

1 Introduction
Synthesis of compounds that change drastically their optical properties upon complexation with metal ions is of practical importance, since they can be used for metal ion sensing and as optical switches [1]. In this respect, the photochemical and complexation properties of aza-crown ethers modified with chromophore groups are of major practical interest and have been part of our studies in the last decades [2–6]. On the other hand, the photochemical properties of 2-substituted 1,3-indandiones are well known [7–9], as well as their good complexation ability with the transition metal ions [10–12]. The synthesis of various 2-substituted-1,3-indandiones has initially been stimulated by their known anticoagulant and pharmacological properties [13–15]. Further, the chemistry of 2-acyl-1,3-indandiones has been thoroughly studied, since they served as useful synthons for the preparation of variety of tricyclic heterocycles [16,17].
In the last few decades, the interest in 1,3-indandiones has moved from pure synthetic chemistry to the field of material science [18]. As a very strong electron acceptor, 1,3-indandione is a part in new dipolar molecules exhibiting interesting optical properties and used as dopants in novel functional materials [19–21]. A famous example is N,N-dimethylamino-benzylidene-1,3-indandione (DMABI) and its derivatives, whose photophysical and non-linear optical properties were extensively studied [9,20,22–27].
Lastly, our interest was focused on the photochemical and complexation properties of 2-cinnamoyl-1,3-indandiones [28], which are easily obtained by Claisen-Schmidt condensation between 2-acetyl-1,3-indandione (2AID) and the corresponding aromatic aldehyde [29]. In this way, the complexation properties of 2AID are preserved due to the presence of the β-dicarbonyl fragment. Additionally, the absorption band of the formed 2-cinnamoyl-1,3-indandiones is shifted to the visible region of the spectrum, due to the elongated π-system, and gives rise to interesting optical properties [30]. These features motivated our studies in the search of new optical sensors and/or extracting agents for metal ions among the 2-cinnamoyl-1,3-indandiones. There are several reports on analytical applications of various 2-acyl-1,3-indandiones for optical detection of some lanthanide ions [31,32] or extraction of transition metal ions (Fe3+) [33].
2 Results and discussion
2.1 Synthetic schemes
In the current study, we aimed to synthesize a new 2-cinnamoyl-1,3-indandione derivative conjugated with N-phenylaza-15-crown-5, compound 2 (Scheme 1), and investigate its optical and complexation properties. As a chromophoric system we used 2AID, the structure and optical properties of which are well known [7,34]. Moreover, the crystal structure, optical and physicochemical properties of the dipolar compounds containing electron-donor group conjugated to the 2AID, such as 1 and DMABI are also reported [35,36] and show interesting possibilities for practical application [24,27,37,38]. Existing in the exocyclic enolic form (depicted in Schemes 1 and 2), the 2AID reacts as a strong vinylogous acid that may easily participate in condensation reaction. Therefore, in order to obtain compound 2 (which is the crown ether analogue of compound 1) we performed the condensation reaction of 2AID with 4-(aza-15-crown-5)benzaldehyde in piperidine (Scheme 1), employing the known procedure for the synthesis of 2-cinnamoyl-1,3-indandiones [29]. However, the described reaction did not yield compound 2, but rather the starting compound was recovered. That is why the condensation reaction between the 2AID and the 4-(aza-15-crown-5)benzaldehyde was performed in acetic acid (Scheme 2), similarly to a procedure, already used by us, for the synthesis of N-phenylaza-15-crown-5 derivatives of barbituric acid and its analogues [2]. Unexpectedly, the reaction resulted in compound 4, instead of compound 2. The structure of compound 4 was established by NMR and X-ray data and will be described further on.
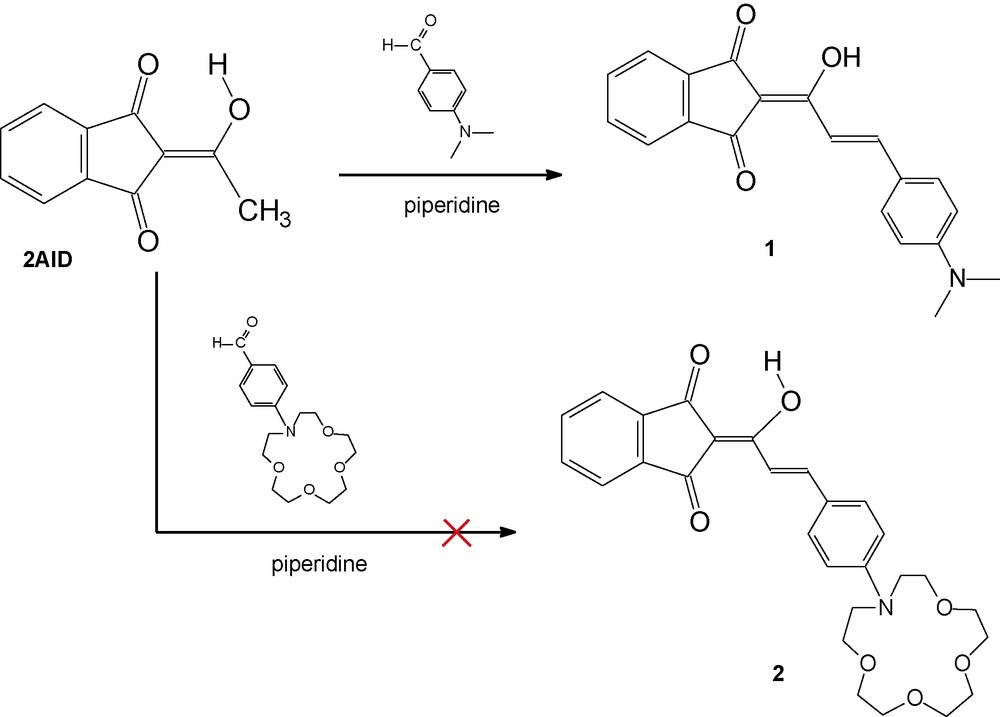
Condensation between 2AID and p-(N,N-dimethylamino)benzaldehyde and 4-(aza-15-crown-5)benzaldehyde in piperidine.
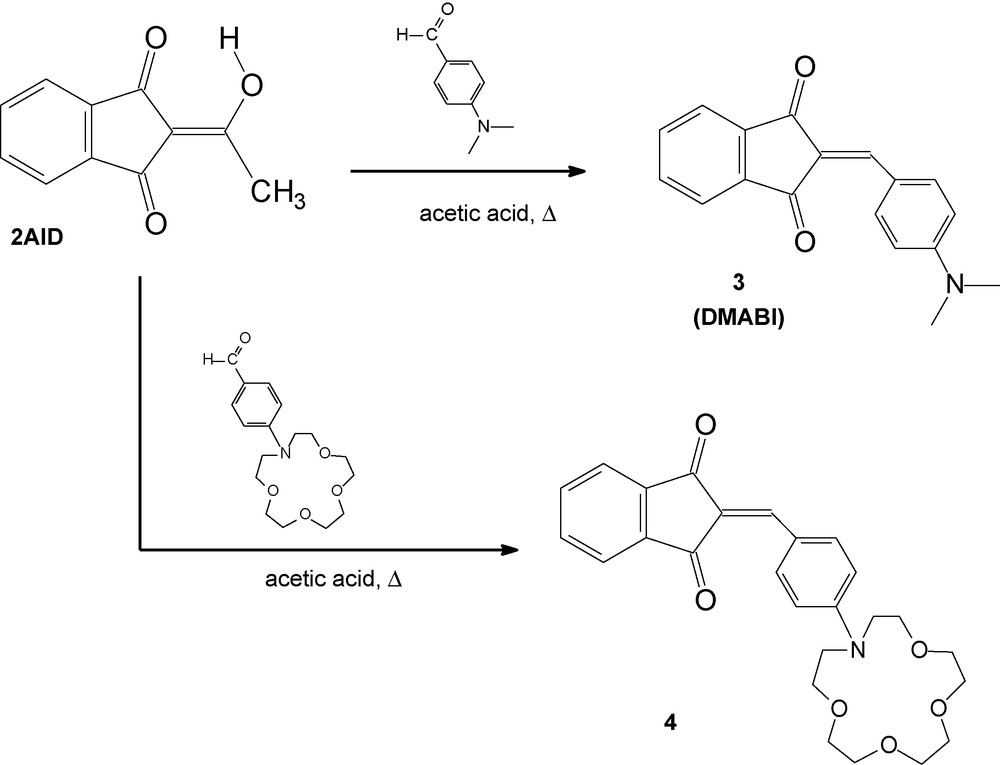
Reaction products from condensation between 2AID and p-(N,N-dimethylamino)benzaldehyde and 4-(aza-15-crown-5)benzaldehyde in acetic acid.
In order to prove that the condensation reactions between the 2AID and p-(N,N-dialkylamino)benzaldehydes proceed in different ways depending on the acidity of the reaction medium, we performed the reactions in piperidine and in acetic acid (according to Scheme 1 and Scheme 2, respectively) using p-(N,N-dimethylamino)benzaldehyde as well. When the reaction of 2AID with p-(N,N-dimethylamino)benzaldehyde was carried out in piperidine, compound 1 was obtained. Further, the reaction of 2AID with p-(N,N-dimethylamino)benzaldehyde was carried out in boiling acetic acid, similarly to the reaction for preparation of 4. The product obtained was proved to be 3 (DMABI). It should be noted that the isolation yields of the newly prepared compounds are moderate (ca. 40%). Compounds 1 and 3 were characterized by elemental analyses, IR and NMR spectroscopy. The spectroscopic data (IR and NMR) are in good agreement with the structures, corresponding to 1 and 3 (DMABI), as depicted in Schemes 1 and 2, respectively. These results clearly show that a deacetylation step takes place when the condensation reaction is performed in acetic acid.
Based on the results obtained from the described experiments and the structural data on the isolated products, we assumed that the condensation of 2AID with p-(N,N-dialkylamino)benzaldehydes in acetic acid is accompanied by deacetylation of the obtained intermediate. The mechanism we suggest is depicted in Scheme 3.
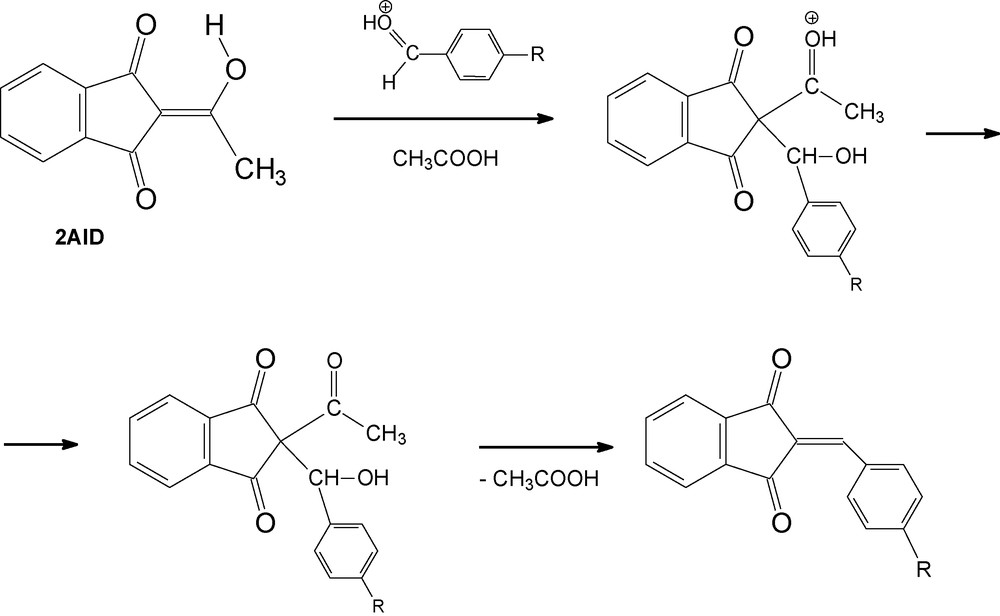
Suggested reaction mechanism of the acid-catalyzed condensation between 2AID and p-(N,N-dialkylamino)benzaldehydes.
To the best of our knowledge, this is the first example for a 2-acyl-1,3-indandione deacetylation observed in an acidic medium as a subsequent step of acid-catalyzed condensation with aldehydes.
2.2 Structure and optical properties
The crystal structure of compound 4 was solved by means of single-crystal X-ray diffraction. The compound 4 crystallizes in the monoclinic P21/n space group. There are four molecules in the unit cell. ORTEP view of the crystal structure of 4 and the way of crystal packing are depicted in Fig. 1a and b, respectively. List of bond lengths and bond angles is available as Supplementary Material. The C-N bonds from the crown ether ring [N1-C17 1.449(4) and N1-C26 1.455(4) A°] are in character, whereas the N1-C1 bond length [1.363(3) A°] shows that there is moderate π-electron conjugation between the phenyl ring and the crown ether. The same holds for the C4-C7 bond [1.422(3) Å] connecting the phenyl ring with the indandione fragment. The data also show that the 2-benzylidene-1,3-indandione fragment in the molecule is planar. The crown ether fragment is almost perpendicular to the aromatic rings. One of the C-atoms from the crown ether, C20, shows a high disorder and is represented by two possible occupations, shown as C20A and C20B. Two types of intermolecular interactions are observed – weak π-π-stacking interactions between the aromatic fragments of two adjacent molecules (approx. distance 3.35 Å) and weak C-H...O contacts involving a third molecule.

ORTEP view of the crystal structure of compound 4 (the ellipsoids represent 50% probability) (a), and view of the crystal packing with selected distances (b).
The crystal structure of compound 4 was used as a starting geometry in the quantum chemical calculations. Other conformers were also modeled and optimized but were found to be energetically less favourable and therefore were not considered further. The optimized gas-phase structural parameters of the studied crown ether are compared with the experimental data (see the Supplementary Material). Two different basis sets were used for optimization of the non-coordinated crown ether and for the studied complexes, namely 6-31G** and LanL2DZ. The former basis set was used for the K+, Na+, Ca2+ and Mg2+ complexes while the LanL2DZ basis set was applied for the Ba2+ and Sr2+ complexes. The calculated data for the crown ether, compound 4, using both basis sets are in very good agreement with the experimental ones. The structural changes caused by coordination of alkaline and alkaline-earth metal ions were also studied by means of quantum chemical methods. The optimized structure of the non-coordinated crown ether and those of its complexes with K+, Na+, Mg2+, Sr2+ and Ba2+ are presented in Fig. 2. As could be seen from the figure, the coordination of metal ions affects not only the conformations of the crown ether ring but also its orientation relative to the indandione fragment. Upon coordination, the crown ether fragment is rotated about the C1-N1 bond and moves closer to the hydrogen atoms of the phenyl ring. The calculated distances between the metal ion and the donor atoms of the crown ether are given in Table 1. The C4-C7 and C1-N1 bond lengths of those connecting the phenyl ring with the indandione fragment and with the crown ether moiety, respectively, are also given in order to estimate the changes in the degree of conjugation between both fragments as a result of coordination with the metal ion.
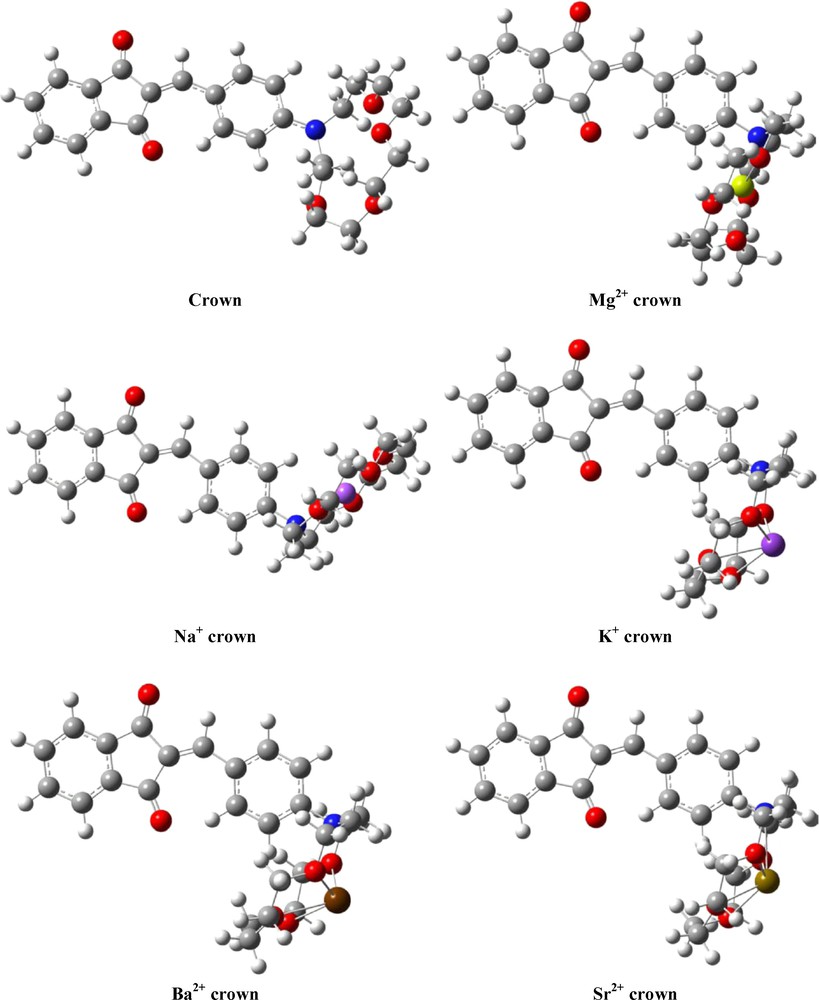
B3LYP optimized structures of compound 4 and its K+, Na+, Sr2+, Mg2+ and Ba2+ complexes.
Calculated (B3LYP) bond lengths and distances in the crown ether complexes. The calculated (B3LYP/6-31G**) C1-N1 and C4-C7 distances in the compound 4 are 1.379 and 1.436 Å, respectively.
| Distance [Å] | Na - complex (6-31G**) | K - complex (6-31G**) | Mg - complex (6-31G**) | Ca - complex (6-31G**) | Sr - complex (LanL2DZ) | Ba - complex (LanL2DZ) |
| M-O3 | 2.229 | 2.675 | 2.144 | 2.364 | 2.517 | 2.701 |
| M-O4 | 2.367 | 2.696 | 2.174 | 2.353 | 2.520 | 2.709 |
| M-O6 | 2.321 | 2.720 | 2.147 | 2.371 | 2.503 | 2.676 |
| M-O7 | 2.295 | 2.688 | 2.116 | 2.368 | 2.524 | 2.716 |
| M-N1 | 2.515 | 2.930 | 2.236 | 2.494 | 2.676 | 2.885 |
| C1-N1 | 1.455 | 1.452 | 1.471 | 1.467 | 1.478 | 1.476 |
| C4-C7 | 1.459 | 1.458 | 1.462 | 1.462 | 1.468 | 1.468 |
The obtained distances between the metal ion and the donor atoms from the crown ether are compared with the sum of the covalent radii of the corresponding atoms, as available in Cambridge Structural Database (CSD). It is only the Mg-O distances that are in the range of ± 0.4 Å tolerance for establishing a bonding connection. On the other hand, however, the largest elongation of the C4-C7 and N1-C1 bonds is obtained for the Ba2+ and Sr2+ complexes of compound 4 (Table 1). These results can be used as a measure for the loss of conjugation between the electron donor and acceptor parts in the molecule and correspondingly explains the observed optical properties.
The optical properties of compound 4 are characterized with a very strong absorbance in the visible region of the spectrum, λmax = 480 nm in acetonitrile. A bathochromic shift of 14 nm is observed in DMSO, while in cyclohexane the absorption maximum is shifted to 466 nm. The compound shows weak fluorescence with a Stokes shift of 62 nm. The emission band is a mirror image of the absorption spectrum. In order to study the effect of metal ions on the optical properties of compound 4, spectra in presence of K+, Na+, Mg2+, Sr2+ and Ba2+ ions were recorded. The observed changes are very subtle and are better pronounced only in presence of the Sr2+ and Ba2+ ions, as can be seen in Fig. 3. Although the good complexation ability of N-phenylaza-15-crown-5 moiety with the divalent ions is well known, in the studied case it does not lead to drastic change in the absorbance spectra of compound 4. One possible explanation can be that the crown ether moiety is directly bound to a very strong electron acceptor, which is the 1,3-indandione fragment. Therefore, the involvement of the N-atom's lone pair in coordination with the metal ion only slightly affects the electron density distribution in the molecule. The calculated vertical excitation energies of the crown ether 4 and its complexes predict correctly the expected hypsochromic shift upon coordination, but the corresponding transitions in the complexes are with very weak oscillator strength. An additional factor to remark on is the stability of the formed complexes. As has been previously reported on other N-phenylaza-15-crown-5 derivates, their complexes with alkaline-earth metal ions have low stability constants and the observed absorption spectra are, in some cases, with low intensity [5,6]. In order to estimate the stabilization energy of the metal complexes of compound 4, the ΔEST for each of them was calculated with the appropriate basis set. The results for ΔEST, defined as ECOMPL-ECROWN-EION, are listed in Table 2 together with the HOMO and LUMO energies, being additional measure of the complex stability. The higher the absolute value of the calculated energy differences, ΔEST, the higher is the expected stability of the complex. Moreover, single-point calculations were performed on the optimized structure of the complexes after removing the metal ion. The resulting energies are compared with the optimized structure of the free crown ether. In this way, it is possible to estimate the role of the metal ion coordination in the energy increase due only to the conformational changes, ΔECONF. These data, along with the structural parameters, are used to explain the obtained stability of the complexes, except for the known ion radius and cavity size matching. For example, the largest obtained stability of the Mg-complexes, which contradicts to the known trends, is apparently due to the strongest metal-oxygen bonds obtained in this case. The energy contribution of the structural changes in the ligand, caused by the coordination, plays a little role in the overall stability of the complex, as can be seen from Table 2.
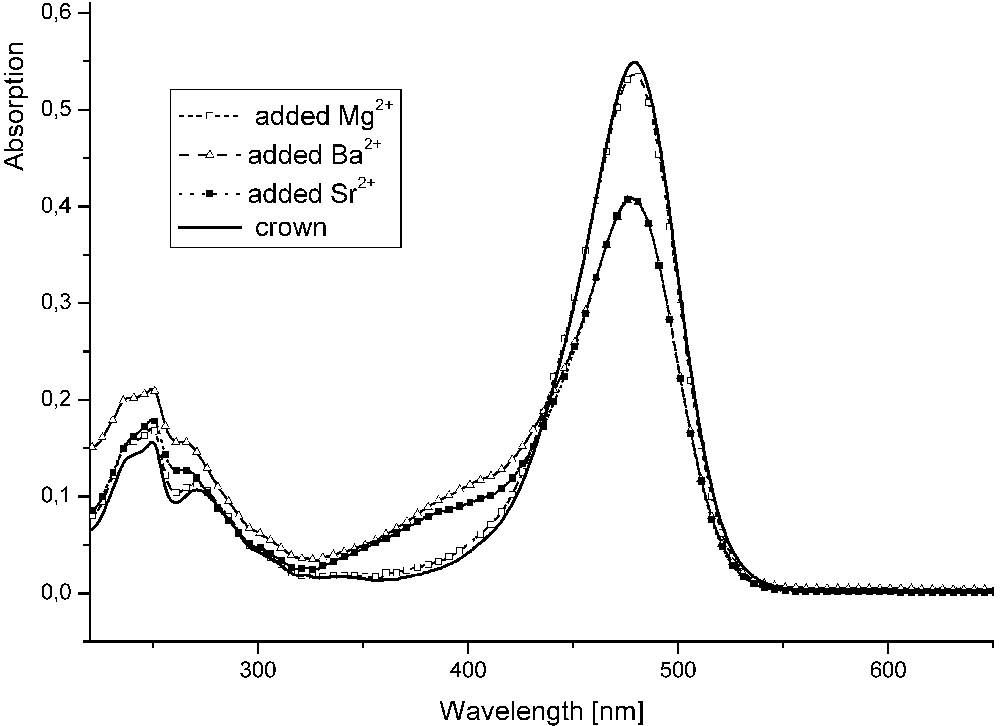
Absorption spectra of compound 4 in acetonitrile; C = 2 × 10-5 M, and in presence of Mg2+, Sr2+ and Ba2+ (perchlorates, C = 10-3 M).
Stabilization energies of the optimized complexes calculated as ΔEST = ECOMPL-ECROWN-EION together with the conformational energy differences ΔECONF due to coordination of metal ion (the same basis set is used for the corresponding species, as indicated in parenthesis).
| Compound (basis set) | ΔEST [kcal/mol] | EHOMO [a.u.] | ELUMO [a.u.] | ΔECONF [kcal/mol] |
| K_crown (6-31G**) | −51.185 | −0.312 | −0.168 | 16.037 |
| Na_crown (6-31G**) | −80.500 | −0.310 | −0.167 | 18.693 |
| Mg_crown (6-31G**) | −304.002 | −0.392 | −0.263 | 33.718 |
| Ca_crown (6-31G**) | −211.576 | −0.396 | −0.262 | 26.385 |
| Ba_crown (LanL2DZ) | −149.533 | −0.395 | −0.263 | 36.123 |
| Sr_crown (LanL2DZ) | −182.418 | −0.396 | −0.267 | 37.575 |
3 Conclusions
The crystal structure of the newly synthesized crown ether derivative of 2-benzylidene-1,3-indandione (compound 4) was determined by means of single-crystal X-ray diffraction. The compound 4 was obtained by aldol condensation of 2AID with the corresponding aromatic aldehyde in acetic acid. The structure of the obtained product indicates that the condensation reaction is accompanied by a deacetylation step. Analogous result was obtained from the condensation of 2AID with p-(N,N-dimethylamino)benzaldehyde in acetic acid yielding compound 3 (DMABI), whose structure was confirmed by the IR and NMR data. When the condensation reaction was performed in piperidine, p-(N,N-dimethylamino)-cinnamoyl-1,3-indandione (1) was obtained. All additional synthetic and spectroscopic experiments suggest that the aldol condensation between 2AID and aromatic aldehydes was followed by unprecedented deacetylation when performed in acetic acid.
The structure of compound 4 and its complexes with alkali and alkaline-earth metal ions were optimized in search of explanation for the observed optical properties. According to the DFT calculations, the divalent metal ions form more stable complexes. In the experimental absorption spectra of Sr2+ and Ba2+ complexes hypsochromic shifts of the absorption maxima were observed. They are, however, of very low intensity, as predicted by the TD-DFT calculations of the vertical excitation energies of the studied compounds. A plausible explanation of the observed very small effect of the metal ion coordination on the optical properties of compound 4 is probably the direct conjugation of the N-phenylaza-15-crown-5 moiety with a very strong electron acceptor, such as the 1,3-indandione. Additional consequence of this structural feature of compound 4 is the low stability of the obtained complexes.
4 Experimental section
4.1 General
The IR spectra were recorded on a Perkin-Elmer FTIR-1600 spectrometer (KBr tablets, and/or nujol mulls). Elemental analyses were performed on a Vario EU III instrument. The UV-Vis absorption spectra were recorded on a JASCO V-570 - UV/Vis/NIR spectrophotometer. The emission spectrum was recorded on a Varian - Cary Eclipse fluorescence spectrophotometer. The NMR measurements were carried out in CDCl3 using a Bruker DRX-250 spectrometer operating at 250.13 MHz for 1Н- and 62.90 MHz - for the 13С-nuclei. The chemical shifts are related to TMS, used as internal reference. 13C-NMR shifts assignment was facilitated by DEPT experiment distinguishing the quaternary carbons from the rest.
All solvents used were Uvasol grade. Acetonitrile was distilled over P2O5 before use. The complexation with metal ions was performed in dry acetonitrile using dried perchlorates: NaClO4, KClO4, Ba(ClO4)2, Sr(ClO4)2, Mg(ClO4)2. The absorption spectra of the complexes were measured in 1:100 ligand-to-metal ratio.
4.2 Synthesis and characterization
4.2.1 2-(4 -Dimethylamino-cinnamoyl) -2H-indane-1,3-dione (1)
Compound 1 was obtained according to a known procedure [29], yield 60%, m.p. 214-2150С. IR (nujol): ν 3400-3500 (νОH), 1700 (νC=O), 1635 (νC=O, νC=C) 1630(νC=O, νC=C). 1H NMR (CDCl3): δ = 3.06 (6H, s, 2xCH3), 6.68 (2H, d, Ph, J = 8.9 Hz), 7.59 (2H, d, Ph, J = 8.9 Hz), 7.63-7.91 (6H, m, 2xCH, 4Ph), 11.7 (s, 1H, OH) ppm. 13C NMR (CDCl3): δ = 40.10 (2C, 2xCH3), 106.38 (1C), 111.71 (2C), 111.79 (2C), 121.76 (1C), 122.12 (1C), 122.58 (1C),131.51 (1C), 133.48 (1C), 134.28 (1C), 138.71 (1C), 140.79 (1C), 146.36 (1C), 152.50 (1C), 174.28 (1C), 188.99 (1C, CO), 197.36 (1C, CO) ppm. Anal. Calcd for C20H17NO3 (319.12): C 75.22%, H 5.37%, N 4.39%; found C 75.11%, H 5.30%, N 4.68%.
4.2.2 2-(4-(Dimethylamino)benzylidene)-2H-indene-1,3-dione, DMABI (3)
A solution of 2AID (0.188 g, 1 mmol) in acetic acid (4 mL) was mixed with solution of 4-(dimethylamino)benzaldehyde (0.149 g, 1 mmol) in acetic acid (4 mL). Additional acetic acid (2 mL) was added and the reaction mixture was refluxed for 5 h. After the completion of the reaction (TLC), the solvent was removed under reduced pressure. The resulting oil was dissolved in dichloromethane (30 mL) and washed with 10% Na2CO3 (10 mL). Organic layer was washed with water and dried (Na2SO4). The solvent was evaporated under reduced pressure and the resulting oil was recrystallized from ethyl acetate thus yielding 0.085 g (31%), m.p. 208-210 °C. IR (CHCl3): ν = 1700 (νC=O), 1655 (νC=O) cm-1. 1H NMR (CDCl3): δ = 3.16 (6H, s, 2xCH3), 6.75 (2H, d, Ph, J = 9.2 Hz), 7.71-7.76 (2H, m, Ph), 7.80 (1H, s, CH), 7.91-7.96 (2H, m, Ph), 8.55 (2H, d, Ph, J = 8.5 Hz) ppm. 13C NMR (CDCl3): δ = 40.11 (2C, 2xCH3), 111.40 (2C), 122.49 (2C), 134.13 (2C), 134.41 (2C), 138.02 (1C), 139.87 (1C), 142.23 (1C), 147.55 (2C), 153.96 (1C), 190.03 (1C, CO), 191.82 (1C, CO) ppm. Anal. Calcd for C18H15NO2 (277.32): C 77.96%, H 5.45%, N 5.05%; found C 77.33%, H 5.68%, N 5.12%.
4.2.3 2-(4-(1,4,10,13-Tetraoxa-7-azacyclopentadecan-7-yl)benzylidene)-2H-indene-1,3-dione (4)
A solution of 2AID (0.188 g, 1 mmol) in acetic acid (4 mL) was mixed with solution of 4-(1,4,10,13-tetraoxa-7-azacyclopentadecan-7-yl)benzaldehyde (0.323 g, 1 mmol) in acetic acid (4 mL). Additional acetic acid (2 mL) was added and the reaction mixture was refluxed for 5 h. After the completion of the reaction (TLC), the solvent was removed under reduced pressure. The resulting oil was dissolved in dichloromethane (30 mL) and washed with 10% Na2CO3 (10 mL). Organic layer was washed with water and dried (Na2SO4). The solvent was evaporated under reduced pressure. The resulting oil was purified by column chromatography (light petroleum-ethyl acetate-acetic acid = 1:1:0.01) and recrystallized from ethyl acetate thus yielding 0.106 g (23%), m.p. 191-193 °C. IR (CHCl3): ν = 1710 (νC=O), 1665 (νC=O), 1120 (δCOC) cm-1. 1H NMR (CDCl3): δ = 3.62-3.64 (4H, br.s, OCH2), 3.65-3.69 (8H, br.s, OCH2), 3.72 (1H, t, OCH2, J = 6.2 Hz), 3.81 (1H, t, NCH2, J = 6.2 Hz), 6.75 (2H, d, Ph, J = 9.2 Hz), 7.70-7.50 (2H, m, Ph), 7.77 (1H, s, CH), 7.89-7.95 (2H, m, Ph), 8.51 (2H, d, Ph, J = 8.3 Hz) ppm. 13C NMR (CDCl3): δ = 53.03 (2C, NCH2), 68.08 (2C, OCH2), 69.92 (2C, OCH2), 70.27 (2C, OCH2), 71.21 (2C, OCH2), 111.55 (2C, Ph), 122.08 (1C, Ph), 122.44 (2C, Ph), 123.04 (1C, Ph), 134.08 (1C, CH), 134.35 (1C, Ph), 138.00 (2C, Ph), 139.80 (1C, Ph), 142.15 (1C, Ph), 147.24 (1C, Ph), 152.20 (1C, Ph), 189.92 (1C, CO), 191.71 (1C, CO) ppm. Anal. Calcd for C26H29NO6 (451.51): C 69.16%, H 6.47%, N 3.10%; found C 68.67%, H 6.25%, N 3.00%.
4.3 Crystal structure determination
Single crystals оf compound 4 were obtained by recrystallization from ethyl acetate. The data were collected at 293(2) K on a Xcalibur3 4-circles diffractometer using a graphite monochromator, MoKα radiation. A reference frame was monitored every 50 frames to control the stability of the crystal and the system revealed no intensity decay. The data set was corrected for Lorentz, polarisation effects, and absorption correction was made by multi-scan method using the CrysAlis Software package.
The structure was solved using direct method with SIR97 software and the refinement was carried out using the SHELXL-97 software package [39]. All non-hydrogen atoms were located from the initial solution or from subsequent electron density difference maps during the initial course of the refinement. After locating the non-hydrogen atoms the models were refined against F2, first using isotropic and finally anisotropic thermal displacement parameters. The hydrogen atoms were treated with the riding coordinate method. Details about the crystal structure and its refinement are given in Table 3.
Crystal data and structure refinement for compound 4.
| Compound 4 | |
| Empirical formula | C26H29NO6 |
| Molecular weight | 451.52 |
| Temperature | 293(2) K |
| Radiation source/wavelength (λ) | MoKα (λ=0.71073 Å). |
| Diffractometer | Xcalibur3 4-circles Diffractometer |
| Crystal system, | Monoclinic, |
| Space Group | P 21/n |
| Unit cell dimensions | а = 12.3150(10) Å |
| b = 12.4090(8) Å | |
| c = 16.4390(14) Å | |
| α=90.00° | |
| β= 111.841(10)° | |
| γ= 90.00° | |
| Volume | 2331.83 |
| Z | 4 |
| Crystal size | 0.2 mm × 0.3 mm × 0.4 mm |
| Theta range for data collection | 3.92° to 28.98° |
| Reflections collected/unique | 7632/4558 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4558/0/297 |
| GooF on F2 | 0.859 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0624, wR2 = 0.1705 |
| R indices (all data) | R1 = 0.1393, wR2 = 0.1959 |
4.4 Quantum chemical calculations
The quantum chemical calculations were performed with full geometry optimization without any symmetry restrictions. All calculations were performed with the Gaussian 03 program package [40]. Geometries of the studied compounds were optimized using the DFT method with the hybrid B3LYP functional [41,42]. For the calculations the 6-31G(d,p) basis set was used, while in case of Ca2+, Sr2+ and Ba2+ complexes the LanL2DZ basis set, which includes Dunning-Huzinaga full double zeta (DZ) basis functions for the first row and Los Alamos effective core potentials (ECP) for heavy elements, was employed. Vibrational frequencies were computed at the same level of theory in order to confirm that local energy minima were attained. Vertical excitation energies were computed with the time dependent DFT (TD-DFT) employing the same functional and basis sets as for optimization.
Acknowledgements
This work has been supported by the Bulgarian National Science Fund, Contract MYX-1502/05.
Appendix A Supplementary material
The list of experimentally determined and theoretically calculated bond lengths and bond angles of compound 4 is available as Supplementary Material.
CCDC-693831 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033; email: deposit@ccdc.cam.ac.uk].