

1 Foreword
In the mid 1980s, Jacques Livage thought that high-resolution solid-state nuclear magnetic resonance (NMR), coupled with magic angle spinning (MAS-NMR), was a necessary equipment for inorganic chemists. He thus played a key role in the purchase of a 400 MHz spectrometer for the department of chemistry of the Université Pierre-et-Marie-Curie (UPMC) in Paris (the magnet is indeed still operational after 22 years!!!) and this was the beginning of the solid-state NMR activity of the sol-gel group in Paris.
Then in 1993, it became evident for Jacques that the group needed its own equipment and he succeeded in gathering enough funding to purchase a 300 MHz spectrometer. This was the starting point for a new research group whose activity was focused on the use of solid-state NMR for the structural characterization of a wide variety of sol-gel derived materials. Currently, one of the main topics developed by this group is dedicated to a better characterization of organic-inorganic interfaces in silica-based hybrid systems.
2 Introduction
Solid-state NMR techniques are now widely developed for the characterization of hybrid materials, not only to probe local atomic environments, but also to explore spatial proximities between various sites. The versatility of this spectroscopic approach relies on the fact that it is possible to play with all NMR interactions (chemical shift, scalar coupling, dipolar and quadrupolar interactions) to specifically address important structural questions. Additionally, solid-state NMR experiments can now be combined with modeling tools and ab initio calculations of NMR parameters based on DFT models to help in the interpretation of the experimental spectra [1].
The aim of this article is to illustrate how solid-state NMR techniques can help to explore organic-inorganic interfaces in silica-based hybrids. The examples that were selected among the author's studies are representatives of key structural questions, still poorly addressed:
- • presence of covalent bonds between the silica surface sites and the organosiloxy moieties after grafting;
- • interactions between templating and inorganic species in mesostructured silicas;
- • or physical state of guest molecules encapsulated in porous silica.
This will allow us to give an overview of the various techniques that can be routinely performed in such systems where 29Si, 13C and 1H nuclei can be probed. The experimental details for each study can be found in the relevant cited references.
3 Functionalization of silica particles
Silica plays an important role in many industrial fields. With its chemical and thermodynamical inertness and its low cost, silica powders are commonly used in a wide range of materials (tires, cosmetics, microelectronic devices, abrasives, catalyst supports, chromatographic columns…). A number of innovative applications are linked to its surface properties, and especially to the quasi infinite possibilities to tailor these properties by functionalization. This is commonly achieved by reacting organoalkoxysilanes or organochlorosilanes on silica, and the unlimited number of organic functions that can thus be introduced at the silica surface allows the materials chemist to precisely adjust the surface property to the desired application.
However, despite the huge amount of synthetic works that involve grafting of organoalkoxysilanes on silica, there are only few accurate descriptions of such anchoring [2]. Indeed, in most cases, the formation of a covalent link between the surface site and the organosilane moiety is taken for granted and not discussed. Actually, only few authors mentioned that the coupling agent could just be physisorbed onto the surface [3]. 29Si NMR, IR or UV spectroscopies are commonly used to study the modification of the silica surface but, neither the efficiency of the grafting onto the surface nor the characterization of the link between the organosilanes and the silica surface is often reported.
NMR should be the perfect tool to probe the covalent grafting by using the J-coupling, which characterizes through-bond interactions between nuclei. This is routinely done on liquid samples. In rigid solids, the isotropic J-coupling value is usually much smaller (tens of Hz) than the other interactions (through-space dipolar coupling, chemical shift anisotropy and quadrupolar interaction when relevant) and was consequently often neglected in solid-state NMR experiments. However, during the last decade, the use of J-derived solid-state NMR techniques largely developed to become extremely valuable tools to describe connectivities in inorganic framework [4]. For isotopes with low natural abundance such as 29Si, the sensitivity of experiments based on the homonuclear J-coupling and thus directly related to the probability of existence of a 29Si–O–29Si spin pair, is low. This is why most of the experiments have been performed on 29Si-enriched samples such as zeolites [5] or silicates [6,7]. It is worth mentioning, however, the early experiments recorded on natural abundance samples by the group of C. Fyfe [8]. However, in the case of grafted particles, the number of interesting 29Si–O–29Si pairs will be related to the number of surface sites and thus will be extremely low. We thus evaluate the possibility to identify covalent grafting of organoalkoxysilanes by simply looking at the isotropic chemical shift values of the organosiloxane species. The reaction of dimethyldiethoxysilane (DMDES) (Me2Si [OEt]2) and methyltriethoxysilane (MeSi [OEt]3) on silica particles in water-rich medium (water/ethanol 25/75 v/v) was studied [9]. The ultimate goal was to bring hydrophobicity to precipitated silica synthesized in an aqueous medium, and it was important to establish the presence of covalent bonds between the organosilane and the silica surface that will ensure better stability of the functionalization.
3.1 Identification of covalent bonds with the surface
3.1.1 Grafting of dimethyldiethoxysilane
DMDES has two functions that can be potentially anchored onto silica particles. In the 29Si cross-polarization (CP) MAS NMR spectra (Fig. 1), in addition to the signals corresponding to the Q units of silica, two resonances at −9.5 ppm and −17.2 ppm correspond to the condensed D1 and D2 units of the organosilane (Dx, Me2Si [OSi]x[OH]2−x) respectively.
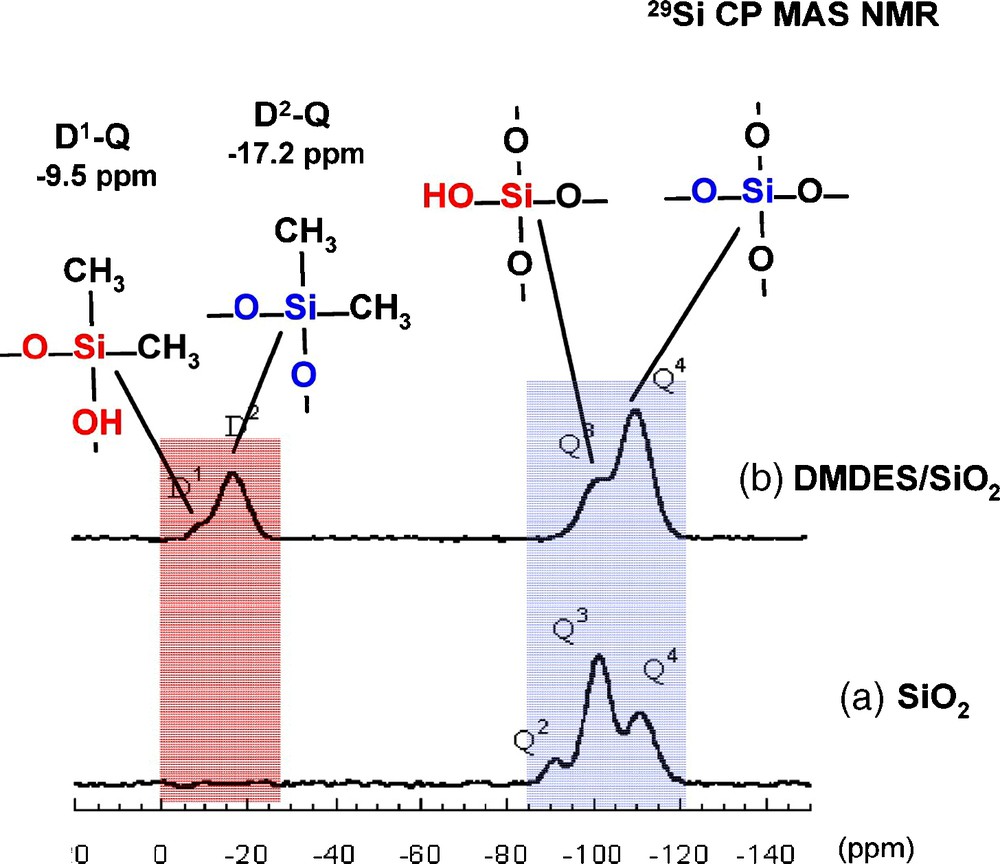
29Si cross-polarization (CP) magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra of a) precipitated silica (SBET = 300 m2/g) and b) silica (2 g) reacted with dimethyldiethoxysilane (DMDES) (1.2 mol/l) in water/ethanol mixture (25/75 v/v).
Hydrolysis of DMDES in solution shows that peaks due to self-condensed species D1–D and D–D2–D, appear between −12.0 ppm and −13.0 ppm and between −18.0 ppm and −24.0 ppm respectively. A typical spectrum is presented in Fig. 2. The assignment of the main peaks reported on the figure has been achieved according to the literature [10,11].
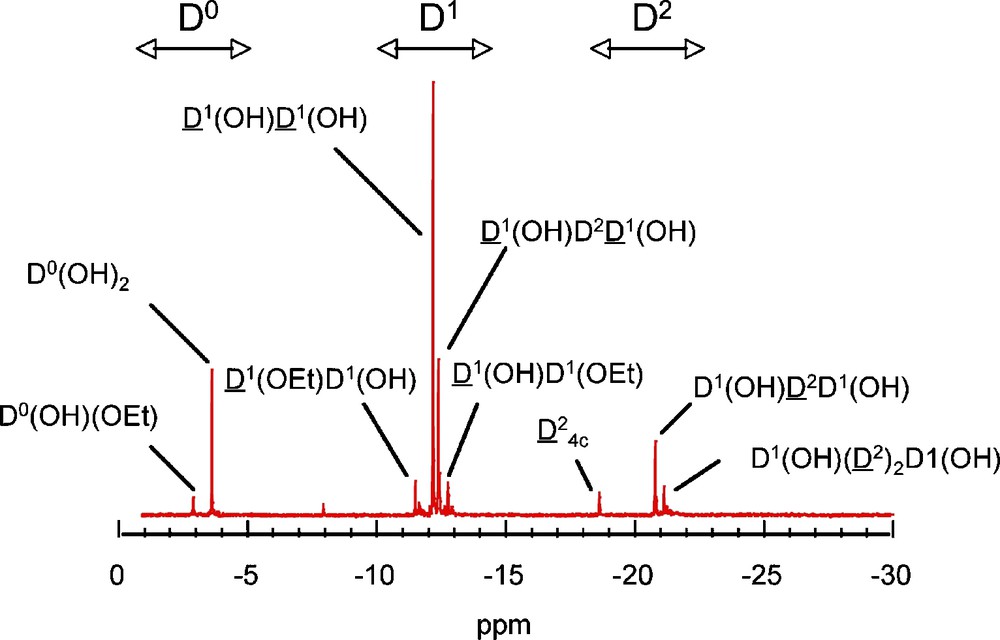
Typical spectrum recorded on a hydrolyzed solution of dimethyldiethoxysilane (DMDES).
The observed differences with the chemical shift values of the DMDES/SiO2 sample clearly indicates the existence of covalent bonds between the organosiloxane and the silica surface, in agreement with what was already observed for the chemical shifts of co-condensed species between D and Q units in solution [12]. In addition, the chemical shift of D1–Q4 and D2–Q4 units in spherosilicate-based networks has also been reported at −8.5 ppm and −16.5 ppm respectively [13]. This is the first time to our knowledge, that in these systems, the presence of covalent bonds is demonstrated and not taken for granted.
3.1.2 Grafting of methyltriethoxysilane
The chemical shift of the T units (Tx, MeSi [OSi]x [OH]3−x), corresponding to the MTES in interaction with the surface of particles are −45.2, −54.4 and −64.3 ppm for T1, T2 and T3 respectively [9]. The difference of the surface species chemical shifts with the self-condensed species in solution (T1–T between −47.0 ppm and −49.0 ppm; T2–T between −55.0 ppm and −58.0 ppm), is smaller than for the D units from DMDES and there is no shift in the case of T3 units (T3–T ∼ −65 ppm). So in the case of the trifunctional organoalkoxysilanes, evidence for the presence of covalent bonds between the organosilane derived species and the silica surface is more difficult to prove, especially for the highly condensed T3 units, for which many possibilities of condensation links between neighboring organosilane molecules exist.
3.2 Description of grafted species
A more detailed study was performed with the DMDES/SiO2 system to see whether the pH used for the reaction could influence the type of grafted species. First, hydrolysis of DMDES was followed in solution at two different pH values (1 and 9) by 29Si NMR. The experimental conditions in terms of concentration were similar to those used for the grafting procedure (1.2 mol/L in water/ethanol mixture (25/75 v/v) for a total volume of 60 ml). A quantitative analysis of the detected species was possible due to the presence of an internal reference (3-[trimethylsilyl] propionic acid-d4 sodium salt) of known concentration in the NMR tube.
At pH = 9 (Fig. 3a), the only species are the three monomers Me2Si(OEt)2−x(OH)x with a relative composition of 40–20–40 for x = 0, 1 and 2, respectively after 200 mn of reaction. As expected, under basic conditions, the hydrolysis rate is low [14]. The kinetics is totally different at pH = 1 (Fig. 3b), where monomers disappear rapidly within 10 mn, leading to the formation of short chains characterized by D1 end groups (−11 to −13 ppm) and D2 units (−20 to −22 ppm), as well as tetrameric D24c cyclic species (−18.6 ppm). The ability of DMDES to form cyclic species is very important in acidic conditions, and indeed this species becomes dominant in solution after two hours of reaction. A typical spectrum is presented in Fig. 2 with the corresponding species [10,11].
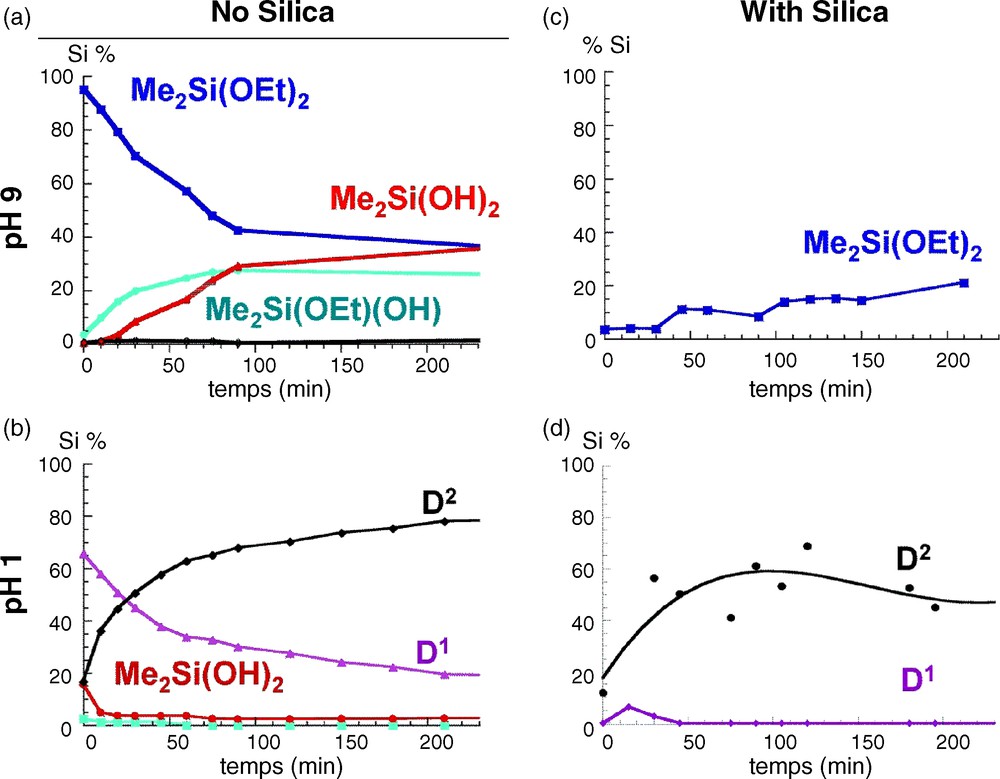
Time evolution of Si species during the hydrolysis of dimethyldiethoxysilane (DMDES) in water/ethanol mixture (25/75 v/v) a) at pH = 9 with no silica; b) at pH = 1 with no silica; c) at pH = 9 in presence of silica and d) at pH = 1 in presence of silica. Quantitative analysis was extracted from 29Si solution state nuclear magnetic resonance (NMR) spectra.
The same experiments were reproduced in the presence of silica (2 g added to the initial 60 ml solution). An important decrease of the total concentration of detected Si sites is observed, less than 20% for pH = 9 and less than 60% for pH = 1, which can be attributed to an adsorption of Si species on the silica surface. In basic medium, most of the species react with the surface, and only the unhydrolyzed monomer remains in solution. The absence of monomers with Si-OH groups strongly suggests their strong ability to interact with the silica surface. Under acidic conditions, cyclic tetramers are dominant in solution with the absence of Si-OH groups similar to what was observed at pH = 9. This large difference in the type of species in solution is indeed reflected in the 29Si CP MAS NMR response of the silica powders grafted at pH = 9 (Fig. 4a) and pH = 1 (Fig. 4b). While for pH = 9, two broad signals at −17 ppm and −9 ppm are observed, at pH = 1, three peaks are observed at −22 ppm, −19 ppm and −9 ppm. Interestingly the intensity of the peak at −22 ppm is enhanced when recording the 29Si NMR spectrum using a one-pulse sequence. The lack of efficiency of the CP sequence for this peak is assigned to a higher mobility of the corresponding species that partially average the heteronuclear 1H-13C dipolar coupling, which is the driving force of the polarization transfer in a CP sequence. This peak can thus be safely assigned to flexible polydimethylsiloxane chains. Analysis of the chemical shift values indicate the presence of short chains of D-units with a large number of covalent bonds with the surface for pH = 9 while longer chains with fewer links are present at pH = 1, in agreement with the species that were detected in solution at the corresponding pHs. Hence, comparison between the 29Si NMR responses shows the existence of different surface species, and allows proposing different coverage scenarios depending on the pH conditions. (Fig. 4).
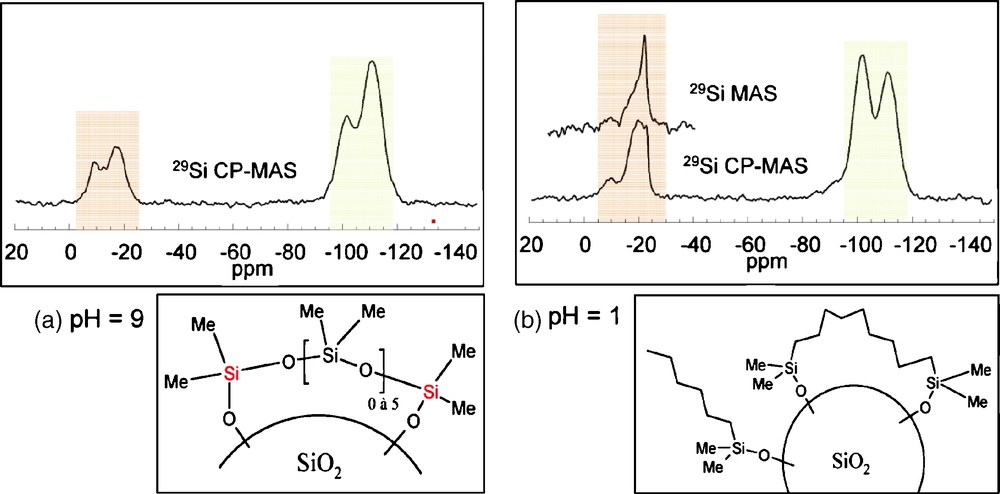
29Si magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra recorded on grafted silica powders and the corresponding proposed surface species for pH = 9 (a) and pH = 1 (b).
4 Interfaces between silica and amphiphilic template molecules
In the last 15 years and still now, a very large number of works in the field of silica-based hybrid materials deals with surfactant templated mesoporous silicates and derived systems. Here, the interactions that the amphiphilic template develops with the siliceous species are essential to dictate the type of surfactant/silica mesophase that will form, and then will transform into a periodic mesoporous silica after surfactant removal.
Solid-state NMR plays also here a key role to better describe the silica/surfactant interface due to its ability to explore spatial proximities between various sites. We are no longer exploring the existence of covalent bonds, but the presence of weaker bonds such as hydrogen bonding or electrostatic interactions. This is achieved by manipulating one main magnetic interaction, namely the through-space dipolar coupling which depends on the internuclear distance. Heteronuclear 1H-29Si dipolar couplings can be probed essentially through one of the most used sequences in solid-state NMR, the CP technique [15], and homonuclear 1H-1H dipolar coupling through the sensitive double-quantum (DQ) [16] experiments. Two examples will be described in the following paragraphs.
4.1 Surfactant/silica interactions
A key NMR technique to investigate the organic/inorganic interfaces is the 2D 1H-29Si heteronuclear correlation (HETCOR) sequence-based on a CP sequence. This was nicely illustrated by Chmelka et al. to investigate the distribution of polyethylene oxide (PEO) and polypropylene oxide (PPO) copolymer blocks within the silica matrix [17], as well as the molecular proximities between the structure-directing surfactant molecules and the crystal-like silicate sheets in layered silicate surfactant mesophases [18]. We have also investigated the silica surfactant interactions in mesophases characterized by 2-D hexagonal p6m symmetry and prepared with cetyltrimethylammonium bromide (CTAB). Extremely valuable information was extracted from 29Si{1H} heteronuclear correlation (HETCOR) experiments [19], on the spatial proximity between the organic moieties and the silica surface. Variation of contact time, tCP (time during which the magnetization transfer occurs between the two dipolar coupled spin systems) can be used to explore increasing internuclei distances. However, these two-dimensional experiments are rather time consuming, especially at moderate static magnetic field.
We have thus developed a sequence-based on a 1H-29Si-1H double CP transfer (Fig. 5a). A first transfer from 1H to 29Si selects the Si sites close to protons, and essentially the silica surface sites. Then a second back-transfer to 1H allows the specific detection of protons that are dipolarly coupled to these surface sites (Fig. 5b). Fig. 5c compares the 1H NMR spectra obtained using this sequence and the classical one-pulse sequence: the intense signal arising from the protons of the alkyl chain disappeared, due to the efficient filtering effect of the double CP sequence that selectively enhances the peak intensity of protons close to the surface Si sites.
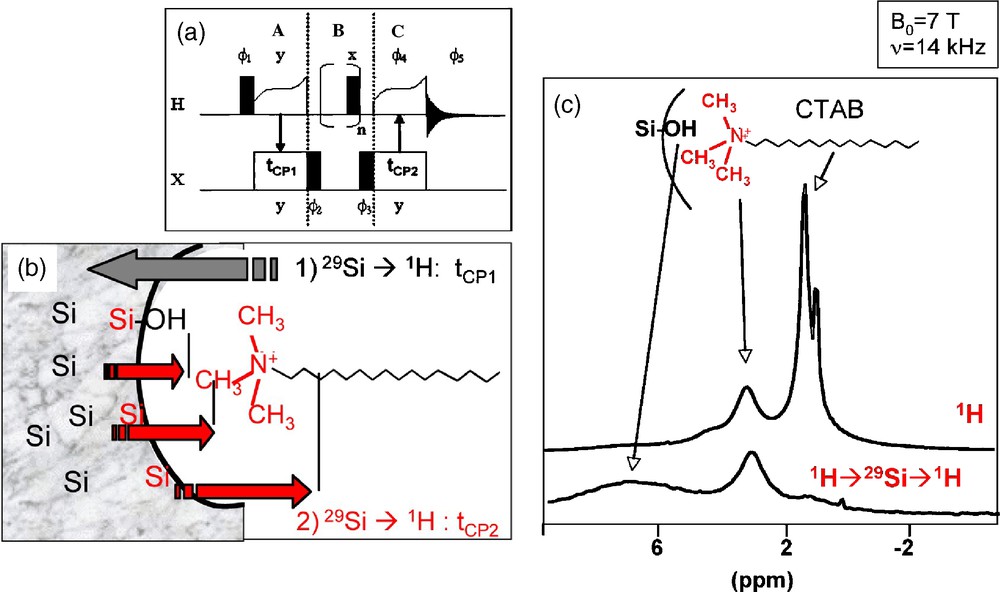
(a) 1H-29Si-1H double cross-polarization (CP) sequence and (b) schematic representation of the magnetization transfers occurring at the silica/surfactant interfaces. (c) Comparison between 1H MAS nuclear magnetic resonance (NMR) and 1H-29Si-1H double CP MAS NMR spectra recorded on a cetyltrimethylammonium bromide (CTAB) templated silica mesostructured sample.
We have chosen to investigate with this new sequence, the CTA+/SiO2 interfaces in two templated mesotructured silica samples characterized by the same 2D-hexagonal symmetry, prepared with the same templating agent (CTAB), but one under acidic conditions (sample CTAB/HCl) and the other under basic conditions (sample CTAB/NaOH). Because of the different synthetic pathways, the proposed interaction mechanisms are different [20]. In the basic medium, a direct S+I− interaction is proposed between the positively charged surfactant (S+) and the negatively charged silica matrix (I−). In the acidic medium, interactions with surfactant micelles are mediated by a negatively charged counter-ion (X−). The interaction would be of type S0X−I+, which accounts for a direct charge neutralization between the counter-ion and the surfactant. In our case, the counter-ion is Cl−, because of the excess of HCl introduced with respect to the Br- ions initially introduced with CTAB.
Fig. 6 compares the 1H MAS NMR spectra recorded on CTAB/HCl and CTAB/OH samples. The single pulse (SP) experiments give very similar spectra with peaks at 0.9 ppm, 1.3 ppm and 3.3 ppm, respectively attributed to alkyl chain CH3 protons, alkyl chain CH2 protons (exception made for N-CH2 in α-position) and polar head N-CH3 and N-CH2 protons. One can, however, notice an additional peak for CTAB/HCl at 4.2 ppm due to the presence of water, that disappears when the sample is dried overnight at 100 °C. The 1H-29Si-1H double CP spectra have been recorded with the first 1H-29Si CP contact time, tCP1 = 3 ms, which corresponds to the quasi exclusive detection of Q3 sites (SiO4 sites with one non bridging O). The effect of 1H spectral editing is evident: the intense signal due the alkyl chain protons is barely detected, and a main difference is now visible between the two samples: a peak centered on 7 ppm due to Si-OH groups is present for CTAB/HCl (Fig. 6c), while no peak of significant intensity is detected for CTAB/OH (Fig. 6e). Indeed, the undefined broad signal of low intensity ranging from 5 to 15 ppm can be tentatively assigned to silanols that are hydrogen bonded to a siloxy group, SiO−•••HOSi [19b]. Comparison between the spectra of CTAB/HCl before and after drying confirms the assignment of the peak at 4.2 ppm due to water. But this water is certainly in interaction with the surface Si sites since the signal can be significantly detected through a CP transfer. These experiments show that the basic CTAB/OH sample is characterized by a majority of Si-O- surface groups while the acidic CTAB/HCl sample exhibits a silanol-rich surface, certainly with extensive H-bonding to adsorbed water.
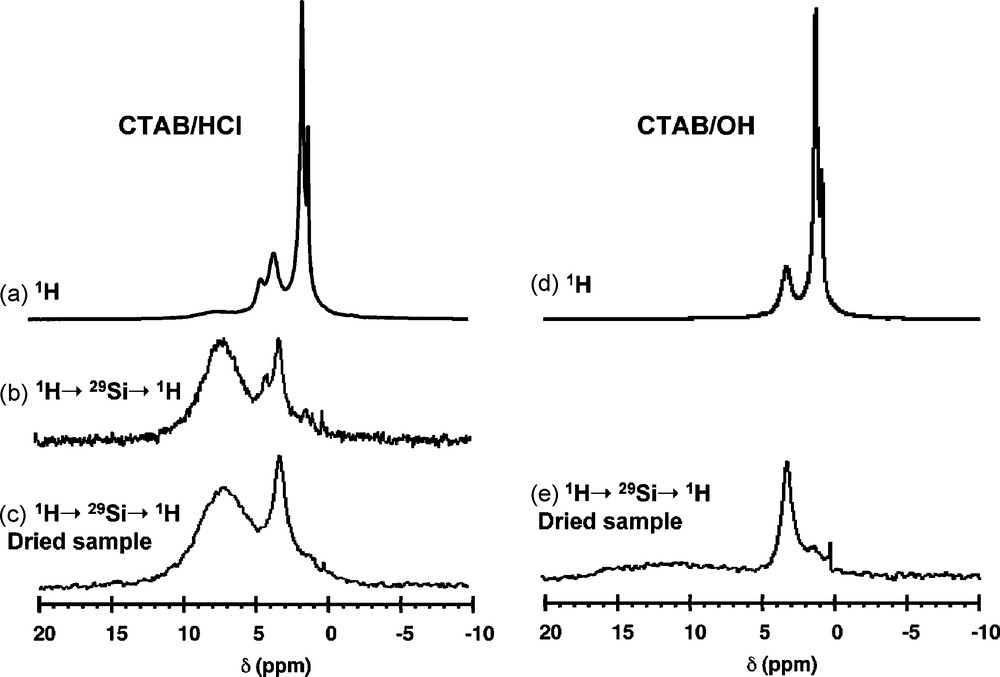
1H magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra recorded on CTAB/HCl and CTAB/OH samples with direct detection (a and d) and with 1H-29Si-1H double CP (b, c and e). νMAS = 14 kHz; tCP1 = tCP2 = 3 ms.
4.2 Localization of organic functions
For many targeted applications, the surface of these templated mesoporous silica is functionalized with organic functions. Two synthetic approaches are followed: either postgrafting via the reaction of organoalkoxysilane or organochlorosilane with the silica surface sites or one-pot synthesis based on the simultaneous hydrolysis of the alkoxysilanes and organoalkoxysilanes in the presence of templating agents. In this last case, one relies on the preferential positioning of the organoalkoxysilane-derived hydrolyzed species at the surfactant/silica interface, to ensure the presence of the organic functions at the silica surface, and not in the walls of the final porous materials.
The specific interactions between the organic functions and the surfactant can be evidenced by using high resolution 1H MAS NMR combined with the most sensitive experiment to probe homonuclear dipolar couplings in solids, the double-quantum (DQ) experiment. For 1H, the BABA or Back-to-Back sequence is often used [21,22]. Templated silicas functionalized with vinyl groups have been studied to characterize the location of the vinyl groups with respect to the polar head groups of the surfactant molecules (CTA+). In the 1H DQ-SQ correlation experiment (Fig. 7), correlation peaks between the vinyl protons and the N(CH3)3+ head-group of CTA+ molecules are observed, which clearly indicate that vinyl groups are located at the surface. Interestingly, correlation peaks are also observed between the vinyl protons and those of the alkyl chains, indicating a close spatial proximity between the vinyl groups and the surfactant molecules.
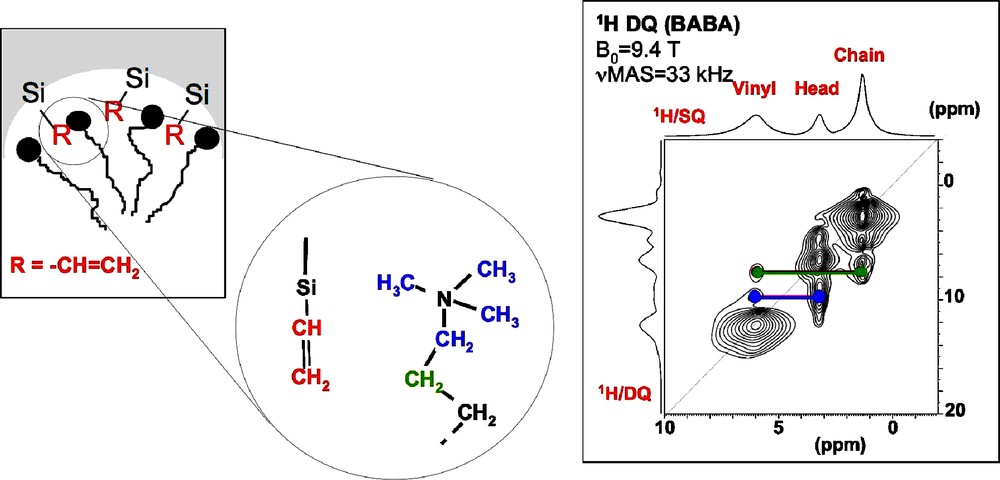
1H double-quantum (DQ) magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra recorded on templated silica modified with vinyl groups.
5 Host-guest interactions in mesoporous silica
Mesoporous silicas with long range ordered porosity appear also very attractive as host matrices for a large variety of organic molecules. The possibility to obtain a well-ordered porous network can facilitate the diffusion of species; the pore size can be tuned by a proper choice of the templating agent and thus be adapted to the size of the guest molecules; as illustrated previously, the surface can be easily functionalized by a large range of organic groups, in order to tune the host-guest interactions.
These materials are currently exploited to develop specific adsorbents for air and water pollutant filtering [23], but little attention has been addressed so far to the behavior of the pollutant within the porous host network. This is obviously an important issue to consider for further materials recycling. Another field of applications that is currently explored is the controlled release of drugs such as ibuprofen [24]. In that case, the possibility to encapsulate large amounts of drugs up to 400 mg/g of SiO2 is definitely a key argument. Additionally, a suitable functionalization of the silica surface can provide a way to control the drug release rate by playing with the drug-surface interactions [25]. In such applications, the interactions between the guest molecules and the silica surface need to be better characterized. However, few NMR studies have been published so far on this topic.
The first study that our group published in this area was the investigation of the behavior of ibuprofen molecules encapsulated in mesoporous silica [26,27]. Despite the large amount of ibuprofen present in the porous structure (≥ 600 mg/g of powder with pore diameters of 35 or 116 Å), the 1H MAS NMR spectra present extremely good resolution for a solid sample with 25 to 40 Hz line width (Fig. 8a) except for the broader peak at 6.7 ppm assigned to Si-OH groups based on a 1H-29Si heteronuclear correlation experiment. Such sharp lines are due to an efficient averaging of the homonuclear 1H-1H dipolar interactions caused by a rapid reorientation of the ibuprofen molecules in the pores. For comparison, the behavior of rigid molecules is illustrated in Fig. 8b with the 1H MAS NMR spectrum of crystalline ibuprofen, showing much broader peaks, but also an additional peak at 11.5 ppm due to the carboxylic group. The absence of this peak for the encapsulated ibuprofen tends to show that the associated protons are involved in a chemical exchange process, possibly with the Si-OH groups of the silica surface. Such high mobility was also found for 2-chloro-4-methoxyphenol (CMP) that was encapsulated in mesoporous silica powders in the course of a study to evaluate the performance of such powders to adsorb pollutants in water [28].
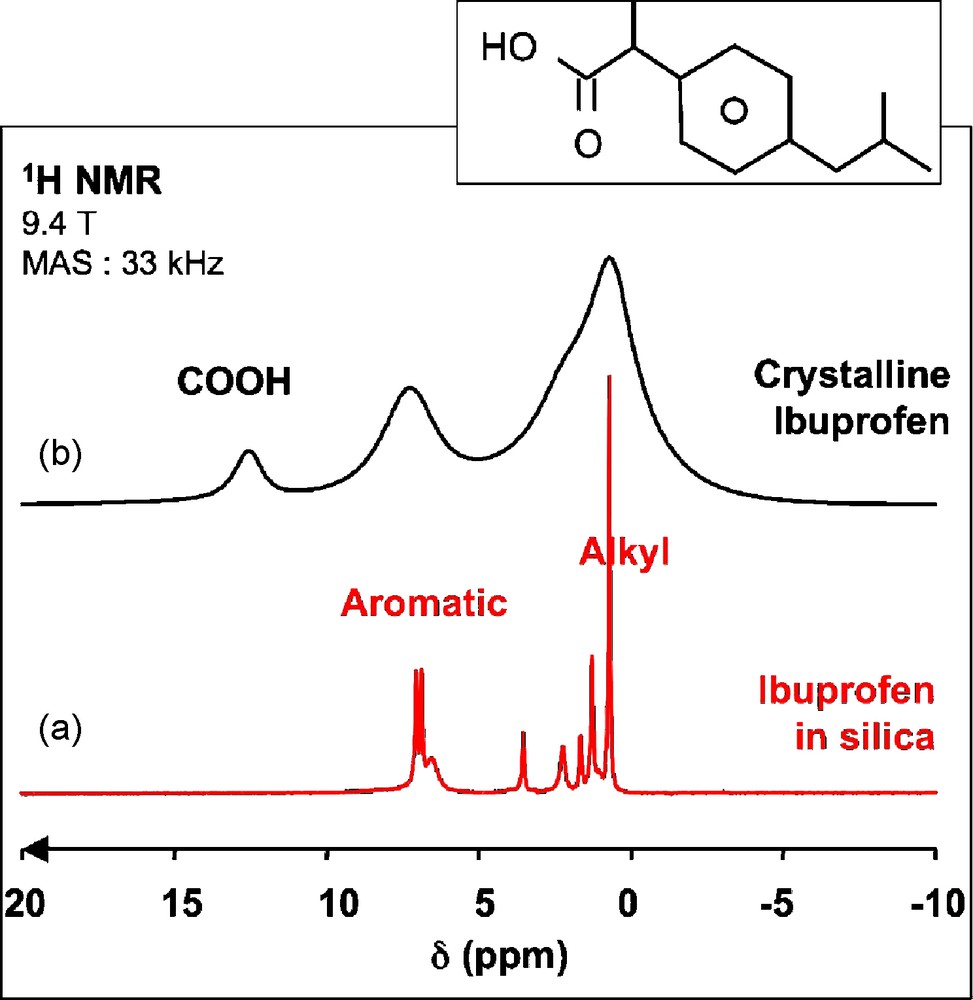
1H magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra of a) ibuprofen confined in mesoporous silica (pore diameter: 116 Å) and b) crystalline ibuprofen (T = 298 K).
A complete study was thus performed to characterize drug-model molecules such as ibuprofen, benzoic acid and lauric acid, encapsulated in mesoporous silica (30 Å pore diameter) and submitted to strong confinement effects [29]. In particular, we show that by a careful choice of the solid-state NMR sequences, it is possible to efficiently characterize these molecules and their interactions with the pore surface. 13C NMR spectroscopy is a powerful tool to characterize and even quantify entrapped and non-entrapped species by using either an SP or CP experiment, but also sequences derived from solution-state NMR and based on J-coupling, which are very efficient with highly mobile molecules.
The effect of confinement can also be studied by recording NMR spectra at lower temperatures. In the case of benzoic acid (Fig. 9a), a broadening of the aromatic peaks is observed with decreasing temperature, but one can still distinguish three signals due to protons in ortho position (6.6 ppm), meta-para positions (7.3 ppm), and due to Si-OH (5.9 ppm). The broadening is due to the presence of a distribution of H sites caused by a reduction in mobility that freezes the benzoic acid molecules in unequivalent positions, as in a glassy state. This was also confirmed by the evolution of the 13C line shapes with temperature, which revealed a similar line broadening. At 238 K, this reduction in mobility allows the CP sequence-based on through-space dipolar coupling to recover enough efficiency as to record a 1H-13C heteronuclear correlation (HETCOR) experiment (Fig. 9b). The presence of a correlation peak between the 13C signal due to C = O and the 1H signal due to Si-OH demonstrates that at this temperature, benzoic acid molecules interact with the silica surface.
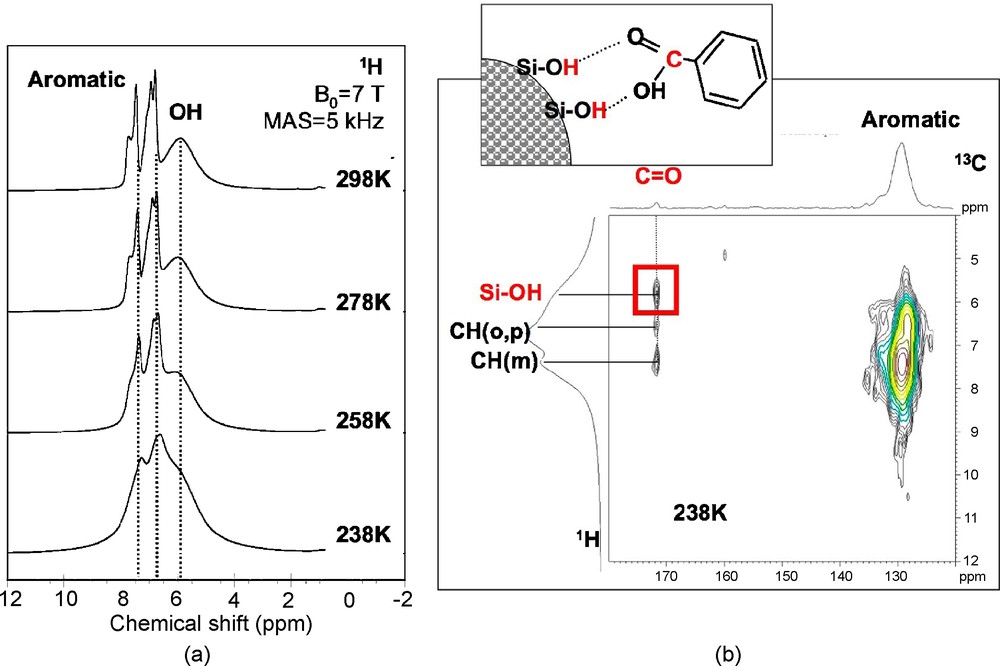
a) Variable-temperature 1H magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra of benzoic acid confined in mesoporous silica (pore diameter: 30 Å) and b) 1H-13C HETCOR experiment recorded at 238 K.
6 Modeling of silica/organic interactions
In parallel to the range of NMR experiments used to better characterize the organic-inorganic interface in silica-based hybrid materials, it is extremely important to use modeling tools to help in the spectra interpretation. A representative model for a hydroxylated surface of amorphous silica obtained using a periodic ab initio approach was recently proposed [30] (Fig. 10a). Spectroscopic data as well as dehydrogenation energies and interaction with water were determined by means of periodic density functional theory (DFT) calculations.
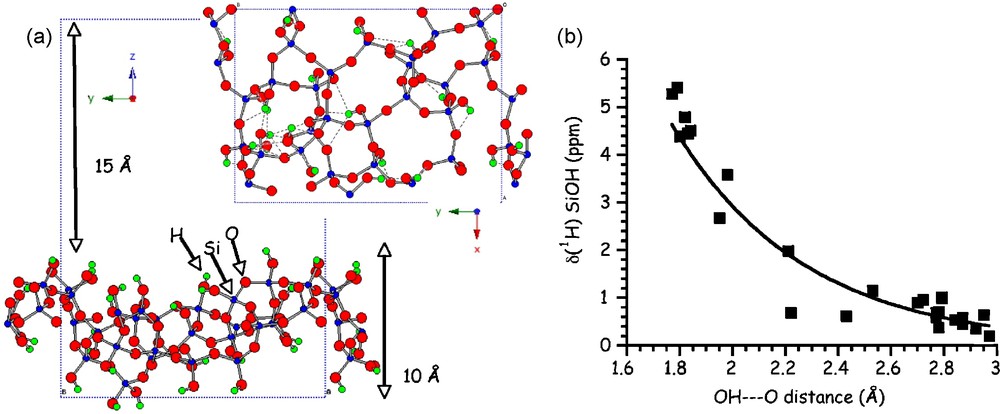
(a) Representation of the side and top view of the hydroxylated silica surface (b) 1H calculated isotropic chemical shift values of silanol sites Si-O-H as a function of OH···O distances.
In particular, NMR parameters were calculated using periodic boundary conditions and a plane wave pseudopotential method recently proposed by Pickard and Mauri [31] that provides data in excellent agreement with experiments in a wide range of solids including silica polymorphs [32] and amino acids [33]. The aim was to check the relevance of the amorphous silica surface model by comparing calculated NMR parameters with experimental values of Si–O–Si bonds in similar systems. The obtained results were also confronted with the relationships previously proposed between NMR properties and local environments like for example correlations between 17O quadrupolar parameters and distributions of structural parameters as the Si–O–Si angle and Si-O distance [34,35].
Moreover, the calculations of 1H and 17O NMR parameters related to the Si-OH surface sites are important since there are currently very few experimental NMR data reported. Hydrogen bonding is a common way of interaction at the organic-inorganic interfaces, and the existence of such weak bonds will however greatly influence the 1H and 17O NMR parameters. Interestingly, calculated δiso(1H) decreases with increasing OH⋯O hydrogen bond lengths (Fig. 10b) as already reported from previous calculations in silicate clusters [36] and from experimental NMR data in organic and inorganic compounds [37–40].
Such a realistic model for this complex amorphous surface appears as a valuable tool to investigate now how an organic molecule can interact with a silica surface. Preliminary results have already been obtained on the adsorption of gaseous and micro-solvated glycine [41].
7 Conclusion
Solid-state NMR is a very versatile spectroscopy to explore organic-inorganic interfaces in silica-based hybrid materials. All NMR interactions, ranging from the isotropic chemical shift to the homonuclear or heteronuclear through-space dipolar interactions can be used as spectroscopic spies to characterize the presence of covalent bonds or weaker bonds such as hydrogen bonding at the organic-inorganic interface. In these studies, 1H NMR plays a key role, and recent technical (ultra fast magic angle spinning experiments) and methodological (efficient homonuclear dipolar decoupling) advances will lead to even higher performance approach to investigate such interfaces in the near future. Additionally, the newly proposed model for amorphous silica surfaces will provide a very valuable tool for first-principle calculations of NMR parameters that will help to validate the assignment of the experimental data.
Acknowledgments
The authors would like to thank all the PhD students, postdocs and collaborators who contributed at various levels to the projects described in this manuscript. Special thanks to Dr. Sophie de Monredon-Senani who worked on the functionalization of silica particles.