act as acceptor to either perpendicularly attacking σ-donors (i.e. base molecules) or symmetry adapted π-donors either from within the stannylene (e.g. lone pairs of planar amido substituents) or through back bonding from filled transition metal d-orbitals (Fig. 3). In that respect, stannylenes behave like carbonyl or phosphine ligands. Indeed, especially with the latter, electronic properties are very well comparable, as seen from similar IR frequencies in substituted carbonyl complexes with PPh3 and different stannylenes [14–16]. Therefore, it is very helpful to start from the concepts already developed for those non-metal ligands and to modify them in order to better understand the bonding in stannylene complexes.
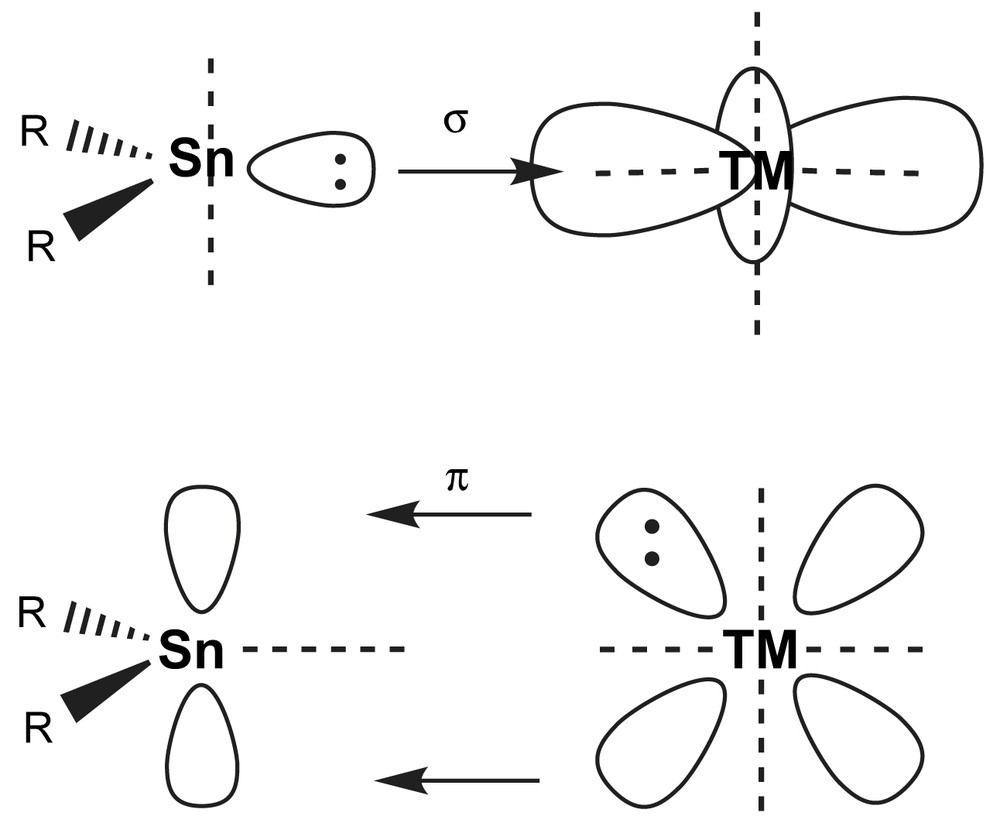
Symbolic MO scheme of the Sn(II)–TM interaction (σ and π) in R2Sn–TM complexes in analogy to carbenes and CO.
It is generally accepted among synthetic chemists that the bond between carbon and TM in R2C–TM complexes is predominantly covalent, exhibiting a high degree of multiple bonding. Recent calculations using the energy decomposition analysis (EDA) on low oxidation state transition metal complexes with Sn(OH)2 as stannylene model show [16] that on going down group-14 the electrostatic contribution to the TM–Grp 14 bond increases, and the π-bonding contribution decreases considerably. According to Mulliken population analyses [17], the TM–Sn bond is determined by σ-donor properties from the group-14 atom and considerable π-back bonding from the TM. In the case of low oxidation state TMs, the donor orbital contains a high contribution of an Sn based singlet s-orbital, thus hosting the lone pair of electrons. DFT calculations also show that within the series R2E–Cr(CO)5 (E = C, Si, Ge, Sn), π-bonding drops dramatically from carbene R2C to silylene R2Si and remains comparable for heavier ligands [18,19]. The strong π-back bonding in carbene complexes has direct effects on the structure of the pentacarbonyl fragment in that the Cr–CO bond in trans position to the carbene is significantly longer than Cr–CO in cis position. The reduced π-bonding in silylenes (E = Si) through stannylenes (E = Sn) is, to a great extent, due to the higher HOMO and especially LUMO orbital energies and not so much to inefficient orbital overlap with the TM d-orbitals [18]. With heavier TM e.g. in the triad Cr, Mo to W, π-back donation increases because of the relativistic destabilisation of the d-orbitals, which means, that they get closer to the high energy LUMOs of the low valent tetrele [18].1 The situation is quite different for N-heterocyclic carbene (NHC) ligands, where π-back donation is considerably reduced [20]. In the X2E–TM(Cl) series (X2E = NHC, R2Si, R2Ge; TM = Cu, Ag, Au), the TM–E bonds show a high degree of coulombian interaction with covalent contributions from σ-donation, but weak π-back donation. DFT calculations show that in heavier NHC analogues, the π-delocalisation across the group-14 element decreases dramatically from silicon to germanium [21]. Interestingly, LCAO–MO–SCF calculations reveal a strong π-interaction only between the two nitrogen lone pairs in tetracyclic stannylenes Sn(NtBu)2SiMe2 (1). The non-bonding orbital on tin is delocalised over the nitrogen atoms and possesses both Sn s- and p-characters [22].
Experimentally, the competition for the tin based empty p-orbital between “external” σ-donors and “internal” π-donation from either lone pairs of electrons of α-heteroatoms or filled d-orbitals of the TM provides one measure for the strength of the TM → Sn back bonding. The coordination of a base-stabilised divalent species (B → R2Sn) to a transition metal complex leading to B → R2Sn–TM does not necessarily induce the release of the base, but stabilises the Lewis base adduct [11]. In hypercoordinated stannylenes such as (Salen)Sn (2), the intramolecular Lewis base coordination found in the free stannylene [23,24] persists in TM complexes [25,26]. Crystallisation of the amido stannylene complex [{Me2Si(NtBu)2Sn(thf)}{W(CO)5}] (1a) from THF solution shows strong bonding of the coordinating solvent to tin, a behaviour not found for the free stannylene. Surprisingly, the TM complex fragment is released when treated with pyridine [27]. The TM complex fragment behaves predominantly as Lewis acid with weak π-back bonding. This is analogous to the stabilisation of X2Sn ← NMe3 (X = F, Cl, Br) adducts on coordination to BF3 [28]. However, π-back bonding is reduced by addition of ethyne (C2H2) or formaldehyde across the Pd–Sn bond in PdSnH2 complexes [{(C2H2(H2E)2)Pd}SnH2] (E = N, P) [29]. The calculations investigate the mechanism of C2H2 coordination on [{dipePd}Sn(CH(SiMe3)2)2] (3a) [30,31]. Mutual perpendicular orientation of the TM and stannylene moieties, as well as the lengthening of the Sn–Pd bond supported by the results of atomic orbital population analyses, point to the thermodynamic importance of π-back bonding. However, the rotation barrier around the Sn–Pd bond is relatively small. It is reduced from 41.4 kJ mol−1 in the base free complex to 25.1 kJ mol−1 after η2-coordination of C2H2 to Sn, showing that π-bonding is not a dominating factor in the total energy of the Sn–TM bond. Electrophilic pre-coordination to the Sn p-orbital has also been found to be an essential step in the activation of E–H bonds through laser flash photolysis studies [32].
The HOMO–LUMO gap in low valent group-14 compounds R2E, which is decisive for the donor–acceptor properties, depends mostly on R substituents [3,33]. The heavier the tetrele (more electropositive) and the more electronegative the substituent, the larger the energy difference between singlet and triplet states [3]. One important effect to stabilise the singlet state is π-donation from the substituent R. Electropositive substituents reverse the energy order when E = C, so that e.g. CH2 has a triplet ground state, whereas SiH2 and CI2 already have a singlet ground state [33].
In summary, besides the classical picture of a dative R2E → TM bond strengthened by covalent π-back bonding, ionic structures play an important role in the stabilisation of the TM–main group metal bond. R2Sn species may be described as good σ-donors, however, with weak π-back donation.
1.3 Heuristic description of the Sn(II)–TM bond
Basically three types of symbolisms for the bonding situation in low valent group-14 TM complexes are used side by side in the literature (see Fig. 4). Some papers describe the Sn–TM bonding as a double bond R2SnTM (Symb. 1) in analogy to the carbene–transition metal complexes R2CTM. This formalism makes use of the isolobal analogy [34] with alkenes R2CCR2, where one of the 6 VE fragments is substituted by the heavier congener R2Sn and the second by a 16 VE TM fragment. Other publications describe Sn–TM bonding as a dative bonding R2Sn → TM (Symb. 2), the arrow going from tin to the transition metal, explaining quite immediately the donor character of the tin lone pair of electrons. Another description, which circumvents the chasm between simple donor–acceptor interaction and multiple bonding, regards only the connectivity and symbolises the bond as single hyphen R2Sn–TM (Symb. 3). One might tend to assign the different descriptions to the background of the author, be it “organometallic” (Symb. 1 and Symb. 2), “organic” (Symb. 1), “coordination chemistry” (Symb. 2) or “main group chemistry” (Symb. 2 and Symb. 3).
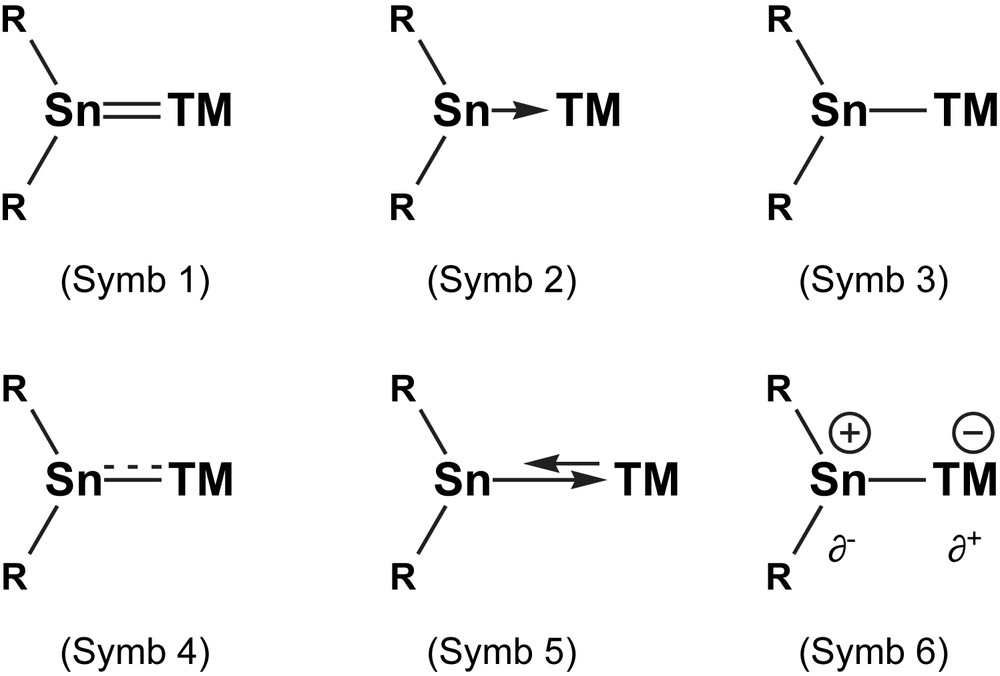
Heuristic descriptions of the Sn(II)–TM bond as found in the literature (symbolisms 1–3) and considering the relative strength of σ- and π-bonding (Symb. 4 and 5) as well as electrostatic contributions (Symb. 6).
Can these types of formalisms be improved? The double bond formalism Symb. 1 overestimates the role of the covalent π-back donation, the donor–acceptor formalism Symb. 2 totally neglects multiple bonding and the simple bond symbolism pretends that the bonding electrons stem from both participating atoms. As usual with simplifying models, the reality can be found between those three formalisms. As seen from the theoretical results, the bonding mode is a superimposition of a σ-dative bond (Sn → TM) and a part of π-back donating character (Sn←TM) with strong ionic participation. Description of the combined bonding behaviour in a simple picture is not easy and needs detailed analyses of the respective compound to find the right balance. Thus, addition of a full-line bonding Sn–TM for strong σ-bonding and a dotted-line bonding Sn⋯TM for partial π-bonding (Symb. 4), a long arrow for σ-donation and a shorter one to account for the weaker π-back donation (Symb. 5) or assigning formal charges (Symb. 6) might be alternatives. In the end the author has to decide individually which symbolism to apply, having always in mind that it reflects only part of the reality. A unifying drawing model cannot be proposed here because all descriptions have their legitimacy.
The authors will try to unify the different formalisms used in the original papers, but in justified exceptions, the originally used formalism will be adopted. The following table lists the most stannylene ligands and complexes discussed in the text. The stannylene is abbreviated with consecutive bold numbers and the complexes with appending small letters.
Table to identify most relevant stannylene ligands (bold numbers) and respective complexes (small letter appendix) discussed in the text. L is used as abbreviation for two-electron, Lpd for polydentate stannylene ligands, L′ denotes other co-ligands specified in the table.
| Stannylene L | No. | Complex LML′n | No. |
| 1 | [{W(CO)5}L(thf)] | 1a | |
| [{Cr(CO)5}L(thf)] | 1b | ||
| mer-[{Cr(CO)3}L3] | 1c | ||
| [{NiCpL}2] | 1d | ||
| [NiL4(μ-OiPr)2] | 1e | ||
| [NiL4(μ-Br)2] | 1f | ||
| [PdL4(μ-Cl)2] | 1g | ||
| 2 | [{W(CO)5}L] | 2a | |
| cis-[{W(CO)4}L(PPh3)] | 2b | ||
| cis-[{W(CO)4}L2] | cis-2c | ||
| trans-[{W(CO)4}L2] | trans-2c | ||
| [{Cr(CO)5}L] | 2d | ||
| 3 | [{Pd(dipe)}L] | 3a | |
| [{CoCp∗(C2H4)}L] | 3b | ||
| [{Pd(dipe)}(μ-C2H2)L] | 3c | ||
| [{Cr(CO)5}L] | 3d | ||
| 4 | [{W(CO)5}L] | 4a | |
| [{Cr(CO)5}L] | 4b | ||
| trans-[{Mn(CO)4(Sn(3,5-tBu2C6H3CMe2CH2)(4,6-tBu2(2-CMe2CH2)C6H2))}(L)] | |||
| 5 | [{Cr(CO)5}L] | 5a | |
| [{W(CO)5}L] | 5b | ||
| 6 | [{W(CO)5}L] | 6a | |
| [{Cr(CO)5}L] | 6b | ||
| [{Fe(CO)4}L] | 6c | ||
| [{Pt(PPh3)2}L] | 6d | ||
| 7 | [{Fe(CO)4}L] | 7a | |
| [{CoCp∗(C2H4)}L] | 7b | ||
| 8 | [PtL4]6− | 8a | |
| [{Fe(triphos)(NCMe)2}L] | 8b | ||
| [{Ru(PPh3)2}(μ,η1:3-L)]2 | 8c | ||
| [{Ni(dppm)}L3]4− | 8d | ||
| cis-[{Pd(dppp)}L2]2− | 8e | ||
| [{Pt(PEt3)2(CNtBu)}L2][{Pt(PEt3)2(C(NiPr)2C2Me2)}L]2− | 8f | ||
| [{Pt(dppp)}L2]2− | 8g | ||
| [{Pt(dppe)(tBuNCPh)PPh3}L] | 8h | ||
| [{Cr(CO)5}L]2− | 8i | ||
| [{Mo(CO)2(η7-C7H7)}L]− | 8j | ||
| 8k | |||
| 9 | [{Ru(bipy)(CO)2}L] | 9a | |
| [{Au(μ-PPh2)}L]2 | 9b | ||
| [{Au(PPh3)}L] | 9c | ||
| [{WCp(CO)3}L] | 9d | ||
| 10 | |||
| 11 | [{W(CO)5}L] | 11a | |
| [{Cr(CO)5}L] | 11b | ||
| 12 | [{W(CO)5}L] | 12a | |
| 13 | [{W(CO)5}L] | 13a | |
| 14 | [{W(CO)5}L] | 14a | |
| [{W(CO)5}{Cr(CO)5}Lpd] | 14b | ||
| [{W(CO)5}2Lpd] | 14c | ||
| [{Cr(CO)5}2Lpd] | 14d | ||
| [{Cr(CO)5}L] | 14e | ||
| 15 | [{W(CO)5}2Lpd] | 15a | |
| 16 | [{WCp(CO)2(cyclo-P(Ph)(NMeCH2)2)}L(OTf)] | 16a | |
| 17 | [{W(CO)5}L] | 17a | |
| 18 | [{W(CO)5}L(thf)] | 18a | |
| [{W(CO)5}L(thf)2] | 18b | ||
| [{{Cp(OC)Mo}2(μ-P(OEt)2)(P(O)(OEt)2)(μ-L)}L] | 18c | ||
| 19 | [PtL3] | 19a | |
| 20 | trans-[{Rh(PPh3)2}L3]2− | 20a | |
| 21 | [{Pt(PPh3)2}Lpd(OPPh3)] | 21a | |
| [PtLpd(thf)2Lpd(thf)] | 21b | ||
| [PtLpdLpd(thf)] | 21c | ||
| 22 | [{W(CO)5}L] | 22a | |
| [{W(CO)5}L(thf)] | 22b | ||
| 23 | [{Cr(CO)5}L(py)] | 23a | |
| 24 | [{Mo(CO)5}L] | 24a | |
| [{Cr(CO)5}L(Fe(CO)4)] | 24b | ||
| [{Cr(CO)5}L(Mo(CO)5)] | 24c | ||
| 25 | [{W(CO)5}7Lpd]2− | 25a | |
| 26 | [{WCp(CO)3}L(Cl)] | 26a | |
| [{W(CO)5}L] | 26b | ||
| 27 | [{MnCp∗(CO)2}6Lpd] | 27a | |
| [{Cr(CO)5}6Lpd] | 27b | ||
| 28 | cis-[{Mn(CO)4(HgMn(CO)5)}L] | 28a | |
| 29 | eq-[{Fe(CO)4}L] | 29a | |
| 30 | trans-[{Co(CO)3}L2] | 30a | |
| 31 | [{Ir(μ-dppm)(CO)}2(μ-S)L2] | 31a | |
| 32 | [{Cr(CO)5}6Lpd]2− | 32a | |
| 33 | trans-[{Co(CO)3}L2] | 33a | |
| 34 | trans-[{Co(CO)3}L2] | 34a | |
| 35 | [{W(CO)5}L] | 35a | |
| 36 | [{Cr(CO)5}2Lpd}] | 36a | |
| 37 | eq-[{Fe(CO)4}L] | 37a | |
| ax-[{Fe(CO)4}L] | |||
| 38 | |||
| 39 | |||
| 40 | |||
| 41 | [{W(CO)5}L] | 41a |
2 119Sn NMR spectroscopy of transition metal stannylene complexes
2.1 On 119Sn NMR chemical shifts and coupling constants
Among the three NMR observable isotopes of tin with I = 1/2, 119Sn is the most abundant and studied isotope (115Sn (natural abundance 0.36%), 117Sn (7.68%), 119Sn (8.58%)) [35]. Because of the good sensitivity of the 119Sn isotope (25.6 times more sensitive than 13C), 119Sn NMR spectroscopy is an excellent analytical tool for the characterisation of tin-containing compounds. Strong 119Sn NMR shift variations are correlated to changes around the tin atom (−2338 [36] to +3301 ppm [37]). The 119Sn NMR shift gives an image of the electronic environment of the tin atom. Ligands and substituents will greatly modify the electronic environment around the tin atom, which is reflected in the chemical shift values. The electronic communication between NMR active nuclei depends on the bond forming orbitals and electron density, for which the NMR coupling constant may act as a measure [10,38,39]. The relatively close gyromagnetic moments of 117Sn (−9.5319 × 10−7 rad T−1 s−1) and 119Sn (−9.9756 × 10−7 rad T−1 s−1) imply that the coupling constants to (i) substituents, (ii) to NMR active transition metal nuclei (e.g. 103Rh, 109Ag, 183W and 195Pt), and (iii) to further Sn nuclei in polytin compounds, show characteristic “double satellites” with similar intensities and a ratio nJ(X–119Sn)/nJ(X–117Sn) of 1.047, which can conveniently be used as diagnostics in solution NMR.
In NMR spectroscopy, the Larmor frequency νA, and magnetic shielding constant σA are linked through the general equation [40,41]:
In general, NMR spin–spin coupling constants (SSCCs) can provide information on the electronic structure along a specific bond. As an example, investigations on hydrocarbons have shown that the 1J(C–X) (X = C, H) coupling constants are correlated to the s-character of the C–X bond orbitals (more precisely in localized molecular orbitals), the C–C bond length, its bond order, its p bond (σ- or π-) character. SSCCs for different isotopes of the same element are scaled by the relative gyromagnetic ratio. 3J(1H–13C) and 3J(1H–1H) coupling constants are often proportional. These results indicate that SSCCs contain useful information on the chemical bond. The electron density around the nucleus can be divided in differents parts and the corresponding Ramsey terms are related to different spin–spin coupling mechanisms [42]. The Fermi-contact (FC) term depends on orbitals with distinct s-character at the coupling nuclei and is the most important contribution for light nuclei like hydrogen. The paramagnetic spin–orbit (PSO) and spin–dipole (SD) terms need orbitals with non-s character at the coupling nuclei. At last, the weak diamagnetic spin–orbit (DSO) term is not dependent on orbital character. Hence, an analysis of the p-character of a bond should be based upon the PSO and the SD terms [43], whereas the strength of the σ-bond mostly influences the Fermi term. Although the mechanisms are well investigated with carbon and hydrogen systems, the contribution of the different mechanisms becomes more complicated with heavier nuclei. Multiple coupling mechanisms with partially opposite positive or negative signs may occur at the same time, making it rather impossible to develop a comprehensive model based on experimental findings alone. However, it has been found in theoretical investigations of Pt–Tl complexes that the coupling constant in these heavy atom systems still depends strongly on Fermi coupling [44]. Besides, at least for low coordination compounds, the solvent surrounding (base adducts and continuum) becomes very important (the more bases, the higher the constant). For heavy nuclei, relativistic effects seem to amplify the different coupling mechanisms and the bond length indeed plays a major role (the shorter the bond, the larger the constant). Less important seem to be the HOMO–LUMO gap and SD coupling.
2.2 Coordination chemical shift
A rather useful qualitative measure in coordination chemistry is the coordination deshielding parameter or coordination chemical shift Δδ. For stannylene ligands, it is expressed as the difference between the 119Sn NMR chemical shift (in ppm) of the R2Sn–TM complex and the corresponding free stannylene R2Sn:
A positive coordination deshielding means that the chemical shift is moved downfield upon coordination, i.e. to higher ppm values or to less chemical shielding. From a simplistic view, one would expect that withdrawing electron density by addition of an electropositive element to the lone pair of electrons would in any case deshield the tin nucleus. Hence, a negative Δδ implies that additional factors must contribute to the overall chemical shift difference. As will be discussed later, it might be useful to differentiate between the s- and p-orbital contribution to the chemical shift, so that it might be possible to define Δδ as a function of a σ-bonding contribution Δδ(σ) with high s-orbital participation, and π-bonding contribution Δδ(π) with predominantly p-orbital participation.
Coordination deshielding has been extensively discussed for transition metal complexes of phosphines [35,45–48]. It is found that both positive and negative coordination deshielding values are realised. A plot of Δδ (31P) versus the chemical shift of the free (non-coordinated) ligand shows a negative slope with positive intercept within classes of phosphines [45]. The intercept is always positive and decreases along the row and going down the group in the periodic table. It may be taken as a measure for the effect of lone-pair coordination (σ-donation), hence the pure “Lewis acid contribution”. There is a bit of confusion about the interpretation of the negative slope. Having in mind the potential π-acceptor ability of phosphines, π-back bonding from the TM would yield an increase of electron density on the main group element. That is expected to cause a shielding of the nucleus, hence moving the signal upfield. The negative slope means that if the stannylene is more deshielded, the π-back bonding strength is stronger and the upfield shift on coordination larger [45]. However, this approach does not take into account that π-back bonding also means the partial population of antibonding MOs, thereby increasing the paramagnetic shift, which is always downfield [35]. Structure–spectroscopy relationships in phosphine and carbonyl complexes show that deshielding correlates with higher π-back donation (increased by weak π-accepting trans ligands, negative charge, filling of TM d subshells). Further, as π-back bonding usually tends to be over-estimated [16], it is doubtful whether its contribution may be strong enough to over-compensate deshielding by σ-donation. Another rationalization for negative Δδ values was deduced from combined 31P and 95Mo NMR studies, where the negative Δδ (31P) values of PX3 (X = Cl, Br) ligands were explained as a consequence of poor σ- and π-bonding abilities [49]. An exhaustive explanation of negative deshielding implies a more sophisticated approach to MO theory.
There are currently no comprehensive reports on the coordination deshielding with low valent tin ligands, despite several articles on experimental tin NMR spectroscopy [38,39,50]. Theoretical treatment of NMR parameters of stannylene TM complexes is still missing. It is therefore helpful to transfer the findings with similar σ-donor systems like phosphines to stannylene complexes. Both phosphines and stannylenes exhibit similar bonding properties, despite the fact that the geometry around the donor atom E (SnR2, PR3) changes from C3v to C2v and hence different orbital symmetries as well as relative orbital energies are involved. Nevertheless, in both systems, σ-donation from E to TM is found. However, the π-back donation from occupied TM orbitals into the empty p-orbital on tin does not have an equivalent on phosphorus, where σ∗ (P–R) orbitals or d-orbitals are discussed to receive the TM electron density [47,51]. It resembles more the back bonding into the π∗ orbitals in CO. As a consequence of the availability of the empty p-orbital and the reduced number of subtitutents, auto-association and base adduct formation are found with stannylenes, but not with phosphines. It is with these findings that one may assume that π-back bonding as contribution to the total bonding may be more important compared to phosphine complexes. However, as discussed above, theoretical calculations find that π-back donation within a main group decreases with increasing atomic number. Since the paramagnetic contribution is decisive for the chemical shift of the heavier nuclei, population of the respective π bonds and the energy gaps between symmetry related MOs become more important. The relative energies are determined by the electronegativity of the substituents, the number and kind of bases on tin, the steric crowding on both TM and tin and the type of TM fragments, besides others. For example, electronegative substituents should lower the energy of the lone pair, at the same time increasing its s-character, and destabilising the π∗(Sn–R) MO. The higher the period of the TM, the better should be the overlap between σ orbital of the TM fragment and the ligand, leading to greater energy separations and hence to less coordination deshielding.
In the following sections, rather than providing a comprehensive review of all reported NMR parameters, we will focus on extracting general tendencies for chemical shifts and absolute coupling constants from selected examples and try to relate them to structural properties, We will look for relationships between the spectroscopic shift parameters δ(R2Sn–TM), δ(R2Sn) and Δδ and chemical properties such as kind and number of base, substituents and TM moiety. We will first consider the spectral dependency on changes at the stannylene for well-defined carbonyl fragments of groups 6 and 8, followed by examination of the base dependency. We will examine further types of complexes, which deserve a separate discussion before briefly looking at absolute scalar TM–Sn coupling constants. A discussion of the relevance of π-back bonding will follow the presentation of NMR parameters.
2.3 Experimental chemical shift dependencies
2.3.1 Dependency of the coordination chemical shift on the chemical shift of the free stannylene (Δδ versus δ(SnR2)): towards a π-acceptor scale for stannylenes?
The variation of Δδ versus δ (119Sn) of the corresponding free stannylene is shown in Fig. 5. The values are strongly scattered because these crude data for different types of stannylenes are measured in different solvents. The plot is less consistent than with phosphines [45], which might be due to variations in the oligomerisation degree and various degrees of base adduct formation with changing substituents, hence causing more dramatic changes on tin than on phosphorus. However, the behaviour parallels that found with phosphine ligands: the slope tends to be negative and the intercept positive. The more negative the chemical shift of the free stannylene (i.e. the more electron rich or the higher coordinated the stannylene) the stronger is the coordination deshielding.
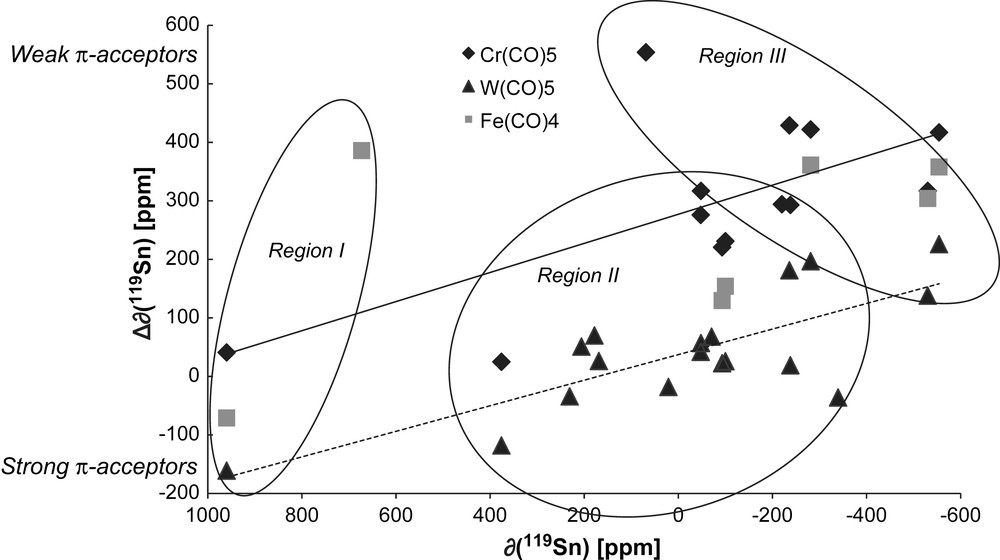
Variation and linear regression of 119Sn coordination chemical shift (Δδ) of an R2Sn–TM (TM = Cr(CO)5 13 data, W(CO)5 14 data) versus the 119Sn chemical shift (δ) of the corresponding free R2Sn. Deshielding is found to the left (high ppm values, downfield). The data for R2Sn–Fe(CO)4 are given but 6 data were not enough for any reasonable correlation. Regions including free-base R2Sn (region I), external base stabilised (region II) and internal base stabilised R2Sn (region III) can be defined. Although it seems easy to separate region I from region (II + III), regions II and III are overlapping.
In the plot, stannylenes may be grouped according to their coordination sphere around Sn. In region I, stannylenes stabilised by steric hindrance (i.e. without base stabilisation; strong deshielding of R2Sn, small to negative Δδ as the R2Sn–TM complexes including Weidenbruch's stannylene (4)), in region II, stannylene complexes with external base stabilisation (intermediate δ(R2Sn), positive Δδ as the B → Cl2Sn–W(CO)5 series described by DuMont) and in region III, intramolecular base-stabilised stannylene ligands are located (strong shielding of R2Sn, positive Δδ as (Salen)Sn–TM complexes).
2.3.2 R2Sn–M(CO)5 complexes (M = Cr, W)
The changing intercept with TM in Fig. 5 implies a strong dependency of Δδ on the TM. Within group-6, the slope is very similar. While for tungsten pentacarbonyl complexes an intercept of 41 ppm is found, the respective chromium complexes show a stronger deshielding ability (277 ppm). The calculated coordination deshielding difference Δ(Δδ) = ΔδCr − ΔδW (228 ppm) may be taken as rough estimate for the prediction of chemical shifts on changing the TM in analogous group-6 complexes. The range of Δδ values differs strongly. For R2Sn–W(CO)5 complexes the observed deshielding values vary between Δδ = +226 ppm for Sn(Salen) (2a) and Δδ = −161 ppm for Weidenbruch's ligand in 4a, while for R2Sn–Cr(CO)5 complexes, the range is found between Δδ = +554 ppm for Sn(SCH2CH2)2NtBu (5a) and Δδ = +29 ppm for the THF adduct to the Veith stannylene ligand Sn(NtBu)2SiMe2 (1a). This behaviour is similar to those of phosphine pentacarbonyl complexes.
Assuming that the global electron density determines the chemical shift, the effective coordination deshielding Δδ may tentatively be regarded as the sum of a deshielding (positive) contribution by σ-bonding (Δδ(σ)) and a shielding (negative) contribution by π-back bonding (Δδ(π)). Deshielding would increase with increasing Lewis acidity of TM and weakness of σ-donor ligands on the TM. π-Back bonding is expected to increase within a TM group with increasing atomic number and decreasing π-acceptor strength of especially the ligand trans to R2Sn. On the stannylene, increase of substituent electronegativity, decrease of substituent π-bonding and absence of base adducts should increase its π-acceptor strength.
In the present compilation, the aryl–alkyl Weidenbruch stannylene (4) and Veith's base-stabilised bis-amido stannylene (1)·THF show the most negative Δδ (TM = {W(CO)5}: −161 ppm (4a) and −118 ppm (1a)). Compound 4a features a sterically stabilised stannylene ligand, of which the empty p-orbital on Sn is prone to accepting π-electron density from the TM. In compound 1, π-back bonding from the electronegative amido substituents is diminished by base addition. However, the base addition should also reduce the effective back bonding from the TM. It seems that more sophisticated models have to be adopted to explain the strong deviations from the expected chemical shift behaviour. One has also to keep in mind that the observed isotropic chemical shift is composed of spatial contributions, highly anisotropic for stannylenes [52], and therefore, changes in one component δii may dominate others. The stronger shielding of R2Sn–W(CO)5 compared to R2Sn–Cr(CO)5 complexes is in accordance with stronger π-acceptor properties of the heavier TM as found also in calculations [18].
The influence of the TM moiety on the coordination chemical shift can be further shown with (Salen)Sn tungsten carbonyl complexes. Starting from [(Salen)Sn]W(CO)5 (2a, Δδ = +226 ppm), and replacing one carbonyl by:
- - a triphenylphosphine ligand leads to cis-[(Salen)Sn]W(CO)4(PPh3) (2b, Δδ = +206.5),
- - a (Salen)Sn leads to cis-[(Salen)Sn]2W(CO)4 (cis-2c, Δδ = +206 ppm) or trans-[(Salen)Sn]2W(CO)4 (trans-2c) (Δδ = +180 ppm).
Positive Δδ values are in agreement with strong σ-donating power of (Salen)Sn (2) towards tungsten. In addition, the cis substituted complexes 2b and cis-2c have very similar Δδ parameters, supporting similar electronic parameters for the phosphine and the (Salen)Sn ligands. Replacing the strong π-acceptor CO in trans position to the stannylene increases the electron density on the TM for π-back bonding to the stannylene. The increased electron density on the stannylene would then be in accordance with the smaller (i.e. more negative) coordination deshielding with respect to the pentacarbonyl or cis-tetracarbonyl derivatives. Furthermore, in complex trans-2c, both stannylenes are engaged in σ-bonding with the same orbitals on W, decreasing for both stannylenes the σ-donating capacities.
2.3.3 R2Sn–Fe(CO)4 complexes
The coordination shift values Δδ in the iron carbonyl system are found between those of the chromium and tungsten carbonyl systems, although they show even stronger variations than the group-6 complexes. This may be partly due to the two isomers with equatorial and axial positions of the stannylene, both of which are observed. Theoretical calculations and experimental observations with different types of ligands have shown that the equatorial position is considered to be preferred to axial for poor σ-donor and strong π-acceptor ligands (see also Section 3.4.2) [53]. If deshielding is considered as the sum of the (positive) σ-donating contribution (Δδ(σ) > 0) and the (negative) π-back contribution, (Δδ(π) < 0), stannylenes situated in equatorial position should lead to smaller Δδ. Unfortunately, in the only case in which both isomers of the same ligand could be isolated, the chemical exchange on the NMR time scale is too fast to assign chemical shifts [54].
2.3.4 Chemical shift of coordinated stannylene against free stannylene
The evolution of δ (119Sn, R2Sn–TM) with δ (119Sn, R2Sn) in Fig. 6 provides a better correlation than that found for Δδ. A relatively consistent linear fit for a given TM moiety is shown despite uncorrected data (different solvents like donor and non-coordinating solvents, and different types of substituents). All three correlations show a positive slope (Table 1).
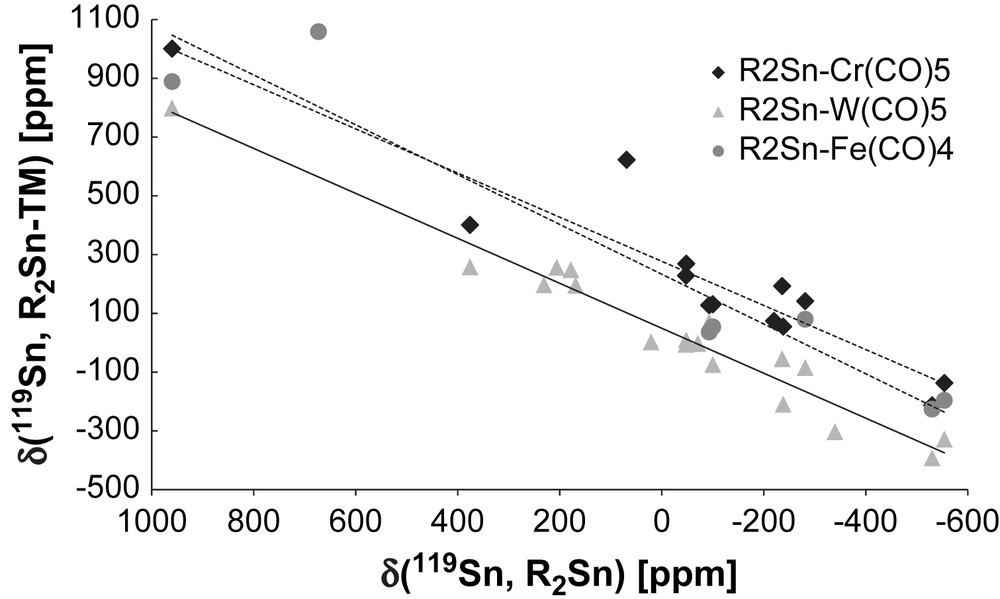
119Sn chemical shifts of the R2Sn–TM complexes (TM = Cr(CO)5 (13 entries)); W(CO)5 (14 entries); Fe(CO)4 (6 entries) versus 119Sn chemical shifts of the respective free R2Sn.
Slope and intercept in the graph δ(R2Sn-TM) versus δ(R2Sn) for R2Sn–TM complexes (TM = Cr(CO)5, W(CO)5, Fe(CO)4), Fig. 6.
| TM(CO)n | A [−] | B [ppm] |
| Cr(CO)5 | 0.75 | 277 |
| W(CO)5 | 0.76 | 49 |
| Fe(CO)4 | 0.85 | 233 |
This means that the change in the electronic properties of tin follows a similar mechanism of all three TM fragments. Furthermore, the similarity of the slopes suggests that the bonding between Sn and TM is similar irrespective of the stannylene ligand. The relative intercepts are indicators of the relative deshielding power of the respective TM fragment: (OC)5W < (OC)4Fe < (OC)5Cr. For the group-6 pentacarbonyl Cr and W complexes, the slope is very similar, while it is steeper for the iron complexes. However, for the 3d metals Cr and Fe, the intercept is similar. It seems that the slope depends on the group, and the intercept on the row. Both are due to varying orbital overlap and reflect the trends seen for phosphine complexes.
For R2Sn–M(CO)5 complex (M = Cr, W) series, the highest 119Sn chemical shift values δTM (119Sn) are attributed to 4–M(CO)5 complexes (δW = 799 ppm (4a), δCr = 1001 ppm (4b)) [55,56] and the lowest to the M(CO)5 complexes of Sn(OC6H3(CH2NMe2)3)2 (6) (δW = −392 ppm (6a), δCr = −213 ppm (6b)) [57]. Concerning the iron tetracarbonyl complexes, the strongest deshielding is found for the non-stabilised highly crowded stannylene (2-tBu-4,5,6-Me3C6H)2Sn–Fe(CO)4 (δFe = 1059 ppm (7a)) [58] and the strongest deshielding for 6c (δFe = −226 ppm) [57], in analogy to the group-6 complexes.
A similar relationship has also been found for phosphine ligands [46]. It has been concluded that besides π-back bonding, steric bulk may induce negative coordination deshielding [46,49]. That means that steric parameters may have measurable impact on the electronic environment of the donor atom (tin or phosphorus).
2.3.5 Base dependency of the coordination chemical shift of B → R2Sn
Base addition to the stannylene tin atom increases its coordination number and electron density, both factors causing an upfield shift of the 119Sn chemical shift [35,38] (Fig. 7). With Sn(IV) complexes, these effects were explained mainly by TM polarisability [59,60]. Increased shielding is often referred to increased coordination number [35] or bond strength [61]. However, it heavily depends on the electronic nature of the R fragment, which adds to tin. Thus, addition of a base B (solvent) or auto-association may shield the 119Sn nucleus by more than 150 ppm. On the other hand, addition of an electropositive element increases the coordination number but decreases the electron density. The general effect here is a deshielding due to withdrawing of electron density (see above) [28]. This has been observed for example with the Veith stannylene (1). The 119Sn NMR of the compound in neat d6-benzene appears at δ = +629 ppm and is shifted at δ = +376 ppm when THF is added. In addition, the complexation of this stannylene to pentacarbonyl fragment M(CO)5 (M = Cr (1b), W (1a)) led to the isolation of a crystalline compound that has been identified by NMR and crystallography as THF·Me2Si(NtBu)2Sn–M(CO)5 [27]. Very recently, studies on benzimidazolin-2-stannylenes C6H4(NR)2Sn bearing functional R side chains (R = alkyl, amine, alkoxy) have shown that their 119Sn NMR shifts compared to that of the corresponding stannylenes without side chains were strongly influenced for ether but only slightly for amine functionalities increasing the polarity of the solvent. This means that the ether side chains interact only weakly with the stannylene tin atom, being replaced by polar solvent molecules, whereas the amine sticks to tin in either solvent [62].
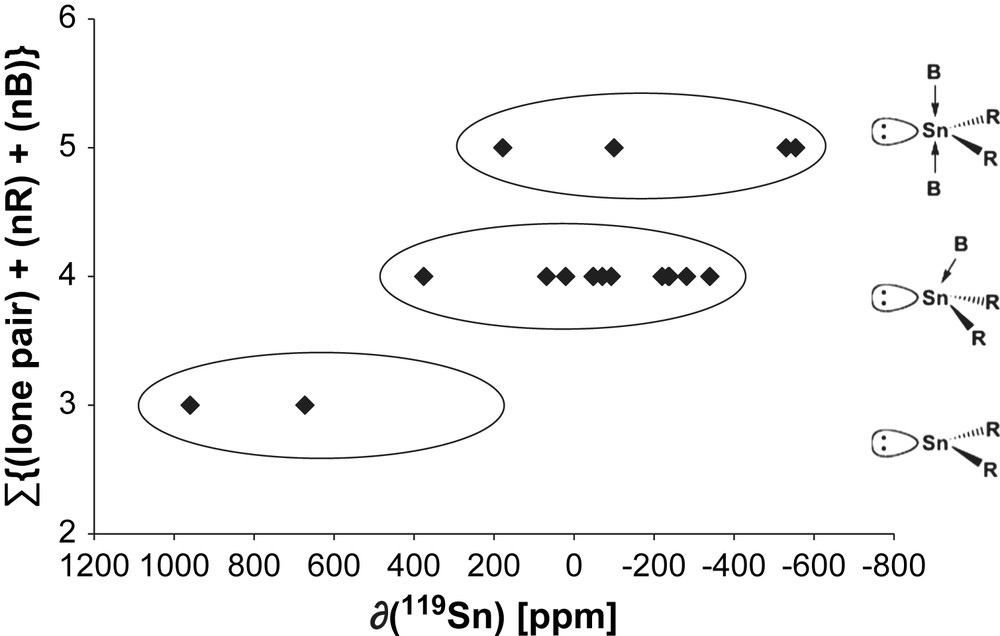
Relationship between coordination number (C.N.) of a free stannylene and its chemical shift δ (119Sn) [ppm].
The influence of the base on the coordination chemical shift has earlier been investigated by Du Mont in a series of B → X2Sn ligands, discussing the influence of the nature of X (chlorine or bromine) and B (phosphine or THF) on Δδ [59]. In the case of B → Cl2Sn–W(CO)5 (B = Et3P, Bu3P, tBu3P), the two linear alkyl phosphines Et3P (Δδ = +42 ppm) and Bu3P (Δδ = +57 ppm) lead to a positive Δδ, but the tBu3P base (Δδ = −18 ppm) to a negative one. The influence of the phosphine can be considered as a superposition of electronic and steric factors. Indeed, the steric demand of the phosphines, expressed by their corresponding Tolman angle, correlates quite nicely with this behaviour. The highly hindered tBu3P (Tolman angle 182°) acts as a less efficient donor than Et3P and Bu3P (132°) [63]. Assuming that the same molecular orbitals are engaged in base stabilisation and π-back bonding from the TM, the lower electron density around the tin atom for the tBu3P adduct may be more than compensated by π-back bonding. The negative Δδ for tBu3P·SnCl2–W(CO)5 would therefore be due to steric reasons, which have electronic consequences. In the respective chromium complexes B → Cl2Sn–Cr(CO)5 (B = Et3P, Bu3P), since π-back bonding should be weaker for chromium than for tungsten complexes [18], the difference is more pronounced (Et3P: Δδ = +276 ppm; Bu3P: Δδ = +317 ppm) [38]. THF as base further increases the Δδ for chromium complexes (Δδ = +429 ppm) [59].
2.3.6 Charged complexes
2.3.6.1 Closo-stannaborane complexes.
The stanna-closo-dodecaborate cluster (SnB11H11)2− (8), has been coordinated to a huge number of TM fragments as summarised in Table 2. The compound is described as a strong σ-donor, which is reflected in positive Δδ values. In addition, it could be qualitatively expressed that the Δδ value of the metallic fragments increases in the following order:
Ir+III (d6 Oh) ≈ Ir+I (d8 Oh) << Cd+II (d10 Td) < Hg+II (d10 Td) ≈ Ni+II (d8 Oh) ≈ Pt+II (d8 planar) < Mo+IV (d2 “Td”) < Fe+II (d6 “Td”).
119Sn NMR parameters of (SnB11H11)TM complexes.
| Complex | δ (ppm) | Δδ (ppm) | JSn–M | Ref. |
| (SnB11H11)2− (8) | −546 | [8] | ||
| (Et3NMe)[Ir+IIIH2(SnB11H11)2(CO)(PPh3)2] | −531 | +15 | [64] | |
| (Me4N)[Ir+III(SnB11H11)2(CO)(C2H4)(PPh3)2] | −530 | +16 | [64] | |
| (Me3NH)6[Cd+II(SnB11H11)4] | −377 | +169 | [65] | |
| (Me3NH)6[Hg+II(SnB11H11)4] | −320 | +226 | [65] | |
| [Ni+II(SnB11H11)6]8− | −319 | +227 | 2JSn–Sn: cis 1930; trans 13,490 | [66] |
| (Bu3MeN)6[Pt+II(SnB11H11)4] | −317 | +229 | 1JPt–Sn = 14,000 Hz | [67] |
| (nBu4N)[(C7H7)Mo+IV(CO)2(SnB11H11)] (8k) | −257 | +289 | [68] | |
| (nBu4N)[CpFe+II(CO)2(SnB11H11)] | −208 | +338 | [68] | |
| (Et3NMe)2[Ir+IH(SnB11H11)2(CO)(PPh3)2] | −502 | +44 | [64] | |
| (Et4N)[Ir+III(SnB11H11)2(CO)(PPh3)2] | −500 | +46 | [64] | |
| (Me4N)3[Ir+I(SnB11H11)2(CO)(PPh3)2] | −497 | +49 | [64] |
It appears that the spectroscopic behaviour is influenced by different factors: the geometry of the complex, the oxidation state and the nature of the metal.
2.3.6.2 Trisamido stannato–TM complexes
The trisamidostannates [MeSi(SiMe2NR)3Sn]− (R = p-tolyl (9); 3,5-xylyl (10)) synthesised by Gade and co-workers have shown a pronounced nucleophilic reactivity and several corresponding TM complexes have been isolated. Some of them, especially containing Sn–Rh and Sn–Ir moieties, have been characterised by 119Sn NMR spectroscopy (see Table 3). A determination of the coordination deshielding is impossible here since the “free” stannylenes are stannates, which exist in the solid state with Na+ or Li+ counter ions coordinated to two amido arms. To start from a parent compound, it would be necessary to have separated ions at least in solution.
119Sn NMR shifts [ppm] and 1JRh–Sn coupling constants [Hz] of MeSi(SiMe2NR)3Sn–RhL∗(L3) and MeSi(SiMe2NAr)2(SiMe2N(η6Ar))Sn–RhL∗ complexes (R = p-tol (9) or 3,5-xyl (10), L∗ = phosphine, isonitrile, L3 = η6-benzene, η6-toluene, η4-NBD, η4-COD).
| MeSi(SiMe2NR)3Sn–RhL∗(L3) | |||||||
| R | L∗ | L3 | |||||
| NBD | COD | η6-C6H5-R | |||||
| δ (ppm) | 1JRh–Sn | δ (ppm) | 1JRh–Sn | δ (ppm) | 1JRh–Sn | ||
| p-tol (9) | PEt3 | −111.7 [69] | 925 | −140.5 [69] | 846 | ||
| PiPr3 | −149.5 [69] | 928 | −187.4 [69] | 829 | |||
| PPh3 | −147.7 [70] | 910 | −176.5 [70] | 806 | R = H −179.7 [70] | 1170 | |
| R = Me −182.8 [70] | 1172 | ||||||
| CNtBu | −103.1 [69] | 910 | −112.2 [69] | 832 | |||
| CNCy | −113.0 [69] | 831 | |||||
| P(OPh)3 | −146.3 [69] | 790 | |||||
| 3,5-xyl (10) | PEt3 | −113.0 [69] | 934 | −146.4 [69] | 858 | ||
| PiPr3 | −192.8 [69] | 831 | R = Me −233.1 [69] | 1331 | |||
| PPh3 | −146.4 [70] | 910 | −176 [69] | 802 | R = H −175.5 [70] | 1159 | |
| R = Me −175.4 [70] | 1165 | ||||||
| CNtBu | −102.2 [69] | 910 | −113.9 [69] | 839 | |||
| CNCy | −115.1 [69] | 838 | |||||
| P(OPh)3 | −152.7 [69] | 1048 |
The 119Sn NMR data of the complexes MeSi(SiMe2NR)3Sn–RhL∗(L3), (L∗ = phosphine, isonitrile, L3 = η6-benzene, η6-toluene, η4-NBD, η4-COD) are summarised in Table 4. It is observed that the 119Sn NMR shift is much more determined by the nature of L3 than by R, which seems to be either too similar or too far from the tin atom [69,70]. The 119Sn chemical shift moves upfield in the following order: δ(NBD) > δ(COD) > δ(η6-C6H5R). The more negative 119Sn NMR shifts for the aromatic ligands are in accord with the higher formal electron count (18 VE compared to 16 VE in diene complexes). Besides, NBD and COD are better π-acceptors, increasing the electropositive character of Rh. For the dienes, steric reasons could additionally be invoked to understand the changes in chemical shift. The regularity of the variation from NBD- to COD-containing complexes could allow for the careful prediction of 119Sn NMR chemical shifts of complexes hitherto not measured with NMR.
119Sn NMR shifts δ [ppm] and coordination chemical shifts Δδ [ppm] of stannylene carbonyl TM complexes.
| Stannylene (R2Sn) | R2Sn | R2Sn–Cr(CO)5 | R2Sn–Mo(CO)5 | R2Sn–W(CO)5 | R2Sn–Fe(CO)4 | R2Sn–Ni(CO)3 | (R2Sn)2W(CO)4 | R2Sn–W(CO)4PPh3 | (R2Sn)2Fe(CO)3 | ||||||||
| δ | δ | Δδ | δ | Δδ | δ | Δδ | δ | Δδ | δ | Δδ | δ | Δδ | δ | Δδ | δ | Δδ | |
| SnCl2·P(tBu)3 [59] | 21 | 2.9 | −18 | ||||||||||||||
| SnCl2·PPh3 [59] | 238 | −15.1 | |||||||||||||||
| SnCl2·PEt3 [59] | −47.5 | 228.5 | 276 | −5.8 | 42 | ||||||||||||
| SnCl2·PBu3 [59] | −48 | 269 | 317 | 9.5 | 57 | ||||||||||||
| SnCl2·THF [59] | −236 | 193 | 429 | −54.6 | 181.4 | ||||||||||||
| SnCl2·THFx [59] | −238 | 55 | 293 | −209.4 | 19 | ||||||||||||
| SnBr2·THF [59] | −70.6 | −2.6 | 68 | ||||||||||||||
| SnBr2·THFx [59] | −217.6 | ||||||||||||||||
| SnRR′a (4) [55,56,71,72] | 960 | 1001 | 41 | 928.5 | −32.5 | 799 | −161 | 889 | −71 | 956 | −4 | ||||||
| Sn(tBuMe3C6H)2 (7) [58] | 673 | 1059 | 386 | 1055 | 382 | ||||||||||||
| Sn(C6H4-CH2PPh2)2 (35) [73] | −7.49 | ||||||||||||||||
| Sn(C10H6-NMe2)2 (17) [74] | 178.3 | 248.7 | 70 | ||||||||||||||
| Sn(C6H4-CH2NMe2)2 (26) [73] | 169 | 195.4 | 26.4 | ||||||||||||||
| Sn(iPr3C6H2)2 (41) | 1483 | ||||||||||||||||
| Sn{(PhNC(Me))2CH}Cl (40) [75,76] | −281.18 | 141.68 | 422 | −84.33 | 196.85 | 80 | 361.18 | ||||||||||
| Sn(Cl){(4-tBu-2,6-[P(O)(OEt)2]2-C6H2)] (11) [77] | −100 | 131 | 231 | −74 | 26 | 54 | 154 | ||||||||||
| Sn(F){(4-tBu-2,6-[P(O)(OEt)2]2-C6H2)] [77] | 43.2 | ||||||||||||||||
| Sn(Cl){(tBuOCH2)2C6H3} (12) [61] | 206 | 257 | 51 | ||||||||||||||
| Sn(Cl){(MeOCH2)2C6H3} (13) [61] | 231 | 197 | −34 | ||||||||||||||
| [Sn(OtBu)2]2 (14) [78] | −93 | 127.7 | 220.7 | −70.5 | 22.5 | 36.37 | 129.67 | 79.3 | 172.3 | ||||||||
| −108.3 | −117.1 | ||||||||||||||||
| [Sn(OSiPh3)2]2 [78] | −339.5 | −303 | −36 | ||||||||||||||
| [Sn(OSiMe3)2]2 [78] | −220 | 74.2 | 294.2 | 35.3 | 255.3 | ||||||||||||
| 24.0 | 244.0 | ||||||||||||||||
| [Sn(OtBu)(OSiPh3)]2 [79] | −225.08 | −24.13 | 201.05 | ||||||||||||||
| Sn(Salen) (2) [23–26] | −554 | −137 | 417 | −328 | 226 | −196 | 358 | −348 | 206 cis | −347.5 | 206.5 | ||||||
| −374 | 180 trans | ||||||||||||||||
| Sn(OC6H3(CH2NMe2)3)2 (6) [57] | −530 | −213 | 317 | −392 | 138 | −226 | 304 | ||||||||||
| Sn(OH)2·THFx[80] | 9.5 | −198 | |||||||||||||||
| Sn(SCH2CH2)2NtBu (5) [81] | 68.9 | 622.8 | 553.9 | ||||||||||||||
| Sn(NtBu)2SiMe2·THF (1·THF) [27] | 376 | 401 | 25 | 258 | −118 |
a R = 2,4,6-tBu3(C6H2); R′ = 3,5-tBu2(C6H3)C(CH3)2CH2.
Because of the negative charge on tin, in these complexes the stannylenes may act as strong (close to pure) σ-donor ligands.
2.4 Coupling constants
Direct (1J) Sn–TM and geminal (2J) 119Sn–117/119Sn scalar coupling constants provide further insight into the kind of bonding in R2Sn–TM complexes. As was pointed out before, simplistic models developed mainly for proton and carbon coupling may not be sufficient to account for the observed variations of coupling constants. Nevertheless, we try to give representative examples for classes of stannylene ligands providing direct coupling constants with NMR active TM isotopes and briefly discuss geminal coupling constants in (R2Sn)2TM bis-stannylene complexes. 119Sn–TM coupling constants in stannylene complexes have been reported for magnetically active TMs such as 103Rh, 183W and 195Pt, which at the same time are favourable spin-1/2 nuclei. The variation with TM moiety and Sn–substituents provides an additional probe for gaining further insight into Sn–TM chemical bonding. The presence of signals 119Sn NMR spectrum arising from TM coupling is a very practical indicator to know if/how the stannylene is bonded to the TM. The relative magnitude of the 1JSn–TM coupling constant in general increases in the order TM = Rh < W < Pt, which follows the absolute products of the gyromagnetic constants for 119Sn with 103Rh, 183W and 195Pt (8.499; 11.118; 57.272 × 1014 rad2 T−2 s−2). That points to similar coupling mechanisms, hence bonding situations between the stannylene ligand and the respective TM. The values for the two silver complexes do not fit into this row, their constants appear to be higher values than expected from the product of the gyromagnetic ratios (10.802 1014 rad2 T−2 s−2).
2.4.1 1J (119Sn–183W) coupling constants
The magnitude of the coupling constant is considerably higher for end-on coordinated stannylene ligands compared to e.g. stannyl ligands (covalent σ-bonding, TM moiety as substituent). For example, the triphenyl stannyl ligand in [{Cp(OC)2W}(P(OPh)(NMeCH2)2)(SnPh3)] exhibits a 1J (119Sn–183W) coupling constant of 404.6 Hz, whereas in the corresponding stannylene complex [{Cp(OC)2W}(cyclo-P(Ph)(NMeCH2)2)(SnPh2)(OTf)] (16a), the value rises up to 610.1 Hz. However, compared to other coupling constants found in stannylene complexes, this value seems low (see Table 5). Another example for small 1J (119Sn–183W) coupling constants is provided by the metallostannylene ligand in [{(OC)5Cr}(Sn{WCp(CO)3}(4-tBu-2,6-P(O)(OEt)2)2-C6H2)] with only 131 Hz [82]. In the respective ferrocenyl bridged complex [{(OC)5W(Sn(4-tBu-2,6-(P(O)(OEt)2)2-C6H2)C5H4}2Fe] (15a), 1099 Hz is measured [83]. The coupling constant with stannyl ligands strongly depends on substituents. In the series of substituted methylchlorostannyl complexes [{Cp(OC)3W}SnMe3−nCln] it increases with the chlorine substitution degree (n = 0: 150.5 Hz; n = 2: 596.8 Hz) [84]. Similar tendencies are found for rhodium [85] and platinum [86] complexes. The large coupling constants for the stannylene complexes can be explained by the Fermi contact mechanism, which implies correlation of the magnitude of the coupling with the s-character of the bond forming atomic orbitals. In general, the observed 1J (119Sn–183W) coupling constants for stannylene complexes range from 500 to 1700 Hz. At the lower end of the scale, a “non-classical” stannylene is found in form of a dimeric “inidene” like W–Sn–W three-centre bond and may therefore be seen as exception. The low value for the cationic complex 16a may be due to special bonding mechanisms at the 15 VE tungsten fragment. “Classical” stannylene bonding situations are found with aryl/alkyl, halogeno, chloro, sulfido and alkoxo/siloxo substituents (see Table 5). The carbon based substituents exhibit small coupling constants between 800 and 1100 Hz. Replacing one aryl ligand in e.g. 15a by chlorine raises the coupling constant to 1372 Hz in [{(OC)5W}(SnCl(4-tBu-2,6-(P(O)(OEt)2)2-C6H2)] (11a) [77]. Sulfido stannylenes have constants between 1200 and 1320 Hz, the amido stannylene complex 1 1204 Hz, the base coordinated halogeno (Cl, Br) stannylenes between 1350 and 1610 Hz, salen-stannylenes around 1450 Hz. Compared to alkoxy stannylenes, which show coupling constants between 1460 and 1660 Hz, the aryloxo stannylene complex 6a exhibits a weak coupling of 1010 Hz [57]. The highest reported value (to the best of our knowledge) is found for the siloxo stannylene compound [{(OC)5W}(Sn(OSiPh3)2)] with 1660 Hz [78]. Considering the strong couplings with oxo and halogenido stannylenes, the value with the mixed dimeric stannylene ligand in [{(OC)5W}(Sn(μ-OtBu)(Cl))2] is unexpectedly low (1192 Hz) [27].
δ (119Sn) and 1J (119Sn–183W) of selected R2Sn–W complexes group according to substituents on tin.
| Compound | δ (119Sn) [ppm] | 1J (119Sn–183W) [Hz] | Ref. |
| [{(OC)5W}4(Sn2(μ-OEt)2)] (nBu4N/PPh4)b | 1169/1169 | 540/523 | [87] |
| [{Cp(OC)2W(cyclo-P(NMeCH2)2)}(SnPh2(OTf))] (16a) | 335.32 | 610.1 | [88] |
| [{(OC)5W}Sn(2,4,6-iPr3C6H2)2] (41a) | 1483 | 838 | [89] |
| [{(OC)5W}Sn(o-C6H4(CH2PPh2))2] (35a) | −7.49 | 894 | [90] |
| [{(OC)5W}Sn(2,4,6-tBu3C6H2)(CH2C(CH3)2-3,5-tBuC6H3)] (4a) | 799 | 940 | [56] |
| [{(OC)5W}Sn(8-Me2NC10H6)2] (17a) | 248.7 | 976 | [74] |
| [{{(OC)5W}(Sn(4-tBu-2,6-P(O)(OiPr)2)2C6H2)C5H4}2Fe] (15a) | 88 | 1099 | [83] |
| [{(OC)5W}Sn(MeOCH2)2C6H3(Cl)] (13a) | 231 | 1249 | [61] |
| [{(OC)5W}Sn (tBuOCH2)2C6H3(Cl)] (12a) | 206 | 1289 | [61] |
| [{(OC)5W}(SnCl(4-tBu-2,6-P(OEt)2O)2-C6H2)] (11a) | −74 | 1372 | [77] |
| [{(OC)5W}2(Sn(Cl)(μ-OtBu))2] | 226 | 1192 | [27] |
| [{(OC)5W}Sn(SC2H4)2NtBu] (5b) | 380.2/196.3c | 1196.0/1285.5c | [14] |
| [{(OC)5W}Sn(SC2H4)2O] | 71.5c | 1259.8c | [14] |
| [{(OC)5W}Sn(SC2H4)2S)] | 39.8c | 1262.0c | [14] |
| [{(OC)5W}Sn(SC2H4)2NMe] | 43.6c | 1300.0c | [14] |
| [{(OC)5W}Sn(SC2H4)2NMe)]cpy | 73.0c | 1304.0c | [14] |
| [{(OC)5W}Sn(SC2H4)2O)] | 69.6c | 1313.9c | [14] |
| [{(OC)5W}Sn(THF)(NtBu)2SiMe2)] (1a) | 227 | 1204 (C6D6/THF) | [14] |
| [{(OC)5W}SnCl2(PEt3)] | −5.8 | 1350 | [91] |
| [{(OC)5W}SnCl2(THF)] (18a) | −54.6 | 1440 | [91,92] |
| [{(OC)5W}SnBr2(THF)] | −2.6 | 1440 | [91] |
| [{(OC)5W}SnCl2(PtBu3)] | 2.6 | 1470 | [91] |
| [{(OC)5W}SnCl2(PnBu3)] | 9.5 | 1490 | [91] |
| [{(OC)5W}SnCl2(THF)2] (18b) | −209.4 | 1594 | [91,92] |
| [{(OC)5W}SnBr2(THF)x] | −217.6 | 1610 | [91] |
| cis-[{(OC)4W}(SnSalen)2] (cis-2c) | −348 | 1456 | [26] |
| [{(OC)5W}(SnSalen)] (2a) | −328 | 1458 | [26] |
| [{(OC)5W}(Sn(2,4,6-(Me2NCH2)C6H2O)] (6a) | −391.2 | 1010 | [57] |
| [{(OC)5W}(Sn2(OtBu)4)] (14a) | −70.5 | 1463 | [15] |
| [{(OC)5W}Sn(OC2H4NMe)2] | −208.2a | 1483.2 | [14] |
| [{(OC)5W}2(Sn2(OtBu)4)] (14c) | −92.4 | 1485 | [15] |
| [{(OC)5W}{(OC)5Cr}(Sn2(OtBu)4)] (14b) | −84.0 | 1492 | [15] |
| [{(OC)5W}Sn(OC2H4)2NtBu] | −248.6a | 1535.6 | [14] |
| [{(OC)5W}Sn(OSiPh3)2] | −303 | 1660 | [78] |
a Autodimerisation.
b μ-Sn(OEt)2.
c In pyridine.
The rising coupling constants with increasing electronegativity of the substituents on tin (C < S < Cl/Br < O) is in accordance with an increasing s-character for more electronegative substituents. For B → X2Sn–W(CO)5 complexes, it has been found that increasing the number of bases (X = Cl (18): B = THF: 1440, (THF)2: 1594 Hz) or the basicity (X = Cl, B = PEt3: 1350, B = PtBu3: 1470; B = PnBu3: 1490 Hz) increases the direct 183W–119Sn coupling constant [59]. With increasing electron density on tin – equivalent to becoming less electronegative – according to Bent's rule, the p-percentage in the Sn–R bonds increases, leaving more s-character to the lone pair. Both effects lead to an increasing Fermi contact. In the dimeric alkoxo stannylene complexes [{(OC)5TM}n{(OC)5W}Sn2(OtBu)4] (n = 0 (14a); n = 1, TM = Cr (14b), W (14c) [15,27], systematic investigations show a relationship between 1JSn–W coupling constants and structural parameters: 1JSn–W increases with the number of coordinated TMs and decreasing period (n = 1 (14a): 1463 Hz; n = 1, TM = W (14c): 1485 Hz; n = 1, TM = Cr (14b): 1492 Hz).
2.4.2 1J (119Sn–103Rh) coupling constants
The 103Rh isotope is extremely useful because of its 100% natural abundance. Thus, although the 119Sn signal multiplicity provides direct information on the number of coupled TM nuclei, experimental values are unfortunately scarce still. Most examples reported for stannato complexes might not be representative for the Rh–Sn(II) bonding (see Table 6). A small direct coupling constant with terminal stannylene ligands is found for the chloro adduct of Lappert's stannylene 3 (497 Hz), higher values are found for adducts to the ligand Sn(N(SiMe3)2)2 (19) (560–928 Hz). The anionic tris-stannaborato complex 20a exhibits an intermediate constant of 760 Hz [93]. In the R2Sn–Rh complex series, the influence of the Rh fragment on the Sn–Rh coupling and therefore on the bond itself becomes evident. While weak coupling is observed for complexes with phosphines as co-ligands (J = 497–760 Hz), the more electron deficient COD-containing Rh complexes exhibit considerably higher coupling constants (815–928 Hz).
δ (119Sn) and 1J (119Sn–103Rh) of selected Rh stannylene complexes. Cp∗Rh complexes with formal hybridisation states2 of Rh and Sn and coordination chemical shifts Δδ in brackets.
| Compound | δ (119Sn) [ppm] | 1J (119Sn–103Rh) [Hz] | Ref. |
| cis-[RhCl(PPh3)2{Sn(CH(SiMe3)2)2}2] | 434 | 497 | [7] |
| trans-[Rh(CO)(PPh3)2{SnCl(N(SiMe3)2)2}2] | −60 | 560 | [7] |
| [Rh(CO)(dppe){SnClN(SiMe3)2)2}2] | 36 | 739 | [7] |
| trans-[Rh(PPh3)2(SnCB10H11)3]2− (20a) | −276 | 760 | [93] |
| [Rh(cod)(CN(Me)C2H4NMe){SnMe(N(SiMe3)2)2}2] | 31 | 815 | [7] |
| [Rh(cod){μ-Sn(N(SiMe3)2)2}2{SnCl(N(SiMe3)2)2]2 | 144/118 | 840/650 | [7] |
| [Rh(cod){Sn(N(SiMe3)2)2}2(μ-Cl)] | −4 | 860 | [7] |
| [Rh(cod)(PEt3){SnMe(N(SiMe3)2)2}2] | −12 | 928 | [7] |
| [{Cp∗Rh}2(μ-CO)2(μ-Sn(NtBu)2SiMe2)] (dsp3–sp3; Δδ = −125 ppm) | 504 | 382 | [94] |
| [{Cp∗Rh}2(μ-CO)2(Sn(NtBu)2SiMe2)] (dsp3–sp2; Δδ = −242 ppm) | 371 | 470 | [94] |
| [{η3-Cp∗(OC)2Rh}(Sn(NtBu)2SiMe2)] (sp3–sp2; Δδ = −641 ppm) | −22 | 809 | [94] |
| [{η5-Cp∗(OC)Rh}(Sn(NtBu)2SiMe2)] (sp2–sp2; Δδ = −604 ppm) | 25 | 928 | [94] |
An instructive example for the successful application of heteronuclear NMR spectroscopy coupling information for the elucidation of the reaction sequence is provided by a series of Cp∗Rh containing complexes synthesised from Veith's stannylene 1 with Cp∗Rh(CO)2 (monomeric or dimeric) (see Fig. 8) [94]. The reaction in solution yields monomeric R2Sn–[Rh] complexes or R2Sn–[Rh2] complexes. All complexes could be clearly distinguished and assigned to defined structures by the multiplet structure of the 119Sn resonances and magnitude of the coupling constants. Taking the Fermi contact as the determining factor, the coupling constant of the four new stannylene complexes increases with the participation of s-orbitals in the Rh–Sn bond (see Table 6). In the bridging dirhodium stannylene complex [{Cp∗Rh(μ-CO)}2(μ-1)] 381.5 Hz has been recorded. Formally, sp3 hybridisation can be assigned to tin (25% s-orbital) and dsp3 hybridisation on Rh (fivefold coordination; 20% s). In the unsymmetrically coordinated dirhodium complex [{Cp∗Rh(μ-CO)}2(1)] with a terminal stannylene (sp2, 33% s; Rh: dsp3), 470.5 Hz is observed, while in the two mono-rhodium compounds [{(η3-Cp∗)Rh(CO)2}(1)] (Rh: sp3) and Cp∗Rh(CO)(1) (Rh: sp2) 808.7 and 927.7 Hz are measured. In fact, the unstable η3-Cp∗ complex, which for phosphines and carbene complexes is only postulated on kinetic and theoretical data [95], has been identified with the help of spectroscopic investigations.
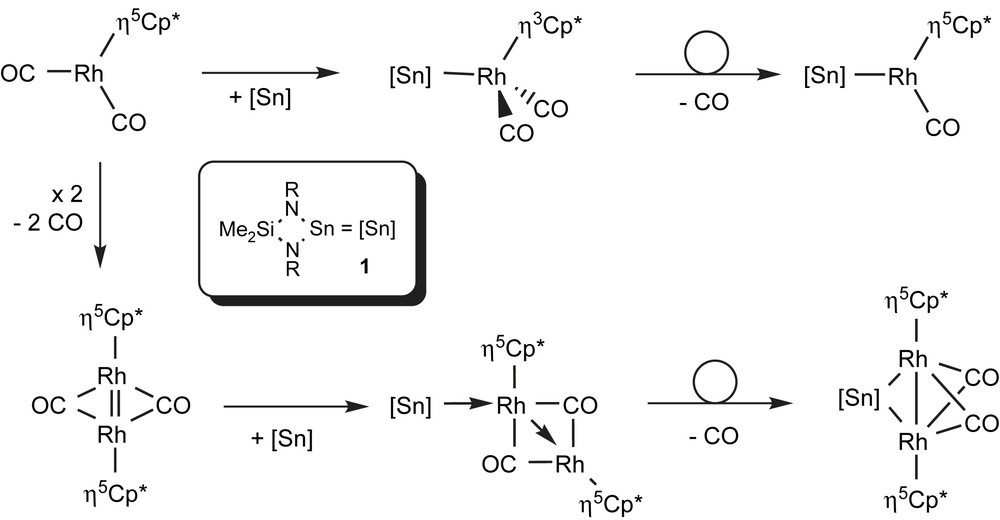
Scheme of the reactions in the [Cp∗Rh(CO)n]m/Sn(NtBu)2SiMe2 (1) system (n = 1, 2) [94].
It is observed that the NBD- and COD-containing 16 electrons 9/10–Rh complexes have smaller 1JSn–Rh coupling constant than the respective 18e electron (η6-C6H5R) Rh complexes in trisamido stannato complexes MeSi(SiMe2NR)3Sn–RhL∗(L3) [69,70].
Unfortunately, there are not enough data available to generalise the findings for rhodium complexes.
2.4.3 1J (119Sn–195Pt) coupling constants
Direct 119Sn–195Pt coupling constants are very large, usually found between 12,900 and 28,000 Hz for stannylene complexes (see Table 7). The tendencies as found for tungsten and rhodium are less evident. Phosphine complexes seem to lead to weaker coupling. The value for the diphosphine mono-distannylene complex 21a (8400 Hz) is surprisingly low compared to the much stronger couplings of the corresponding bis-distannylene complexes 21b and 21c (22,050 and 27,874 Hz, resp.). The strongest coupling is found for the homoleptic tris-stannylene complex [Pt{Sn(N(SiMe3)2)2}3] (19a, 27,874 Hz) [96].
δ (119Sn), 1J (119Sn–195Pt) of selected Pt–stannylene complexes.
| Compound | δ (119Sn) [ppm] | 1J (119Sn–195Pt) [Hz] | Ref. |
| [Pt(PPh3)2{Me2C(CH2NC6H4N(CH2tBu)Sn)2}] (21a)a | 239.1 | 8400 | [97] |
| [Pt(PPh3)3(Sn(acac)2)] | −601 | 12,891 | [98] |
| [Pt{SnB11H11}4]6− (8a) | −317 | 14,000 | [67] |
| [Pt{Sn(NtBu)2SiMe2}4(μ-Cl)2] (1g) | 7.17 | 14,037 | [99] |
| [Pt(PPh3)2{Sn(N(SiMe3)2)2}2] | 963 | 16,650 | [7,100] |
| [Pt(PPh3)2{2,4,6-(Me2NCH2)3C6H2O)2Sn}] (6d) | −8.39 | 17,901 | [57] |
| [Pt{CMe2(CH2NC6H4N(CH2tBu)Sn)2}2(thf)3] (21b) | 197.0 | 22,050 | [97] |
| [Pt{(m-C6H4(CH2NC6H4N(CH2tBu)Sn)2)2(thf)}] (21c) | 228 | 22,300 | [97] |
| [Pt{Sn(N(SiMe3)2)2}3] (19a) | 885 | 27,874 | [96] |
a thf solution: OPPh3 substituted.
2.4.4 1J (119Sn–109Ag) coupling constants
1J(107/109Ag–119Sn) constants have been reported occasionally: in the N-(propyl)-2-(propylamino)troponiminatotin(II)chloride tris(pyrazoyl)boratosilver(I) adduct 5234 Hz [101], and the respective azide 4866 Hz [102].
2.5 Conclusion of the NMR discussion
The δ (119Sn) chemical shift data and 1J (119Sn–TM) coupling constants for a variety of R2Sn–TM complexes have been collected and investigated to extract tendencies. For carbonyl complexes of groups 6 and 8, the coordination deshielding Δδ (119Sn) shows a roughly linear dependency on the chemical shift δ (119Sn) of the respective free stannylene depending on the TM moiety. The positive intercept and negative slope (the coordination deshielding is stronger for more negative chemical shifts of the free stannylene) parallel the behaviour found with phosphine ligands. For Sn(salen) ligands, a strong π-acceptor ligand trans to the stannylene causes strong deshielding. Than another stannylene ligand. Better correlations are found for the dependency of the chemical shift of the ligand δ (119Sn, TM–SnR2) versus that of the free stannylene δ (119Sn, SnR2). The values of the (positive) slope seem to be characteristic for the group, whereas those of the (positive) intercept seem to be characteristic for the period of the TM. Negative Δδ values for base-stabilised complexes show the influence of steric parameters on the chemical shift.
The coordination chemical shift may be regarded as being composed of several additional components. In one model, σ- and π-contributions may be separated (Δδ = Δδ(σ) + Δδ(π)), where Δδ(σ) comes from Lewis acid coordination and is always positive, whereas Δδ(π) denotes the partition of π-back bonding, which may be assumed to cause shielding of the 119Sn nucleus (negative Δδ). Some data support this model, especially the strong negative Δδ for aryl–alkyl stannylene complexes. However, as with phosphine ligands, where similar trends are observed, the explanation of negative Δδ values is not straightforward. In another model, Δδ may be regarded as being composed by participation from TM coordination, substituents R and bases B (Δδ = Δδ(TM) + Δδ(R) + Δδ(B)), which all show strong anisotropy. A small relative change in a strong contribution may alter completely the observed isotropic chemical shift in solution. Solid-state NMR and theoretical investigations are necessary to gain better insight into the mechanisms causing the NMR shift variations.
In the graphs depicting chemical shift data, a strict linear correlation of chemical shift data for all R2Sn–TM complexes is not possible. This is mainly due to solvents with very different polarity and donor strengths used during the NMR studies. Indeed, measurements in polar solvents can shift the δ appreciably towards lower values for the same stannylene, pointing to at least partial coordination in solution. Another factor of uncertainty is the chemical nature of the ligand R, which can be strongly different (π-donor, cluster, oligomerisation…), which renders stannylene complexes with different substituents difficult to compare.
The relative magnitudes of the coupling constants with respect to the TM in general increases in the order 103Rh < 183W < 195Pt, which follows the absolute products of the gyromagnetic constants. The direct coupling 1J (119Sn–TM) constant in stannylene complexes R2Sn–TM is higher than that found in stannyl complexes R3Sn–TM. It increases with several factors like substituent electronegativity, number of added bases and p-orbital participation in the Sn–R bonds. These findings are in accord with a strong determination of Sn–TM NMR coupling by the Fermi contact mechanism.
3 Solid state structures
3.1 Scope
In this part, we will give an overview of structural features of transition metal complexes with terminal stannylene ligands of general formula Bn → R2Sn–TM. The aim is to:
- - collect the range of reported Sn(II)–TM bond lengths depending on the TM,
- - discuss the influence of the variation of R substituents and base adduct formation (B) at tin on the Sn–TM bond,
- - discuss the impact of TM coordination on the stannylene geometry,
- - discuss the impact of the stannylene on the TM environment
- - discuss the TM-stannylene bonding compared to other two-electron donors like phosphines (Fig. 9).
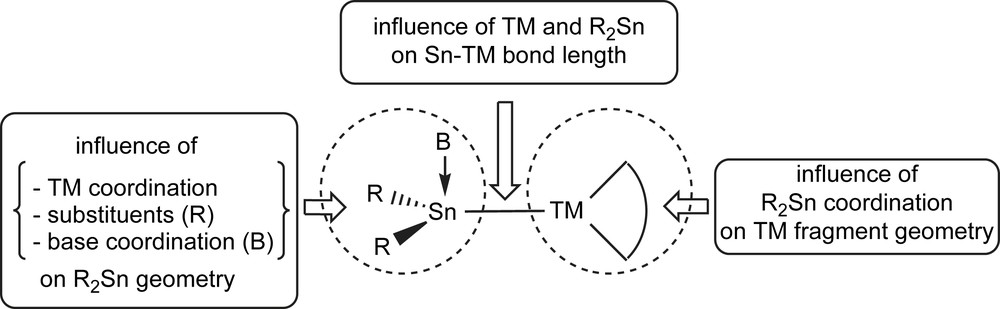
Scope of the structure discussion.
The “terminal” stannylene ligands included in this survey, are bis-alkyl, -amido, -chalcogenido (mainly alkoxo), -halogenido, -metallo stannylenes, trisamido stannates(II), porphyrino- and salen-stannylenes, both homo- and heteroleptic. We have also considered cluster compounds such as stannaboranes, Zintl-ions and multinuclear oxo-/hydroxo tin compounds, because they provide exohedral lone pairs of electrons to which TM moieties are prone to coordinate and therefore are regarded as terminal stannylenes as well.
Furthermore, complexes with bridging stannylene ligands and TM moieties other than those discussed above are regarded as substituents to the TM, not as ligands as, for example, the bridging SnCl2 ligand in [{(OC)(Cl)Ir}2(μ-dppb)2(μ-SnC12)] [103] or the tricyclic fragment in [{(OC)5W}Sn{W2(CO)10}] (22a) [104].
A certain ambiguity is connected with trihalogenido tin moieties (i.e. “X3Sn”) bound to TMs. Those moieties can be viewed from two points: either as neutral stannyl ligand SnX3, or as halogenido adduct to a dihalogenido stannylene (X → SnX2)−. The TM–Sn bond length for the radical in X3Sn–TM can formally be seen as covalent bond, while the stannylene (i.e. X− → X2Sn → TM) is better described as coordinative bond. One could expect clearly separated ranges of Sn–TM bond lengths for the two bonding modes, as seen for e.g. the Sn–N bond lengths in “covalent mode” (amido substituent) in Veith's stannylene (1) (d(Sn–N): NR2: 2.09 Å) [105] and the N-protonated derivative (“donor mode”, amine base adduct) [Sn(Cl){N(tBu)SiMe2N(tBu)(H)}] (NR3: 2.35 Å) [106]. Such separation is not found for most of the X3Sn–TM compounds. It might mean that either all complexes belong to the same bonding type or that the differences are not that pronounced. It might also be expected that the geometry around tin should be more tetrahedral for the SnX3 mode and more pyramidal for the (X → SnX2)− description. However, strong deviations from perfect tetrahedral or planarity (see for example in THF → Cl2Sn–W(CO)5 (18a) [92]) are found, reducing the usefulness of this argument. In the same way, other R3Sn (R = Ph [107], R2N [108]) moieties should be regarded, but are as well only discussed occasionally (Fig. 10).
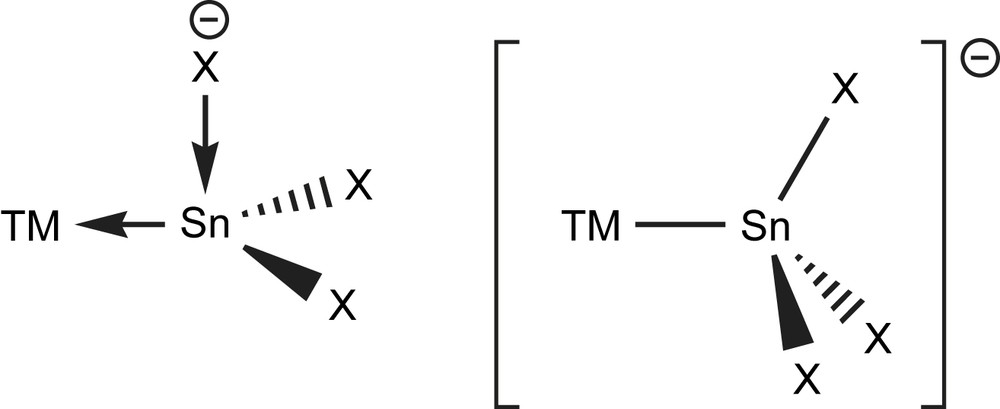
Formal description of the “stannylene” base adduct (X → SnX2)− (trigonal planar, left) and “stannyl” mode SnX3 (tetrahedral, right) of trisubstituted, negatively charged tin ligands.
3.2 Impact of the TM on the TM–Sn bond
3.2.1 Variation of the transition metal
The TM–Sn(II) coordinative bond length varies considerably with the period and group of the TM in the periodic table. The evolution of the TM–Sn(II) coordinative bond lengths with the transition metal is depicted in Fig. 11 (see also Tables 8 and 9) regardless of the spectator ligands on the TM, the substituent R or base B on tin.
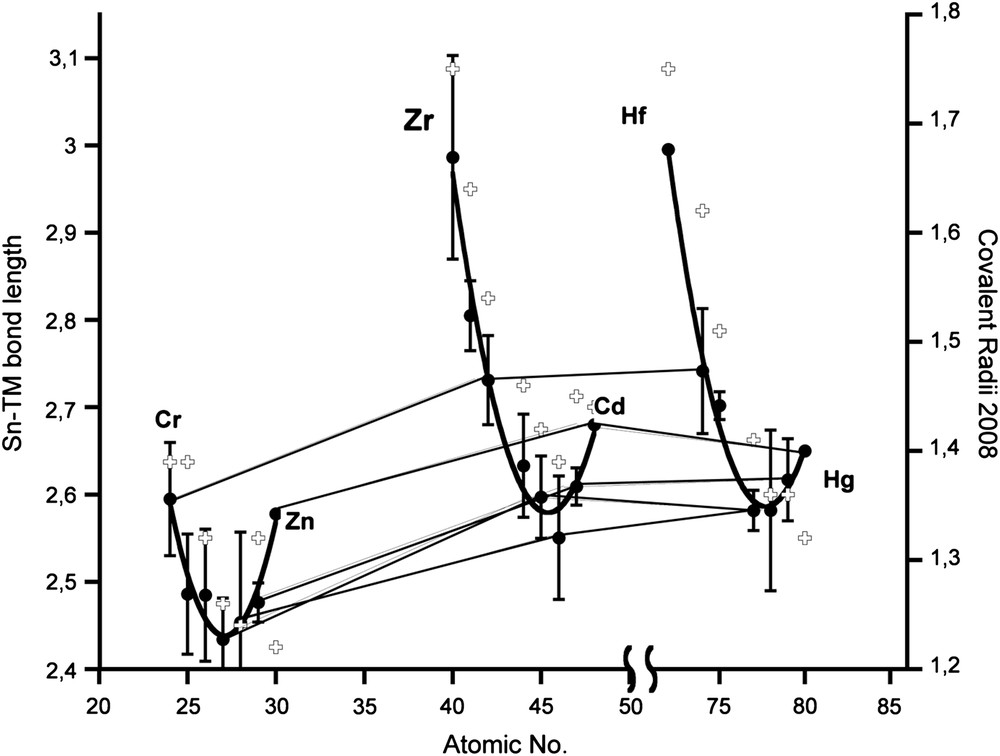
Variation of the TM–Sn bond length depending on the atomic number of the TM along with covalent radii (crosses) from Ref. [109]. Given are the mean values (dots) along with the respective longest and shortest bonds reported for each TM–Sn pair. A parabolic fit is given for each row. Values within the rows of groups 6, 9, 10, 11 and 12 are connected with solid lines between the periods. For statistical values see Table 8.
TM–Sn(II) bond lengths according to TM: average, minimum and maximum values with number of compounds in brackets (see also Fig. 11).
| Element (No. of Samples) | Ti | V | Cr (36) | Mn (9) | Fe (53) | Co (7) | Ni (14) | Cu (2) | Zn (1) |
| Distance [Å] Average | – | – | 2.595 | 2.507 | 2.485 | 2.434 | 2.454 | 2.477 | 2.578 |
| Min Max | 2.5302.654 | 2.4382.700 | 2.4092.587 | 2.3872.510 | 2.3502.544 | 2.4542.499 | |||
| Element (No. of Samples) | Zr (4) | Nb (2) | Mo (10) | Tc | Ru (10) | Rh (4) | Pd (8) | Ag (3) | Cd (1) |
| Distance [Å] Average | 2.987 | 2.805 | 2.731 | – | 2.633 | 2.597 | 2.551 | 2.608 | 2.680 |
| Min Max | 2.8703.039 | 2.7602.85 | 2.6802.770 | 2.5742.721 | 2.5502.633 | 2.4802.670 | 2.5882.660 | ||
| Element (No. of Samples) | Hf (1) | Ta | W (24) | Re (2) | Os | Ir (5) | Pt (11) | Au (3) | Hg (1) |
| Distance [Å] Average | 2.996 | – | 2.742 | 2.702 | – | 2.582 | 2.582 | 2.617 | 2.650 |
| Min Max | 2.6702.820 | 2.6862.718 | 2.5592.610 | 2.4902.640 | 2.5702.681 |
Shortest and longest TM–Sn distances in stannylene complexes according to the TM [in Å] (R = 2,4,6-tBuC6H2; R′ = CH2CMe2(3,5-tBuC6H3); R″ = ).
| TM | d(Sn–TM)min | Compound | d(Sn–TM)max | Compound | ||
| Cr | 2.513 [6], trans-SnR2 | [{(OC)3Cr}(Sn(NtBu)2SiMe2)3] | 1c | 2.654 [111] | [{(OC)5Cr}Sn(py)tBu2] | 23a |
| Mo | 2.68 [112] | [{Cp(OC)Mo}2(SnCl2)(μ-SnCl2)(μ-P(OEt)2)(P(O)(OEt)2)] | 18c | 2.77 [113] | [{(OC)5Mo}(Sn(μ-OtBu)3In)] | 24a |
| W | 2.67 [114] | [{(OC)5W}7{Sn7(μ3-OH)(μ3-O)3(OEt)3}]2− | 25a | 2.82 [115] | [{Cp(OC)3W}Sn(Cl)(o-NMe2C6H4)2] | 26a |
| 2.82 [74] | [{(OC)5W}Sn(8-(Me2N)C10H7)2] | 17a | ||||
| Mn | 2.428 [116] | [{Cp∗(OC)2Mn}6{Sn6O4(OH)4}] | 27a | 2.55(30) [117] | [cis-{(OC)5MnHgMn(CO)4}(porphyrine)] | 28a |
| 2.531 [72] | trans-[{(OC)4Mn}(RR′Sn)(SnR′R″)] | 4c | ||||
| Fe | 2.409 [118] | [{eq-(OC)4Fe}Sn(O-2,5-tBu2C6H3)2] | 29a | 2.587 [119] | [{(MeCN)2(triphos)Fe}(SnB11H11)] | 8b |
| Ru | 2.574 [120] | [{(Ph3P)2Ru}(μ,η3:1-SnB11H11)]2 | 8c | 2.721 [121] | trans-[{(OC)2(bipy)Ru}{Sn(N(p-tol)SiMe2)3SiMe}2] | 9a |
| Co | 2.39 [122] | [{Cp∗(η-C2H4)Co}Sn(2-tBu-4,5,6-MeC6H)2] | 7b | 2.51 [123] | trans-[{(CO)3Co}2{(Sn(Cl)(Co(CO)4)2}2]− | 30a |
| 2.39 [124] | [{Cp∗(η-C2H4)Co}Sn(CH(SiMe3)2)2] | 3b | ||||
| Ni | 2.35 [125] | [{CpNi}2(Sn(NtBu)2SiMe2)2] | 1d | 2.545 [126] | [{(dppm)Ni}(SnB11H11)3]4− | 8d |
| Pd | 2.48 [30,31] | [{(dipe)Pd}(Sn(CH(SiMe3)2)2] | 3a | 2.67 [30,31] | [{(dipe)Pd}(μ,η1:1-C2H4)(Sn(CH(SiMe3)2)2] | 3c |
| 2.578 [67] | cis-[{(dppp)Pd}(SnB11H11)2]2− | 8e | ||||
| Pt | 2.49 [127] | [Pt{Sn(N(SiMe3)2)2}3] | 19a | 2.64 [128] | trans-[{(Et3P)2(tBuNC)Pt}(SnB11H11)2]2− | 8f |
The values roughly follow the recently published tendencies for the covalent bond length in TMs as deduced from CSD data base analysis [109]. Strong deviations are found for early and late 5d TMs. However, due to the small number and limited variation of the periphery (ligands on TM and substituents R in R2Sn), a general behaviour for those TM complexes cannot be deduced. The variation with group number in each period follows a parabolic profile with minima at groups 9–11. A bimodal distribution as found for the 3d TMs of groups 7–9 in (Mn, Fe, Co) cannot be proved for stannylene complexes, which may be due to the small number of complexes [109].
Within one group, the TM–Sn bond length evolution follows the known tendencies for TMs: from 3d to 4d TM, the bond length increases between 4% and 6% and remains basically the same on going from 4d to 5d TM (1% and below), which is due to the lanthanide contraction [110].
The range of the Sn–TM bond length for each TM is listed in Table 8, examples for the extreme values in Table 9 for R2Sn–TM complexes with more than 6 entries. The biggest difference Δd between minimum and maximum bond length is found for nickel (0.195 Å), the smallest with molybdenum (0.09 Å). Although regularities cannot be found (Δd: Mo < Co < Mn < Cr < Ru <W < Pt < Fe < Pd < Ni), group-10 TMs tend to be found to the right.
3.2.2 Variation of the coordination sphere around the TM
It is impossible to attribute a relationship between the ligands (e.g. CO, phosphines, diphosphines) on TM and the TM–Sn distances. Although the co-ligands on TM strongly influence the TM–Sn bond, it is difficult to assign a set of spectator ligands to long or short bonds. However, Cp systems are preferably found with short distances, carbonyl and phosphine ligand moieties are flexible and found with both extremes.
3.2.2.1 Group-6 pentacarbonyl complexes
Two stannylene ligands in trans position around the TM result in relatively short Sn–TM bonds. In the homoleptic square planar platinum tetrastannaborane complex [Pt+II(SnB11H11)4]6− (8a) (2.566; 2.560 Å) [67] with stannylene ligands in trans positions, the Pt–Sn bond is shorter than in complexes bearing the π-acceptor ligands isonitrile trans-[{(Et3P)2Pt(CNtBu)}(SnB11H11)2] [128] (8f, 2.590 Å) or carbene trans-[{(Et3P)2Pt(C(NiPr)2C2Me2)}(SnB11H11)]2− [129] (8g, 2.606 Å), or in the phosphine complexes [{(dppp)Pt}(SnB11H11)2]2− [130] (8h, 2.596 Å) and [{(dppe)Pt}(SnB11H11) (tBuNCPh)] [131] (8i, 2.60 Å). The Cr–Sn distance for the two stannylenes in trans position in mer-[{(OC)3Cr}(Sn(NtBu)2SiMe2)3] (1c) is also shorter than that of the stannylene trans to the CO ligand (2.512 versus 2.544 Å) [6].
Metal–metal bonds trans to σ-donor/π-acceptor ligands cause very short TM–L bonds, if L is a π-acceptor like CO [132]. It is therefore in this class of compounds, where the strongest π-back donating effect to stannylene ligands should be found. In fact, the SnCl3− fragment trans to the Ir–Ir bond in 31a (2.61 Å) is longer than the one in cis (2.574 Å) in the same molecule [133]. The same tendency is observed for the Au–Sn bond (2.681 Å) in the dimeric complex [{Au(μ-PPh2)}2{Sn(N(o-tol)SiMe2)3SiMe}2] (9b) [134] with close Au–Au contacts compared to the monomeric stannato complex trans-[{(Ph3P)Au}{Sn(N(o-tol)SiMe2)3SiMe}] (9c) (2.565 Å) [134].
The influence of geometric parameters is exemplified by different rotamers in the dichromium and ditungsten distannylene complexes [{(OC)5Cr}2{Sn2(OtBu)4}] (14d) [15] and [{(OC)5W}2{Sn2(OtBu)4}] (14c, see Table 10) [27]. If the terminal OtBu group on tin is eclipsed to one CO ligand in cis position, the resulting TM–Sn distance is 1.3% (W) or 1.5% (Cr) longer compared to the alkoxo ligand in staggered conformation (see Fig. 12). The changes are more pronounced for chromium because the {Cr(CO)5} fragment is closer to the stannylene than tungsten. Hence, {W(CO)5} in the mixed complex 14b is more prepared to adopt the unfavoured eclipsed rotamer. Addition of another TM to the second Sn atom anti with respect to the central Sn2O2 plane also causes an elongation of the bond, which demonstrates that the tin atoms are not independent in these dimeric stannylenes [15,27].
Sn–TM bond lengths for bis-alkoxo stannylene complexes [{(OC)5W}m{(OC)5Cr}n{Sn2(OtBu)4}] (Fig. 12) [15,27].
| Compound | Sn–Cr [Å] | Sn–W [Å] | |
| [{(OC)5Cr}{Sn2(OtBu)4}] | 14e | 2.576 (staggered) | |
| [{(OC)5Cr}2{Sn2(OtBu)4}] | 14d | 2.59 (staggered) | |
| 2.62 (eclipsed) | |||
| [{(OC)5W}{Sn2(OtBu)4}] | 14a | 2.72 (staggered) | |
| [{(OC)5W}2{Sn2(OtBu)4}] | 14c | 2.72 (staggered) | |
| 2.76 (eclipsed) | |||
| [{(OC)5W}{(OC)5Cr}{Sn2(OtBu)4}] | 14b | 2.61 (staggered) | 2.74 (eclipsed) |
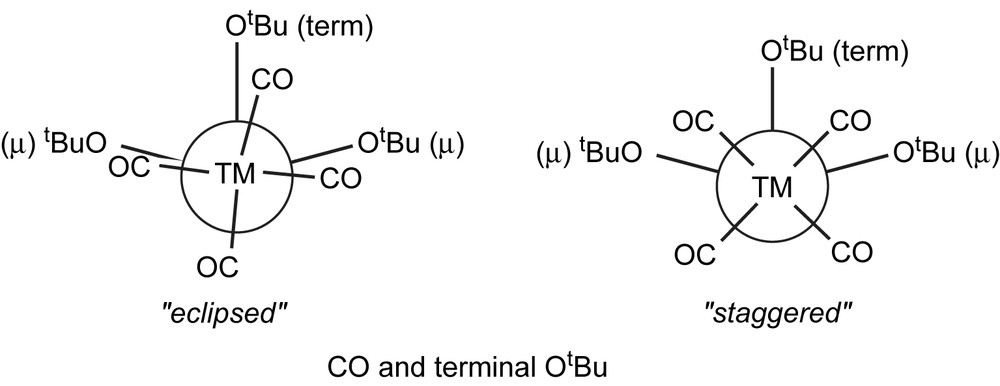
Schematic drawing of the molecular structures of 14d, 14b [15,27] and 14c [27] highlighting the eclipsed and staggered rotamers of the {TM(CO)5} (TM = Cr, W) groups at the dimeric alkoxo stannylene [Sn(OtBu)(μ-OtBu)]2 (14) (Table 10).
3.3 Impact of the stannylene ligand on the TM–Sn bond
3.3.1 Type of stannylene
It is difficult, if not impossible, to predict from Section 3.2 if a given stannylene will cause long or short TM–Sn bonds. Lappert's stannylene Sn(CH(SiMe3)2)2 (3) forms complexes with very short (3b (Co), 3a (Pd)) and long bonds (3c (Pd)). (SnB11H11)− is found at the lower end (8c (Ru)), but more at the long end (8b, 8d, 8e, 8h). Amido stannylenes tend to give short bonds (1c, 1d, 19a).
A correlation between the type of stannylene (e.g. amido, alkyl, aryl, alkoxo stannylene) and the TM–Sn bond length is not evident. Comparing the group-6 pentacarbonyl fragments with such different stannylenes as alkoxo (salen (2), Janus (Sn(OtBu)3In (24)), dimeric (Sn2(OtBu)4 (14), amido (Veith stannylene (1)), alkyl and aryl (Lappert 3 and Weidenbruch stannylene 4) and tin cluster compounds (Zintl ion in 32), the regions of the classes of stannylene complexes overlap considerably so that a trend is not obvious. This is examplified with R2Sn–Cr(CO)5 complexes in Table 11.
Bond lengths in R2Sn–Cr(CO)5 complexes with different types of stannylene ligands (Ar = 2,4,6-tBu3C6H2; Ar′ = 3,5-tBu2C6H3).
| Stannylene ligand | Compound | d(Sn–Cr) [Å] | Ref. |
| Sn(CH(SiMe3)2)2 (3) | 3d | 2.562 | [135] |
| Sn(μ-OtBu)3In{Mo(CO)5} | 24c | 2.573 | [9] |
| Sn(salen) (2) | 2d | 2.578 | [26] |
| Sn6{Cr(CO)5}52− | 32a | 2.605; 2.61 | [136,137] |
| Sn(μ-OtBu)3In{Fe(CO)4} | 24b | 2.636 | [9] |
| Sn(ArCH2CMe2)Ar′ (4) | 4b | 2.61 | [55] |
3.3.2 Electronegativity of substituents
If the electronegativity of substituents is considered, Sn–TM distances reflect the impact of the electronic change on the tin atom. A rough estimate of the electronic influence is given in Fig. 13, where the Cr–Sn bond length of selected R2Sn–Cr(CO)5 complexes is plotted against the electronegativity of the α-atom of the substituent R. The more electron-withdrawing, the shorter becomes the TM–Sn bond. The same trend is observed in the chloro bis-stannylene complexes trans-[{(OC)3Co}{SnClnX3−n}2]. They show decreasing Co–Sn distances with increasing number of chlorine substituents (X = {Co(CO)4}, n = 1 (30a): d(Co–Sn)= 2.509 Å [123]; n = 2 (33a): 2.468 Å; n = 3 (34a): 2.443 Å [138]). This finding is in accordance with Bent's rule, which says, that more electronegative substituents cause higher p-orbital contribution in their bonds, leaving more s-character for the lone pair of electrons, thus shortening the donor radius [139] (Table 12).
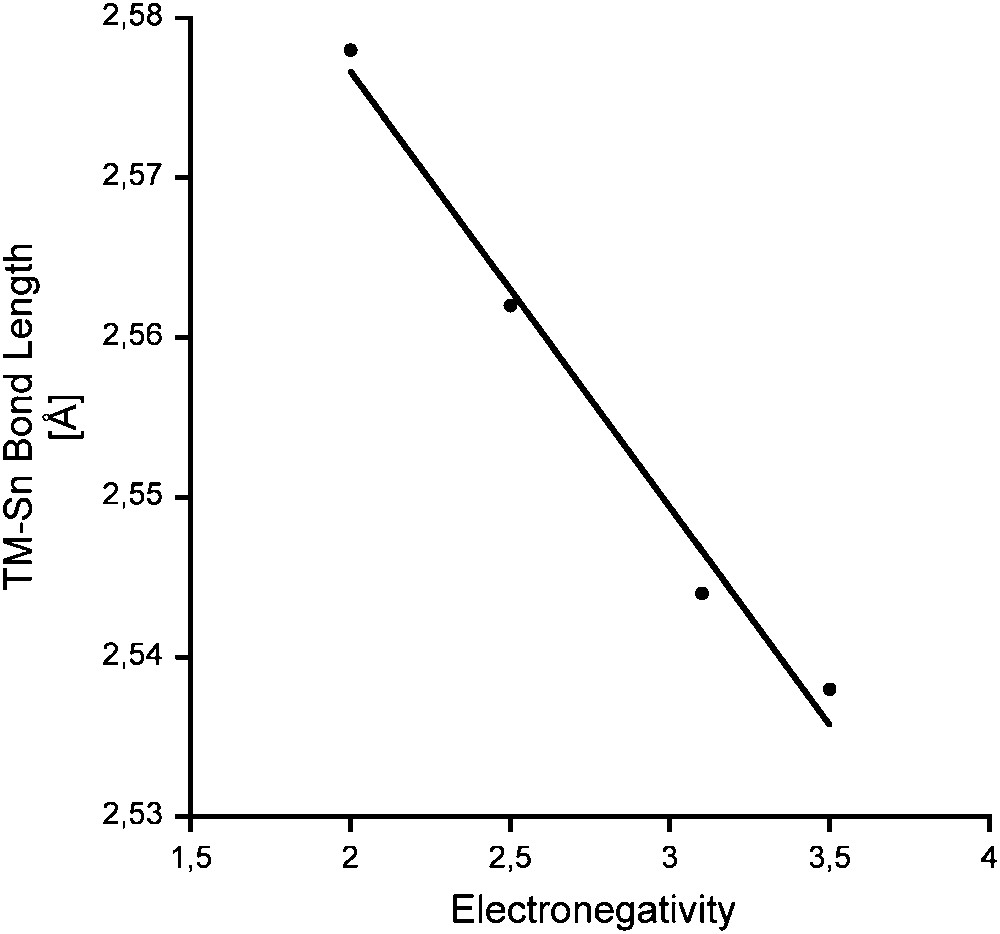
Sn–Cr bond length versus electronegativity of selected R2Sn–Cr(CO)5 complexes (Table 12).
3.3.3 Base adducts
Bases add perpendicular to the plane formed by the R2Sn–TM fragment, (see Fig. 14) through a lone pair of electrons. The overall impact around tin is an increase of electron density, coordination number and concomitant steric crowding. A higher electron density at tin should increase the σ-basicity and lead to longer Sn–TM distances. Moreover, steric repulsion, hybridisation (higher p- or even d-participation) and blocking of π-back bonding emphasise that trend.
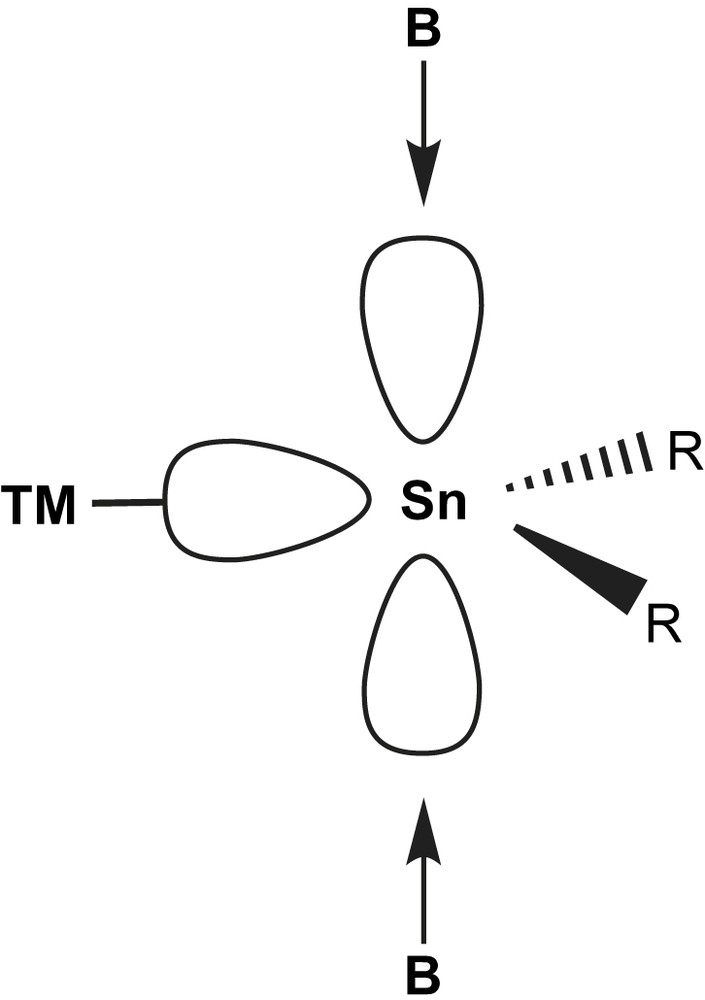
Schematic representation of the base adduct formation on R2Sn–TM complexes.
3.3.3.1 External bases
If we compare the same type of stannylene ligands and similar TM fragments, an increase of the TM–Sn distance is indeed observed. This is shown for pairs of chromium pentacarbonyl complexes in Table 13.
Comparison of Bn → R2Sn–M(CO)5 complexes (M = Cr, W; n = 0, 1, 2; d [Å]).
| Stannylene | M | Compound | d(M–Sn) (0 bases) | d(M–Sn) (1 base) | d(M–Sn) (2 bases) |
| Sn(CH(SiMe3)2)2 | Cr | 3d | 2.562 [135] | ||
| SntBu2 | Cr | 23a | 2.654 [111] (py) | ||
| Sn(NtBu)2SiMe2 | Cr | 1c | 2.544 [6] (trans-CO) | ||
| Sn(NtBu)2SiMe2 | Cr | 1b | 2.605 [27] (THF) | ||
| Sn{W(CO)5}2 | W | 22a | 2.702 [141] | ||
| Sn{W(CO)5}2 | W | 22b | 2.723 [104] | ||
| SnCl2 | W | 18a | 2.712 [92] | ||
| SnCl2 | W | 18b | 2.737 [92] |
Calculations on H2Sn–Pd complexes show that addition of weak Lewis bases like formaldehyde to the empty tin p-orbital bends the Sn substituents slightly away from the entering base (Sn is situated 0.18 Å above the plane TM/H2) with only marginal increase of the H–Sn–H angle from 95.349° (base free) to 95.739° (base adduct) but clear elongation of the Sn–Pd bond (2.5189 to 2.5492 Å) [29]. The latter was interpreted as consequence of the loss of π-back bonding due to base coordination to the Sn(p) orbital. Real structures show that the side-on coordination of a C2H2 molecule to the Pd–Sn bond in [{(dipe)Pd}(μ,η1:1-C2H2)(Sn(CH(SiMe3)2)2] (3c) causes an extraordinarily long Pd–Sn distance of 2.67 Å [30,142].
3.3.3.2 Internal bases
Another influence is seen by the bonding mode of the base to tin (inter-/intramolecular; chain length, i.e. ring size). Increasing the ring strain in the R2Sn–W(CO)5 intramolecular amino stabilised complexes [{(OC)5W}Sn(o-NMe2CH2C6H4)2] (26b) [73] (methylene bridge) and [{(OC)5W}Sn(8-(NMe2)C10H7)2] (17a) [74] (naphthyl bridge) elongates the bond from 2.749 Å to 2.822 Å.
The influence of the donor atom has scarcely been investigated. The phosphine analogue to the amine-based complex 26b, [{(OC)5W}Sn(o-PPh2CH2C6H4)2] (35a), exhibits a slightly longer Sn–W bond (2.762 Å) [73]. However, the sterical hindrance is increased at the base atom. With the ether side chain in [{(OC)5W}Sn(2,6-(ROCH2)C6H3)2] (R = Me (13a), tBu (12a)), the Sn–W distances are similar (2.732 Å; 2.7655 Å) [61]. Unfortunately, the analogous derivative with phosphonic acid ester side chain has only been characterised with chromium [{(OC)5Cr}(SnCl(4-tBu-2,6-(P(O)(OEt)2)2-C6H2)] (11b) [77]. Changing the isopropoxy bridge in [Ni{(Sn(NtBu)2SiMe2)2(μ-X)}2] (X = OiPr; 1e) [143] by bromine in 1f (X = Br) [99] elongates the bond slightly (2.455 to 2.463 Å).
3.3.4 Charge
Charge on either the TM or Sn should influence the TM–Sn bond considerably, especially in the light of the results from energy partition calculations, where a considerable, if not the largest, contribution comes from electrostatic attraction (vide supra). Oxidising the TM (resp. Sn) should reduce (resp. increase) back bonding and increase (resp. decrease) coulombic attraction between Sn and TM (but increase attraction to substitutents R). However, systematic electrochemical investigations accompanied by structural elucidation of the products have not yet been performed mainly due to the decomposition of the products formed during cyclic voltammetry [83,144]. Therefore, it is difficult to extract the influence of charge. Similar systems may be compared but have the drawback that electronic and steric changes occur at the same time. However, from reported data, there seems to be only little influence on the bond length (see Table 14 for Sn–Cr). The Sn–Cr distances do not show any tendency and are even overlapping.
Chromium pentacarbonyl complexes with differently charged stannylene ligands.
| Stannylene | Compound | Charge | d(Sn–Cr) [Å] | Ref. |
| {{(OC)5Cr}5Sn6}2− | 32a | −2 | 2.61 | [136] |
| {Sn2(OtBu)6Ba} | 36a | −1 | 2.65 | [9] |
| {(OC)5TM}nSn2(OtBu)4 | n = 0 (14e) | 0 | 2.58–2.62 | [15] |
| n = 1; TM = Cr (14d), W (14b) |
Changing the formal charge on the TM is experimentally accompanied by drastic changes in the TM coordination sphere. In the {TM(CO)5} 16 VE fragments the group-6 metal is formally uncharged. In complexes with the {CpTM(CO)3} 17 VE fragments, TM is formally positively charged. The stannylene Sn(o-Me2NCH2C6H4)2 (26) [145] forms complexes with both fragments. In the pentacarbonyl complex 26b, a Sn–W bond length of 2.749 Å is found [73]. In 26a, the same ligand is coordinated to {CpW(CO)3}, for which a positively charged complex would result [115]. However, a chloride anion coordinates to the tin atom and leads to a neutral complex. The chloride base even causes structure distortion by replacing one pendant amine side chain. However, a metal–metal bond shortening based on the increased coulombic interaction could be expected, but an elongation is observed (d(Sn–W) = 2.820 Å). The deviation may be due to the superposition of several effects (charge, steric demand, type of base). A relatively long Sn–W bond is also found in the triamido stannato(II) complex [{Cp(OC)3W}(Sn(N(p-tol)SiMe2)3SiMe)] (9d) (2.783 Å) [121]. However, these two findings should not lead to the general assumption that the 17 VE fragments may lead to longer metal–metal distances. In the similar ionic compound [{WCp(CO)2(cyclo-P(Ph)(NMeCH2)2)}SnPh2(OTf)] (16a), the Sn–W bond length is found at the lower end of the typical Sn–W range (d(Sn–W) = 2.709 Å) [88]. This may be due to higher ionic character of the stannylene (triflate as weakly coordinating counter ion) and/or the replacement of the strong π-acceptor CO by a phosphane. While neutral {Mo(CO)5} complexes exhibit Mo–Sn bond lengths between 2.724 and 2.77 Å, the negatively charged zwitterionic [{(η7-cycloheptatrienyl)(OC)2Mo}{SnB11H11}]− (8k) exhibits a short distance of 2.712 Å [68]. The {Co(CO)3}+ complexes with trans-stannato(II) ligands [SnCln{(Co(CO)4}3−n] (30a, 33a, 34a) [123,138] show long Sn–Co bond lengths (2.44–2.51 Å), those with Cp fragments (“neutral”) shorter Sn–Co bonds (2.393–2.44 Å) [122,124,146,147].
3.4 Impact of R2Sn ligand on the coordination sphere around the TM
The TM–CO distance in carbonyl complexes is an indicator for the sum of σ-donor and π-acceptor strength of the carbonyl ligand [132,148]. Since a ligand L in trans position to the carbonyl competes for the same orbitals, the TM–C bond length provides a good indirect measure for the donor/acceptor abilities of L. If a strong variation of the ratio donor/acceptor strength would occur with modification of the stannylene ligand L, the carbonyl trans to the stannylene would be more influenced than those in cis position. The same concept is behind the use of carbonyl stretching frequencies in IR spectroscopy. The substitution of a strong π-acceptor/weak σ-donor by a weaker π-acceptor causes a bathochromic shift of the respective absorption. However, σ- and π-effects may be separated by neither method. It would be very useful to perform systematic investigations on the influence of the stannylene ligand on physical properties of the TM directly, such as 95Mo, 103Rh, 183W or 195Pt NMR (for 95Mo NMR studies with phosphine ligands see Ref. [49]) or 57Fe Moessbauer spectroscopy, accompanied by careful calculations, to separate the electronic effects. These measurements would also support the interpretation of the chemical shifts and coupling constants in 119Sn NMR spectra.
3.4.1 R2Sn–M(CO)5 complexes (M = Cr, Mo, W)
The plot of the TM–C bond lengths for carbonyl ligands cis to the stannylene ligand against those trans to Sn reveals a linear relationship. Because both positions are influenced the same way, irrespective of TM and stannylene (base adduct, cluster, charge…), the π-bonding along the Sn–TM bond should be similar. The trans-TM–C bond is always shorter than the cis-TM–C bond as seen from the positive intercept (0.065; mean ratio d(Sn–TM) cis/trans = 0.97(1)). This is in agreement with poor π-back bonding of R2Sn ligands (Fig. 15).
Plot of the TM–C bond lengths of carbonyl ligands cis- versus trans-Sn in group-6 pentacarbonyl complexes.
The stannylene bonding parameters may be compared to more common two-electron donor ligands to better classify the donor/acceptor properties. In the series of L–TM(CO)5 complexes (TM = Cr, Mo, W), a decreasing π-acceptor strength of L is observed in the order L = CO > PCl3 > C(NR2)2 > P(OR)3 > PPh3 > NR3. For the complexes with Veith's stannylene 1b and 1a, the alkoxo stannylene 14b [15] and salen 2d and 2a, TM–COtrans bond lengths are found close to the values found for the C(NR2)2, P(OR)3 and PPh3 complexes (see Table 15). From this comparison, the TM–Sn π-back bonding should be comparable to medium π-acceptors such as PPh3, and less to strong acceptors like CO and PCl3 [149] or pure σ-donors like R3N or ethers. This is in accordance with IR spectra of e.g. 1b and 14b, where the A1 vibrational mode of the C4v symmetric {TM(CO)5} moiety is found at the same wavenumber as for the respective PPh3 derivative, with a clear hypsochromic shift in respect to [(OC)5TM(THF)] [15].
Comparison of the influence on the TM–Ctrans distance on variation of the ligands in pentacarbonyl group-6 complexes (TM = Cr, W; L∗ = ).
| trans ligand | Cr–Ctrans dist [Å] | Compound | W–Ctrans dist [Å] | Compound |
| CO | 1.92 | [Cr(CO)6] [150] | 2.02–2.05 | [W(CO)6] [151] |
| PCl3 | 1.898 | [{(OC)5Cr}(PCl3)] [152] | 2.02 | [{(OC)5W}(PCl3)] [152] |
| Carbene | 1.868 | [{(OC)5Cr}C(N2C10H8)] [153] | 2.003 | [{(OC)5W}(C(N(Ph)NC(Ph)N(Ph))] [154] |
| PPh3 | 1.870 | [{(OC)5Cr}(PPh3)] [155] | 2.005 | [{(OC)5W}(PPh3)] [156] |
| P(OPh)3 | 1.861 | [{(OC)5Cr}(P(OPh)3)] [157] | 2.040 | [{(OC)5W}L∗] [158] |
| NR3 | 1.823 | [{(OC)5Cr}(N(C2H4)3CH)] [159] | 1.946 | [{(OC)5W}(NMe2CH2Ph)] [160] |
| Stannylene | 1.86 | [{(OC)5Cr}{Sn(thf)(NtBu)2SiMe2)}] (1b) [27] | 1.99 | [{(OC)5W}{Sn(thf)(NtBu)2SiMe2)}] (1a) [27] |
| 1.87 | [{(OC)5Cr}{(OC)5W}(Sn2(OtBu)4)] (14b) [15] | 2.01 | [{(OC)5Cr}{(OC)5W}(Sn2(OtBu)4)] (14b) [15] | |
| 1.86 | [{(OC)5Cr}Sn(salen)] (2d) [26] | 1.99 | [{(OC)5W}Sn(salen)] (2a) [26] |
The Sn–TM–Ceq angle in the Grp-6 stannylene complexes is very close to 90° with small statistical deviations [15], which means that the “umbrella effect” [10], which describes the bending of the equatorial CO ligands towards the hetero ligand, is for most complexes not observed. Calculations have indicated that such distortion is due not to crystal packing but to enhanced carbonyl–tetrelene interaction. It stabilises the HOMO [18] but the effect decreases on going down from carbenes to stannylenes [17].
3.4.2 R2Sn–Fe(CO)4 complexes
In pentacoordinated compounds where a trigonal bipyramidal coordination geometry is adopted, the site preference for a given ligand depends on its σ-donor/π-acceptor strengths and the electron configuration of the TM. For strong σ-donors L with d8 TM complexes, the axial position is more stable. Strong π-acceptors should prefer the equatorial position forming stronger bonds, which often goes with shorter bonds. Thus, the equatorial preference in d8 complexes increases with increasing π-acceptor strength (NR3 < PR3 < CO) [2]. However, since strong σ-donors often at the same time are strong π-acceptors, the prediction of the preferred site is not straightforward.
In R2Sn–Fe(CO)4 complexes, the R2Sn ligand competes with the strong π-acceptor CO ligand for the equatorial site. From a statistical point of view, the axial position is preferred for R2Sn ligands (12 versus 4 structure reports). The Fe–Sn bonds are slightly longer in the axial position (d(Sn–Fe)ax = 2.48 Å; d(Sn–Fe)eq = 2.46 Å, see Fig. 16). The axial Fe–C distances become very similar to those in Fe(CO)5 if the stannylene occupies the equatorial position. In complexes with the stannylene in axial position, the Fe–C distance trans to Sn is shortened (Fe–Cax = 1.77 Å). Irrespective of the position of the stannylene ligand, the equatorial Fe–C distances are considerably shortened (Fe–Ceq = 1.78 Å) with respect to Fe(CO)5 (Fe–Ceq = 1.83 Å).
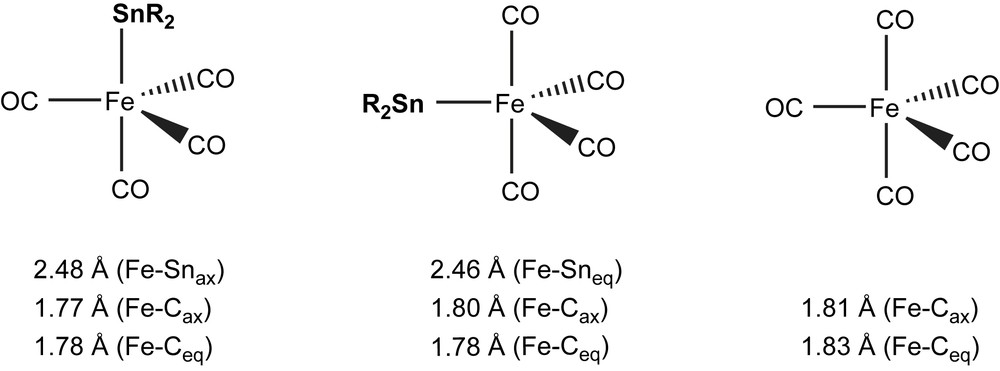
Structural features of axial (left) and equatorial (centre) R2Sn–Fe(CO)4 complexes from CSD analysis compared to Fe(CO)5 (right).
The site preference, shortening of d(Fe–COeq) in axial and equatorial stannylene complexes and of d(Fe–COax) only in the axial isomer, support weaker π-back bonding of stannylenes compared to CO. The shorter Fe–Sn bond in the equatorial isomer is also in accordance with weak π-acceptor strength. This would leave more electron density on iron to strengthen the π-back bonding, hence shortening d(Fe–CO), which is most evident in the axial isomer.
The four eq-R2Sn–Fe(CO)4 complexes all comprise substituents with oxygen as α-atoms [54,118,161]. In the ax-R2Sn–Fe(CO)4 complexes, oxygen [25,54,143,162] and less electronegative substituents (nitrogen [75,163], carbon [71], TM [164]) are found as α-atoms. This also emphasises that more electronegative substituents lower the LUMO energy and produce better π-acceptors and hence are more often found in the equatorial position.
A direct comparison of the axial and equatorial positions is accessible in the complexes eq-[{(OC)4Fe}Sn(OC2H4NMe2)2| (eq-37a) and ax-[{(OC)4Fe}Sn(OC2H4NMe2)2| (ax-37a) [54]. Both isomers are found in equilibrium in solution, with a preference for the axial conformer in non-coordinating (less polar) solvents. According to DFT calculations, the energy difference between both isomers is close to zero (0.3 kcal mol−1) with a very small inversion barrier (1.6 kcal mol−1). However, both isomers could be crystallised separately. The Fe–Snax distance in ax-37a is 0.019 Å longer than the Fe–Sneq distance in eq-37a. The axial carbonyl carbon atom trans to tin is closer to iron (ax-37a: 1.773 Å) than trans to CO (eq-37a: 1.789 Å). The equatorial TM–CO distances are identical within the errors (eq-37a: 1.784 Å; ax-37a: 1.786 Å (1.773–1.795 Å)). These tendencies are in total agreement with those found in the statistical treatment and follow the energy arguments.
Looking from the VSEPR model, the sterically demanding stannylene ligands should prefer the equatorial position. As this model does not take π-bonding into account, the deviation from the mainly steric argument might point to a certain percentage of π-bonding.
In conclusion it can be stated that albeit weaker than CO, stannylene ligands are appreciable π-acceptors.
3.5 Impact of TM coordination on the stannylene
3.5.1 Bond lengths to substituents (Sn–R) and bond angles between substituents (R–Sn–R)
For free stannylenes, a simple correlation between bond angle and electronic characteristics of the substituents cannot be deduced from structure data. This can be shown by comparison of the monomeric alkyl-, amido- and alkoxo-stannylenes SnR2 (R = CH(SiMe3)2 [165], N(SiMe3)2 [166], O(2,6-tBu-4-MeC6H2) [167], see Table 16). The bond angle does not show any correlation with electronegativity or steric demand. The R2Sn(II) covalent bond radius rcov(SnII) = d(Sn–E) − rcov(E), calculated as the difference between the measured tin–substituent distance and the single bond covalence radius rcov(E) for the substituent α-atom, decreases considerably with increasing electronegativity. A strong influence of the substituent on electron distribution on tin might account either for a strong ionic bonding, increasing multiple bonding or a combination of both [21,22,168]. A detailed discussion of these effects is beyond the current review.
Comparison of bond length d(Sn–E), bond angle [°] (E–Sn–E), covalent radius rE and electronegativity EN of E and corresponding “covalence radius” of Sn(II) (rcov(Sn(II)) = d(Sn–E) − rcov(Sn–E)) for selected alkyl, alkoxo and amido stannylenes SnR2 (E = α-atom of substituent R).
| Compound SnR2 (R=) | d(Sn–E) [Å] | [°] (E–Sn–E) | rE [Å] | EN | rcov(SnII) [Å] | Ref. |
| CH(SiMe3)2 (3) | 2.24 | 96 | 0.77 | 2.5 | 1.47 | [165] |
| N(SiMe3)2 (19) | 2.09 | 104.7 (96 gas phase) | 0.70 | 3.1 | 1.39 | [166] |
| O(2,6-tBu-4-MeC6H2) (29) | 2.01 | 88.4 | 0.73 | 3.5 | 1.28 | [167] |
With these difficulties in mind, tendencies from statistical analysis of the structure data are not expected to yield sound information. However, comparing all stannylenes, for which R2Sn and R2Sn–TM structures are available, a surprisingly distinct tendency for the changes in tin–substituent bond lengths (coordinative change in bond length Δd) and substituent–tin–substituent bond angles (coordinative change in bond angle Δ) evolves: the TM–Sn bonds are shortened and the R–Sn–R angles are widened (see Fig. 17). There is no obvious dependency on the number of bases on tin.
Distributions of the changes in Sn–R bond length ([%], left; maximum at −2.25%) and R–Sn–R bond angle ([°], right; maximum at +1.5°) on stannylenes upon TM coordination.
Better resolved results are obtained if the stannylenes are grouped into classes with similar substituents (see Fig. 18). An increase of the bond length is only observed for the halogenido stannylene complexes (SnCl2, SnCl3−, SnBr3−). These are referenced against the gas phase structure of the respective SnX2 monomers due to lack of monomeric structures in condensed phase and may therefore not be representative to estimate the coordination impact in the solid state. Furthermore, it is the only class for which a decrease of the bond angle is observed. A second class shows decrease of the bond angle: the complexes derived from the “Janus” alkoxo stannylenes. The parent compounds show disorder in the solid state due to lacking site-specifity of the two “Janus” faces (group-14 and group-13 elements) [113,169]. This causes less reliable geometric data. Strong deviations for the coordination changes (max/min Δd: +0.089/−0.054 Å; Δ∠: +2.5/−8.0°) yield average values with little confidence. Apart from disorder, the constraint geometry of three bridging alkoxo groups may also contribute to the different behaviour.
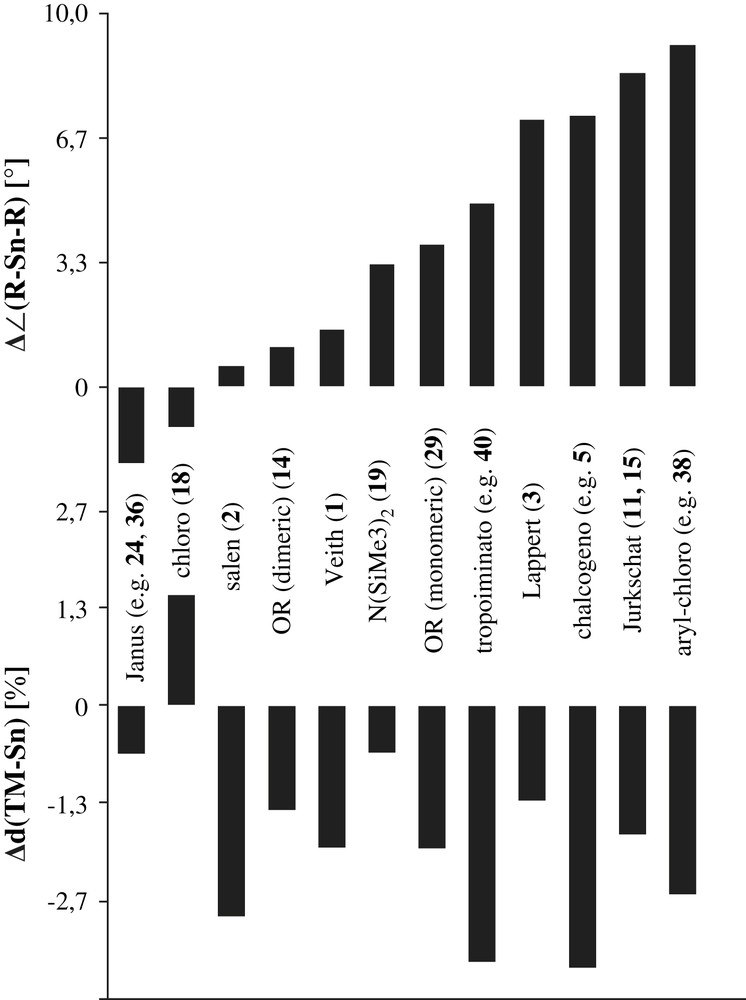
Variation of coordinative change in bond angle ([°], top) and bond length ([%], down) upon TM coordination for several classes of stannylenes.
With the exception of those two classes, the angle increases and the bond length decreases. However, a correlation between the parameters is not found.
These changes in bond lengths and angles can be rationalised from the calculations on the “free”, uncoordinated stannylene (vide supra) and in analogy to calculations on TM complexes of group-15 compounds [170]. In the free R2Sn, the lone pair of electrons possesses a high percentage of s-orbital character. The bonds to the substituents hence possess a high fraction of p-character. The extreme situation is described in the simplified picture of the non-hybridized stannylene (see Fig. 19). On coordination to a Lewis acid, the p-character in the lone pair increases with concomitant decrease of the p-character of the bonds towards the substituents, which shortens the Sn–R bonds and widens the R–Sn–R bond angle. This observation has been formulated in Bent's rule: “Atomic s-character concentrates in orbitals directed towards electropositive substituents. Lone-pair electrons are regarded as electrons in bonds to very electropositive atoms” [139]. For PMe3, the s-character of the lone pair decreases from 51.2% in the free molecule to 13.7% in [(OC)5Cr(PMe3)] [170]. Another model leads to the same conclusion: removing electron density by Lewis acid coordination increases the electrostatic attraction between the more electronegative substituents and tin, hence decreasing the bond length.
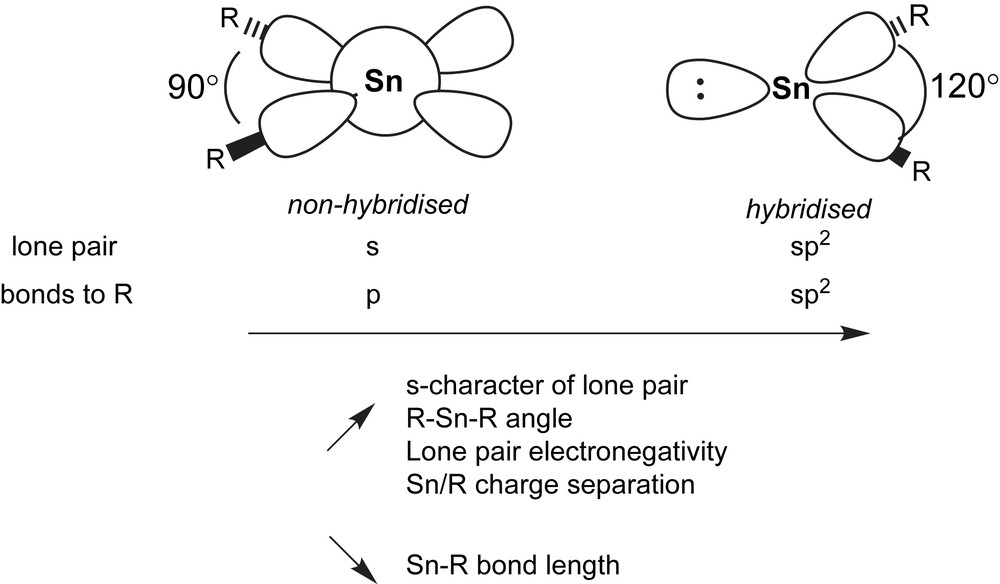
Schematic representation of the changes around Sn upon TM coordination.
Further graphs bewteen bond angles R–Sn–R and R–Sn–TM, TM–Sn–base and R–Sn–base show randomly scattered data pairs and seem to depend strongly on packing or steric demand.
3.5.2 Impact on base adduct formation
Base adduct formation increases the coordination number. In a simplified model, this can be attributed to change in hybridisation from sp2 (or unhybridised, coordination number (c.n.) 3) to sp3 (c.n. 4) and dsp3 (c.n. 5, see Fig. 20),3 creating distorted tetrahedral or trigonal bipyramidal coordination polyhedra around tin. A decrease of the s-character (sp2 via sp3 to dsp3) should increase the Sn–R bond length, whereas the R–Sn–R bond angle should decrease upon addition of the first, but increase again with addition of the second base molecule. Further effects should account for an increase of the Sn–R bond length: the global electron density around tin should increase, thus diminishing the electrostatic attraction between tin and the more electronegative substituents; steric encumbering; break down of potential Sn–R multiple bonding. Addition of a second base trans to the first one should increase the bond length of the latter by competition for the same orbital. Unfortunately, reports on compounds with all three states (zero, one and two base molecules) are missing, so incremental steps have to be regarded.
TM complexation increases the overall Lewis acidity on tin, therefore increasing the tendency to form base adducts and decreasing the distance between tin and base. On the other hand, TM coordination allows for cutting smaller pieces out of stannylene aggregates, which can be regarded as intramolecular base adducts (vide supra).
The bond length d(Sn–base) does not show any consistent change upon coordination. For example, for the intramolecular bis-amino stabilised complexes Sn(Cl)(o,o'-(Me2NCH2)2C6H3) (38) [171], Sn(OC2H4NMe2)2 [54], Sn(o-(Me2NCH2)C6H4)2 (26) [115], Sn(O(2-MeC9H5N))2 (39) [172], and the PO adduct [Fc(Sn(2,6-(P(O)(OiPr)2-4-tBu-C6H2)2] (15a) [83], the first two stannylenes show a decrease of the tin–nitrogen bond (−1 to −5%), whereas in the latter three stannylenes, bond elongation (+1 to +8%) is observed upon TM coordination. In the porphyrino stannylene 28 [117,163] shortening, and in Sn(Salen) [26], elongation is observed. However, irrespective of the TM moiety, the coordinative changes for the same stannylene are in the same direction.
The addition of one base causes a deformation of the effectively planar surrounding of tin in the base free complexes. The tin atom is moved out of the plane TM–substituent (distance “n” in Fig. 20) by 0.2–0.7 Å, forming more a trigonal pyramid than a tetrahedron. According to the donor atom, the average values for “n” differ to various degrees. Addition of a second base (of the same kind) pushes the tin atom back into the plane (deviation from plane: 0.0–0.07 Å). The Sn–base bond length increases by 3 to 11% in the average. An illustrative example is provided by the series of mono- and di-thf complexes of [(OC)5W{SnCl2(thf)n}] (n = 1 18a; 2 18b), where the Sn–O distance increases from 2.22 to 2.36 Å [92].
A correlation between the changes in Sn–R bond length or R–Sn–R bond angle with the number of added base molecules is not found. This might be due to the very different types of bases, be it amine/imine, phosphane, esters of phosphonic acid, ether, halogenido, by intra- or intermolecular adduct formation. However, if selected stannylenes are considered, it comes out that addition of one base has no considerable effect on the bond length, while bond angles slightly increase (see Table 17). Complexes with base free stanylenes Sn(N(SiMe3)2)2 (19), Sn(CH(SiMe3)2)2 (3) and the dimetallo tricyclic [{(OC)5W}{Sn(W(CO)5)2}] (22a) show a small bond elongation and angle widening upon addition of one base. With Veith's cyclic stannylene 1, the bond length ranges overlap, whereas the bond angle slightly increases. Going from one to two base molecules in the pair 18a/18b leaves the bond length practically unchanged but increases the Cl–Sn–Cl bond angle.
Comparison of Sn–R bond lengths [Å] and R–Sn–R bond angles [∘] in different classes of stannylene complexes with 0, 1 and 2 added base molecules.
| Complex/no. of base molecules | 0 | 1 | 2 | |||
| d(Sn–R) | ∠(R–Sn–R) | d(Sn–R) | ∠(R–Sn–R) | d(Sn–R) | ∠(R–Sn–R) | |
| [{(OC)4Cr}(Sn(N(SiMe3)2)2)2] [173] | 2.06 | 105 | ||||
| [Pd{Sn(N(SiMe3)2)2}3] [96] | 2.08 | 106.7 | ||||
| [{(Et3P)Pt(μ-Cl)}{Sn(Cl)(N(SiMe3)2)2}]2 [127] | 2.08 | 111.9 | ||||
| [{(η6-C7H8)(η2-cod)Rh)}{Sn(Cl)(N(SiMe3)2)2}] [174] | 2.09 | 108.3 | ||||
| [{TMLn}Sn(CH(SiMe3)2)2] [31,124,135,146,175] | 2.19–2.24 | 97.3–105.3 | ||||
| [{TMLn}Sn(base)(CH(SiMe3)2)2] base = NCS−, [176] OH− [147,177] Cp− [144] | 2.19–2.24 | 99.5–109.3 | ||||
| [{TMLn}Sn(NtBu)2(SiMe2)] [6,178] | 2.03–2.06 | 75 | ||||
| [{TMLn}Sn(base)((NtBu)2SiMe2)] base = amine, [143] Br−, Cl−, [99] OEt−, [143] Cp−, [125] thf [27] | 2.02–2.08 | 75.5–76.6 | ||||
| [{(OC)5W}{Sn(W(CO)5)2}] (22a) [141] | 2.77 | 74.1 | ||||
| [{(OC)5W}{Sn(thf)(W(CO)5)2}] (22b) [141] | 2.79 | 73.8 | ||||
| [{(OC)5W}Sn(thf)Cl2] (18a) [92] | 2.36 | 98.3 | ||||
| [{(OC)5W}Sn(thf)2Cl2] (18b) [92] | 2.37 | 101.8 |
The increase in Sn–R bond length accompanied with a slight increase of the R–Sn–R bond angle is inconsistent with the formally expected decrease of s-character of the Sn–R bond upon base addition. Therefore the findings do not support the change in hybridisation from sp2 to sp3. The deviation from planarity on base addition may therefore be attributed to the steric demand of the base and the electrostatic attraction between the Lewis acid and base pair. The lengthening of the Sn–R bond may also be explained by electrostatic and steric reasons. The latter may also account for the observed increased R–Sn–R angle. From an orbital point of view, the influence of bases on the bonds in the “bonding plane” formed by the TM, Sn and R is small. Hence, addition of donors seems to occur in well-separated orbitals, consistent with the non-hybridised and sp2 description of stannylenes (vide supra).
3.6 Towards an Sn(II) “coordinative radius”
The Sn(II)–TM bond length is often seen as a measure for the strength of the Sn–TM interaction. However, as has been pointed out for phosphine complexes of group-6 pentacarbonyl fragments, geometric findings are no direct measure for thermodynamic properties [170]. Especially the spatial distribution of the orbitals, which form the respective bond, have to be taken into account. Nonetheless, it is instructive and informative to compare bond lengths between a TM and coordinated tin(II). A major difficulty arises immediately when one tries to define an Sn(II) “coordinative radius”. Since the electron distribution around tin is highly anisotropic, the “bonding radius” heavily depends on the relative direction of the bond. Substituents R mainly use Sn orbitals with high p-contribution for σ- and π-bonding (covalent bonding). Bases perpendicular to the SnR2 plane form donor bonds with the empty 5p-orbital (coordinative σ-base bonding). Lewis acids such as TMs bind through an orbital with high s-participation (coordinative σ-acid bonding). In TM complexes, π-back bonding from TM d-orbitals into the empty p-orbital on Sn occurs concurrently (π-coordinative acid bonding), which makes a profound estimate for a single bond length difficult (Fig. 21).
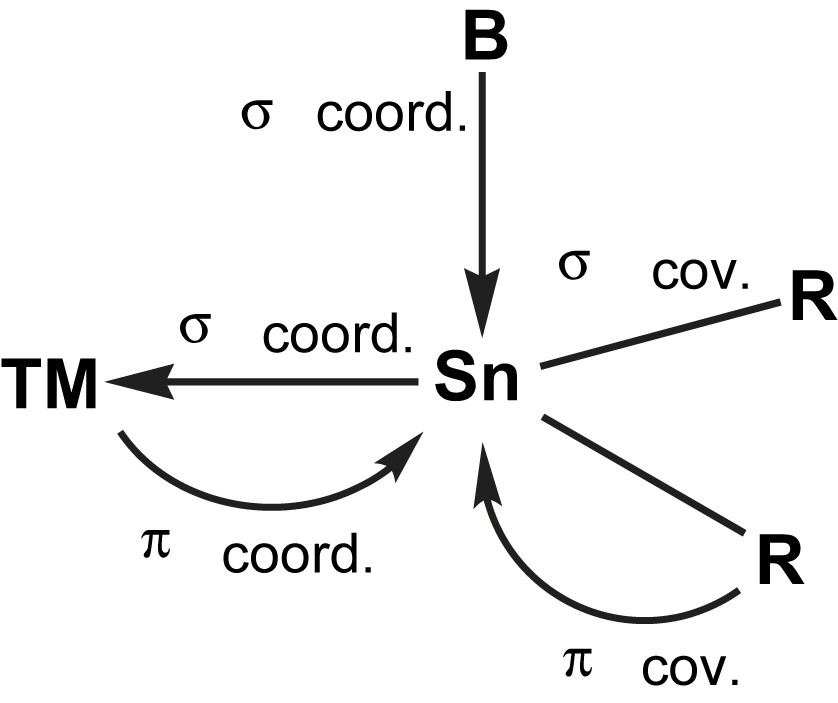
Bonding types with Sn(II) in TM stannylene complexes: covalent σ- and π-bonding between Sn and R, coordinative σ-base bonding (Sn–base), coordinative σ-acid bonding (Sn–TM), coordinative π-bonding (TM–Sn).
However, by taking the average of the difference between the Sn(II)–TM bond length and the covalent radius for the respective TM (for which sufficient data are available), a rather consistent value is obtained. This value might be defined as “Coordinative Lewis Base Radius of Sn(II)” rcoord(Sn(II)) (rcoord(Sn(II)) = d(Sn–TM) − r(TM)). 15 combinations Sn(II)–TM (Cr through Cu; Mo, Ru through Ag; W; Re; Ir; Pt) have been averaged to a value of 1.17 Å with an e.s.d. of 0.03 Å. The minimum and maximum bond lengths are 1.090 (for Mn) and 1.222 Å (for Pt). Considering only first-row TMs with Lappert's stannylene Sn(CH(SiMe3)2)2, thereby minimizing electronic and orbital effects by the substituents, a similar value of 1.18 Å with less deviation (min/max = 1.150/1.205 Å) is obtained. The variation of the radius along the periods is random, so different degrees of multibonding for different groups may be excluded. This rough estimate is smaller than the substituent “covalent bond radius” of 1.47 Å derived from the Sn–C distance in Lappert's stannylene 3 (2.24 Å) and the C(sp3) radius of 0.77 Å. The Sn–O distances for the four mono-thf adducts vary strongly between 2.22 and 2.38 Å (18a: 2.22 [92]; 22b: 2.29 [141]; 1a: 2.37 [27]; 9: 2.38 Å [27]). This is longer than those typically found for covalently bonded alkoxy substituents (e.g. 29 (d(Sn–OAr) = 2.01 Å [167]). An estimate for the THF dative radius from the B–O bond length in Ph3B∗THF results in a value of 0.81 Å (d(B–O) = 1.65 Å [179]; r(B) = 0.84 Å [109]), which yields an “Sn(II) acceptor radius” between 1.38 and 1.54 Å. However, it remains conceptually arguable to define an “acceptor radius” and if it is regarded at all, this value is a very coarse estimate with a huge variation.
The gradation of bond lengths reflects the relative extension of the orbitals involved in bonding around Sn(II): the coordinative donor bond radius rcoord(Sn(II)) is shortest (predominantly s-character; 1.17/1.18 Å), the coordinative acceptor radius (towards bases), and the covalent bonding radius to the substituents rcov(Sn(II)) possess predominantly p-character (1.38–1.54 Å; 1.47 Å).
3.7 Summary to solid-state structures
The Sn–TM bond length d(Sn–TM) in TM stannylene complexes R2Sn–TM follows a parabolic profile along the TM period. It reflects the variation of the TM bond radius recently reported from CSD analysis. The d(Sn–TM) is strongly influenced by steric effects; it shows (i) shortening with increasing electronegativity of R and (ii) elongation by base adduct formation. Weak but existing π-back bonding properties can be assigned to stannylene ligands by analysis of their electronic influence on TM–CO bond lengths in Ψ-octahedral group-6 pentacarbonyl and Ψ-trigonal bipyradmidal tetracarbonyl iron complexes as well as seen by their preference for the axial position in the latter. TM coordination reduces d(Sn–R) and increases (R–Sn–R), which is consistent with reduced s-character of the lone pair, as predicted by Bent's rule and calculated for phosphines. Bases on tin tend to increase (R–Sn–R), but only slightly influence d(TM–Sn). This suggests well-separated sets of orbitals for TM–Sn and Sn–R bonding on one side and Sn–base bonding perpendicular to the TM/R2 plane on the other side. A “coordinative Lewis base radius” rcoord(Sn(II)) of 1.18 Å can be assigned to Sn(II) ligands from analysis of first-row TM complexes with the alkyl stannylene Sn(CH(SiMe3)2)2.
4 Molecular structure–119Sn NMR spectroscopy relationships
Structure–spectroscopy relationships have been investigated for tungsten carbonyl complexes. However, the number of compounds for which both data sets are available is still limited (13), with strongly differing stannylene ligands. Therefore, unambiguous correlations cannot be expected but tendencies can be extracted.
4.1 Direct coupling constant 1J (119Sn–183W) versus bond length d(Sn–W)
A correlation between bond length and coupling constant is a priori not expected. Various calculations pointed out that bond length is not a good measure for thermodynamic bond strength [16], and coupling constants depend not only on hybridisation (vide supra). However, within the limits of a defined set of co-ligands, a correlation might work as shown with stannyl ligands in bent-sandwich {Cp2WR} complexes, where a consistent correlation between 1J(Sn–W) and d(Sn–W) has been found [180].
Having the limitations in mind, it is remarkable to find at least a tendency of 1J against d in R2Sn–W(CO)5 complexes (see Fig. 22). Complexes with short Sn–W bonds show large coupling constants and long distances correlate with smaller coupling constants. For the Weidenbruch stannylene complex RR′Sn–W(CO)5 (4a), the relatively small coupling constant with respect to the measured distance (d = 2.75 Å; 1J = 940 Hz) [56] might support π-bonding as shortening contribution to the bond length. In comparison, the longest distance belonging to the bis-intramolecular base adduct [{(OC)5W}Sn(8-Me2NC10H6)2] (17a) (d = 2.82 Å; 1J = 976 Hz) exhibits a similar 1J value.
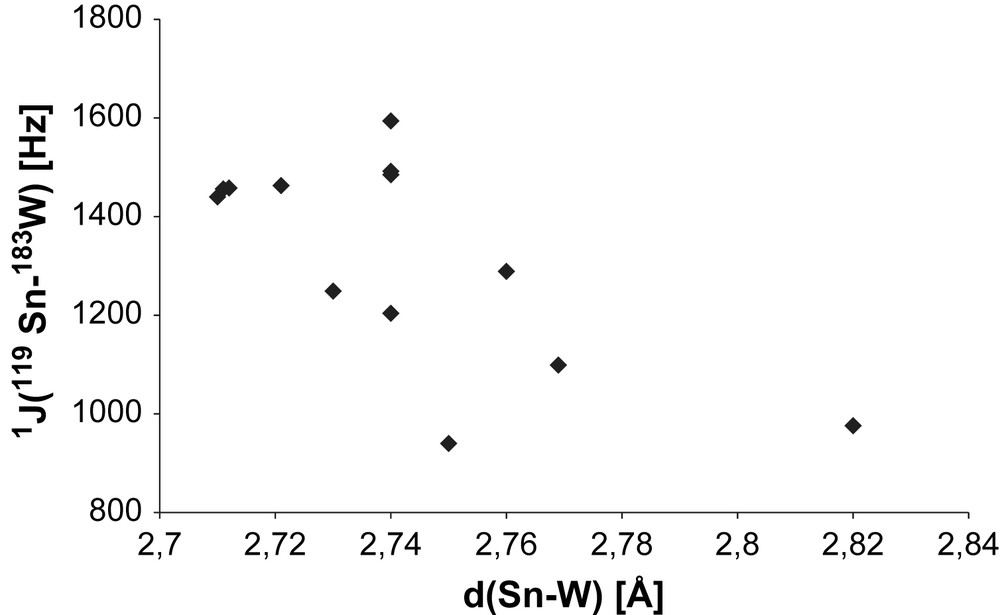
1J (119Sn–183W) versus d(Sn–W) in R2Sn–W(CO)5 complexes.
4.2 Direct coupling constant 1J (119Sn–183W) versus bond angle R–Sn–R
The bond angle R–Sn–R is an indicator for the hybridisation around tin and gives indirect hints on the relative s-character of the lone pair. From spectroscopy, the scalar coupling TM–Sn provides analogous information through the Fermi contact. It may therefore be expected that with increasing angle, the s-character of the lone pair decreases, hence the absolute value of the coupling constant decreases (for structure discussion see chapter 3.5.1).
In the series of dimeric alkoxo stannylene complexes [{(OC)5W}{(OC)5TM′}n{Sn2(OtBu)4}] [15,27], (TM′ = Cr, W; n = 0, 1 (14a–14e)), it is possible to investigate the influence of geometric parameters of tin on the 119Sn NMR parameters with subtle changes of the stannylene ligand. Variation of the stannylene is realised by variation of the coordination on the second tin atom. It is indeed seen that a smaller bonding angle within the Sn2O2 cycle corresponds to a higher coupling constant (74.8°: 1463 Hz; 74.0°: 1485/1492 Hz; see Table 18).
Comparision of d(TM–Sn), R–Sn–R) and 119Sn–183W coupling constant J in dimeric alkoxo complexes [{(OC)5W}{(OC)5TM′}n{Sn2(OtBu)4}] [15,27], (TM′ = Cr, W; n = 0, 1).
| Compound | d(W–Sn) [Å] | (O–Sn–O) [°] | 1JW–Sn [Hz] |
| [{(OC)5W}{Sn2(OtBu)4}] (14a) | 2.721 (staggered) | 74.8 | 1463 |
| [{(OC)5W}{(OC)5Cr}{Sn2(OtBu)4}] (14b) | 2.740 (eclipsed) | 74.0 | 1492 |
| [{(OC)5W}2{Sn2(OtBu)4}] (14c) | 2.721 (staggered) | 74.0 | 1485 |
| 2.757 (eclipsed) | 74.1 |
However, the TM–Sn bond length does not correlate in the expected direction, which emphasises that the bond length depends not only on hybridisation but also on further factors like sterics, packing and electrostatics (see the discussion of eclipsed and staggered conformers in Section 3.2.2.1).
Acknowledgement
D. Agustin and M. Ehses are grateful to Prof. Dr. Michael Veith, Saarland University, Saarbrücken, Germany for giving the opportunity to develop research activities in the field of tin chemistry during post-doctoral stay and beyond (ME) in the frame of the International Research Training Group 532 (GRK532). The authors thank DFG, DFH/UFA and MENESR (International Research Training Group 532 – GRK532) for financial support.
1 Alternative nomenclature for groups 13: “trieles”, 14: “tetreles”, 15: “penteles”; see e.g. N. Wiberg, in: Holleman-Wiberg, Lehrbuch der Anorganischen Chemie, N. Wiberg (ed.), 102. ed., De Gruyter (publ.), 2007 (Chapter XV), p. 861.
2 For a brief discussion of hybridisation with heavy elements see Section 3.5.2.
3 However, as usual with simplified valence bond models, it has to be kept in one's mind that various theoretical investigations show hybridisation with heavier p-block elements to be much less important than for first-row elements and incorporation of d-orbitals in bonding to be very unfavourable for p-block compounds (see e.g. W. Kutzelnigg, Angew. Chem., 96 (1984) 262–286; Angew. Chem., Int. Ed. 23 (4) (1984) 272–295.)