

1 Introduction
Coumarins are compounds that display a special role in nature. Pharmacologically, coumarins and their derivatives are included in the family of the flavonoids and exhibit a variety of interesting biological and pharmacological activity. Therefore, coumarins and their derivatives have raised considerable interest because of their potential beneficial effects on human health [1,2]. They have been reported to possess: anti-HIV [3–6], anticoagulant [7], antibacterial [8,9], anticancer [10], anthelminthic [11], anti-inflammatory [12–14] and antioxidant activities [15–17]. As a result, coumarins and derivatives have been the subject of extensive investigations.
The coumarin nucleus incorporates the styryl carbonyl moiety into a rigid framework. It has been reported that styryl carbonyl derivatives possess appreciable anti-inflammatory activity [18,19]. Moreover, coumarin and related derivatives have been used as inhibitors of lipoxygenase (LOX) and cyclooxygenase (COX) pathways of arachidonic acid metabolism [20]. One of the most well studied coumarin-based anti-inflammatory drugs is cloricromene (8-monochloro-3-b-diethylaminoethyl-4-methyl-7-ethoxy-carbonylmethoxy coumarin), and several synthetic analogues of this compound have been recently reported and tested for their anti-inflammatory and antioxidant activities [21]. In addition, many coumarin derivatives have shown special ability to scavenge Reactive Oxygen Species (ROS) and in processes involving free radical injury [22,23]. In addition, the 4-hydroxycoumarin moiety, which widely spreads among coumarin, is the molecular template used for the synthesis of a variety of analogue derivatives which exhibit important biological activity.
In the course of our continuous interest on the synthesis of coumarin derivatives including 4- hydroxyl coumarin with antibacterial and antioxidant activities [24,25], we have extended our research to the synthesis and biological evaluation of new coumarinyl chalcones using 3-acetyl-4-difluoro boryloxycoumarin coumarin (2) via an easy, efficient and quit method.
2 Results and discussion
The acetylation of 4-hydroxycoumarin to give 3-acetyl-4-hydroxycoumarin (1) was carried out by the method of Dholakia et al. [26] by using glacial acetic acid in the presence of POCl3. The quick reaction led to the desired product without obtaining any product from the intramolecular condensation of 4-hydroxycoumarin [27]. We have observed previously [28] that the reaction between hydroxyacetylcoumarin and boron compounds substantially increases the acidity of Hα atoms. For instance, reactions of 3-acetyl-4-difluoro boryloxycoumarin (2) with triethyl orthoformate, Fischer aldehyde and DMF produces symmetrical and unsymmetrical polymethine dyes in high yields [29] which are identified by their intense absorption in the visible region and fluorescence properties. Here we describe the condensation process of compound 2 with aromatic aldehydes, which affords the α, β-unsaturated ketones from the coumarin series.
Heating 3-acetyl-4-hydroxycoumarin in a mixture of toluene and boron trifluoride etherate gave rise to the boron product 2 with good yield (Scheme 1).
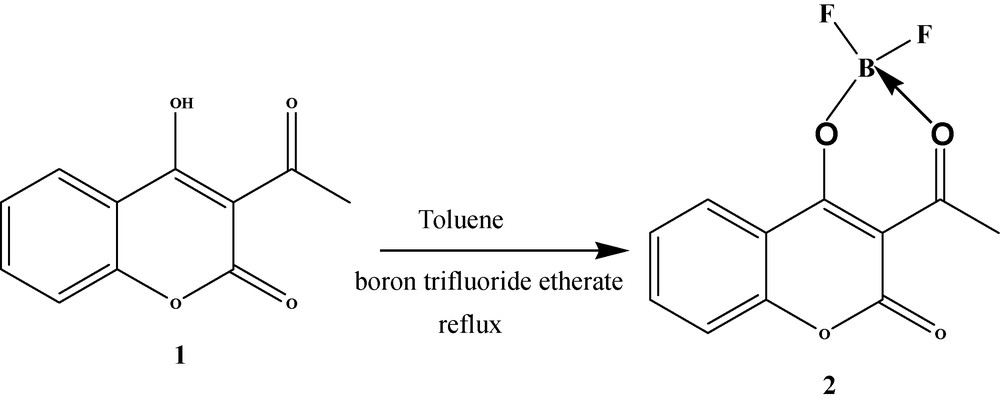
Synthesis of 3-acetyl-4-difluoro boryloxycoumarine.
Compound 2 was characterized by 1H NMR, 13C NMR, and IR spectroscopy. The 1H NMR spectra of 2 display the signal for the protons of the methyl group of the acetyl fragment shifted to downfield than those found for the starting compound 3-acetyl-4-hydroxycoumarin. The carbonyl IR intense stretching band of 3-acetyl-4-hydroxycoumarin observed at 1740 cm−1 remains virtually unchanged for compound 2 while the carbonyl band for acetyl group arises at 1560 cm−1 instead at 1600 cm−1 as is observed in starting compound and additionally this band is less intense for 2 than for 3-acetyl-4-hydroxycoumarin. That fact could be justified by the donor acceptor interaction with boron atom of the acetyl group. In the 1000–1100 cm−1 interval, there are two intense absorption bands (1045 and 1070 cm−1) which can be assigned to C-O-B stretching vibrations.
The coumarinic chalcones 4a-i was synthesized in one step by a new procedure: refluxing 3-acetyl-4-difluoroboryloxycoumarin (2) with aryl and heteroaryl aldehydes in the presence of piperidine in chloroform. Using this reaction conditions, the reaction time is c.a. 1–1.5 h. In previously used methods, the time needed for completing the reaction was much longer, from 24 to 36 h at room temperature [30–32]. Reaction of 3-acetyl-4-difluoroboryloxycoumarin (2) with aryl or heteroaryl aldehydes likely takes place according to the probable rearrangement of 4′ to give 4.
The formation of the new boronate coumarinic chalcone probably takes place at the first stage, and the deciding role in such a reaction path is played by the boron atom coordinated with the nitrogen atom of piperidine rearrangement of the complex 4′, which leads to the coumarin derivative 4. It should be noted that this reaction of condensation is accompanied by elimination of the difluorobopiperidine.
All the resulting products were isolated in solid state, their purity was checked by thin layer chromatography (eluctant: hexane/ethyl acetate, 1/1. v/v) and characterized by NMR and IR spectroscopy. Reactions and obtained products are displayed in Scheme 2 and Table 1.
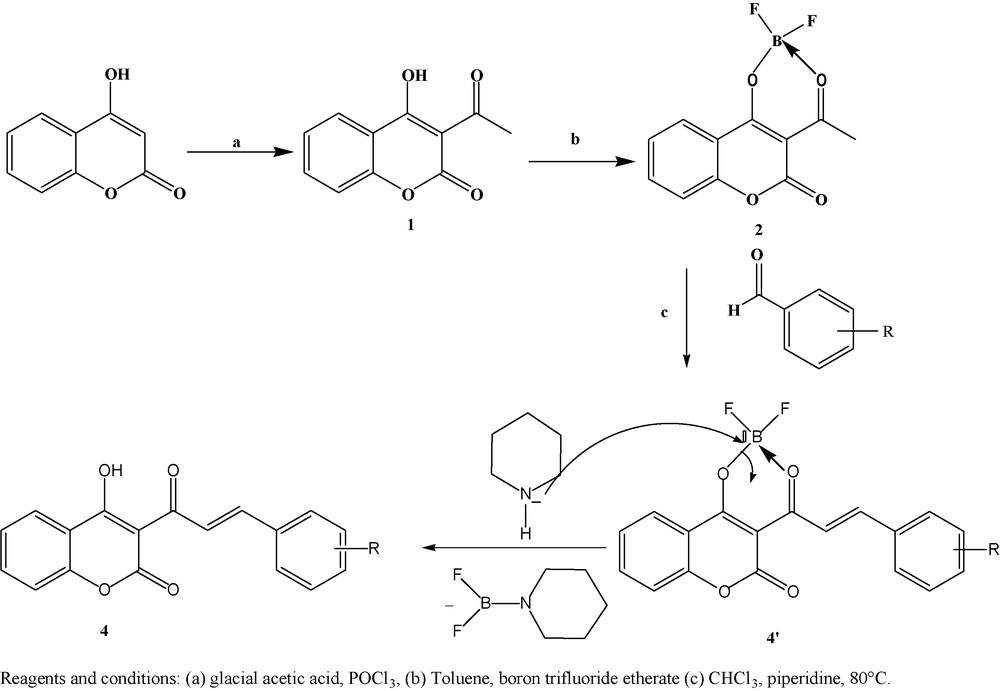
Formation of the coumarinic chalcones 4 by the reaction of 3-acetyl-4-difluoroboryloxycoumarin (2) with aryl and heteroaryl aldehydes.
Obtained products and aryl aldehydes substituent groups.
| 4a-i | Ar |
| a | C6H5 |
| b | pF- C6H4 |
| c | pOCH3C6H4 |
| d | pMe C6H4 |
| e | pBr C6H4 |
| f | pNO2 C6H4 |
| g | 3,4,5 OCH3 C6H2 |
| h | C10H7 |
| i | pN(Me)2 C6H4 |
The IR spectra of compounds 4a–i showed characteristic absorptions for OH, (ketone) CO and (lactone) (CO) stretching in the region 3500–3300 cm−1, 1670–1710 cm−1 and 1720–1750 cm−1, respectively. The spectroscopic characterization of compound 4g is presented as an example. The 1H NMR spectra of 4g shows the aromatic proton as a multiplet between 7.41 and 8.21 ppm, the singlets at 3.74 and 3.85 ppm were assigned as methoxylic protons. The 13C NMR spectrum of 4g in DMSO is constituted by two downfield signals at 162.42 ppm (Ketone CO) and 157.41 ppm (lactone CO) as well as one upfield signal at 55.48 ppm (OCH3). The DEPT spectrum showed that the HC carbons of the coumarinic ring appear at δ 134.5, 131.3, 125.6 and 124.11 ppm. The low field region of the spectra of compounds 4g shows the signals for the aromatic protons between 7.4 and 8.1 ppm and doublet signals for olefin protons in the same region with 3JHH = 13.5 Hz. These coupling constants are in agreement with a disposition for the olefin protons trans to each other. In order to confirm the proposed composition for 4g, its crystals structure was determined by single crystal X-ray diffraction analysis (Fig. 1).
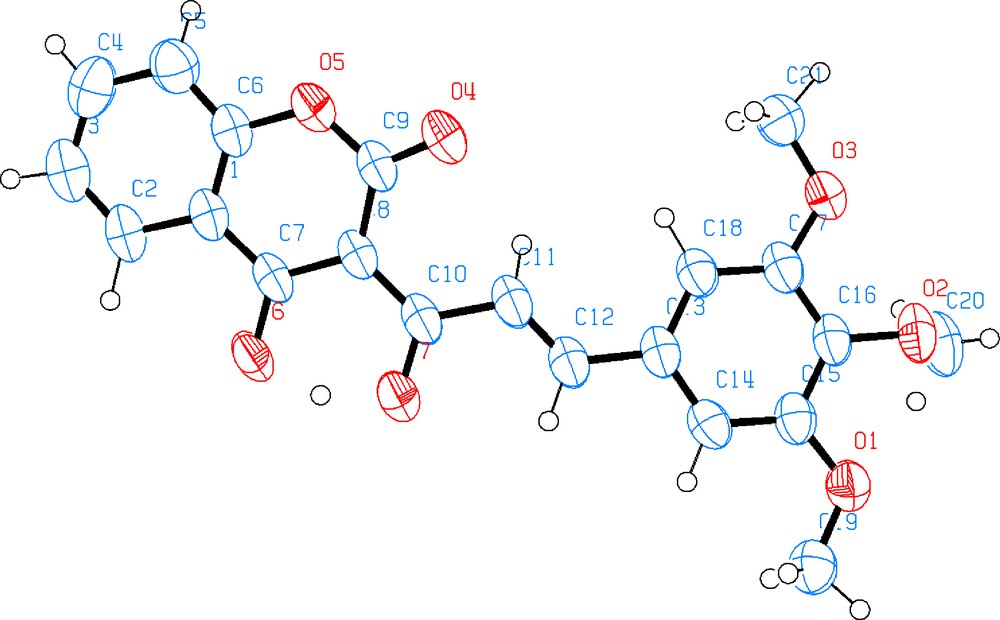
Solid state structure of 3-((2E)-3-(3,4,5-Trimethoxy-phenyl)prop-2-enoyl)-2(H)-chromen-2-one (4g).
The X ray crystal structure confirmed the propose composition for 4g. A more accurate description of the structure was presented in [33] but it is important to point out that along the connection between the coumarin moiety and phenyl ring (C13–C12–C11–C10) the double bond is located on C11–C12 (C11—C12 = 1.325 (5) Å; C10— C11 = 1.467 (5) Å; C12— C13 = 1.475 (4) Å). Therefore, there is a π-bridge for the charge transfer from phenyl ring to coumarin moiety. As well, it is interesting to stress that intermolecular and intramolecular hydrogen bonds are between C2–H···O3 and C19–H···O4 leading to a hydrogen network among molecules (Fig. 2).
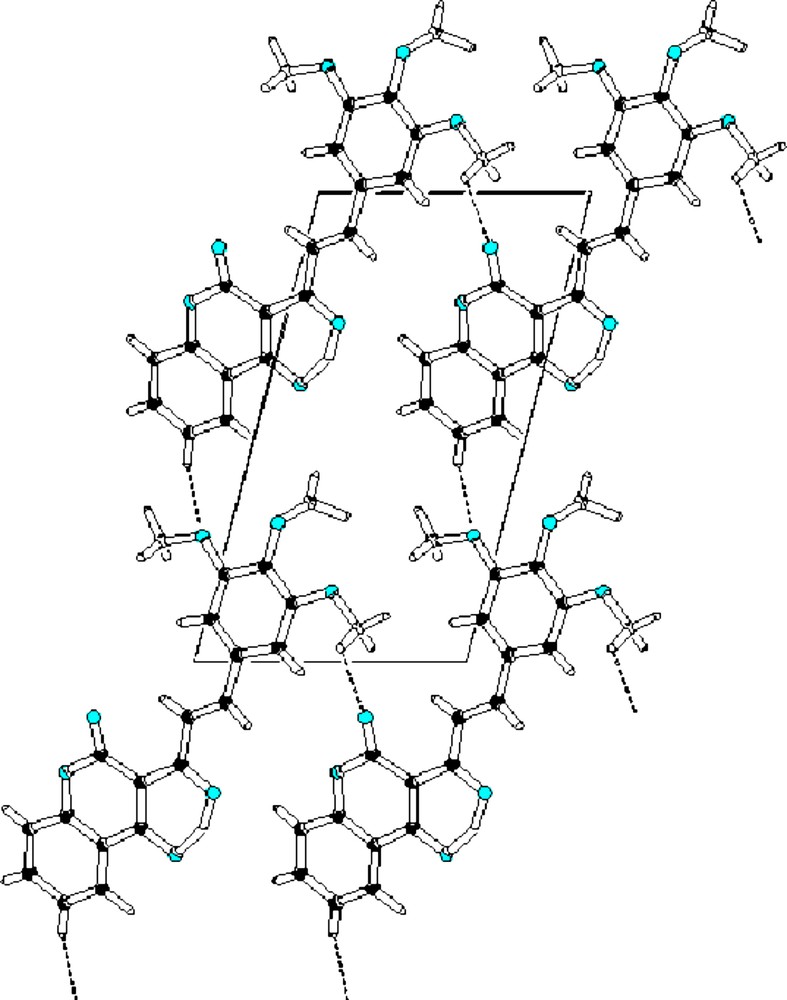
Unit cell view of the crystal down c-axis showing intermolecular and intermolecular hydrogen bonds (dashed lines).
The electronic absorption spectra of compounds 4a–i were obtained in chloroform and are displaying in Fig. 3. The spectrum shows intense narrow bands with a high extinction coefficient. Compounds absorb in the same spectral region but the shape of their absorption spectra differs with the nature of the tether. The spectra of 4b display two bands at around 450 and 600 nm. The spectrum of 4f, in which both chromophores are directly condensed without any spacer between them, shows only one band at 525 nm with a shoulder at short wavelengths. For all the coumarinic chalcones the magnitude of the molar absorption coefficient is typical of π–π transitions (Table 2).
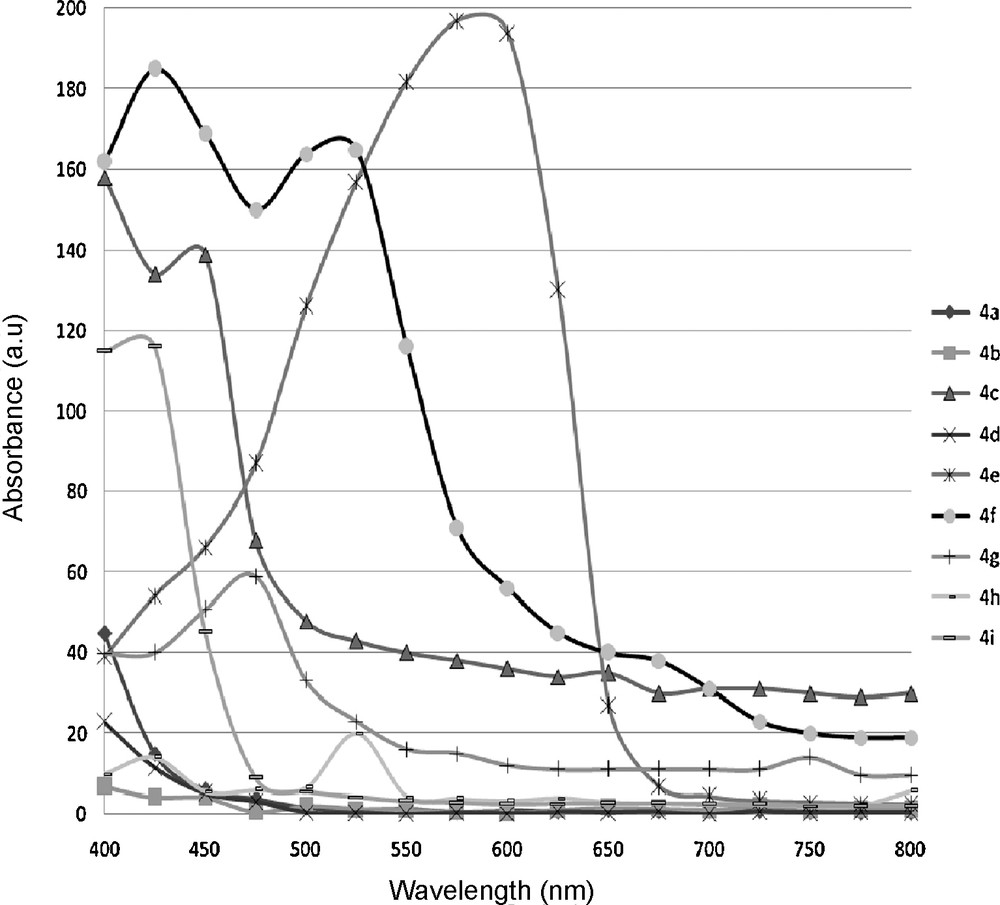
Absorption spectra of the coumarins chalcones in chloroform solution. These compounds were also characterized to determine their UV–vis absorbance spectra, and molar absorption coefficients.
Spectral properties of the coumarinic chalcones 4a-i in chloroform.
| Compounds 4a-i | λmax (nm) | ɛ mol−1 dm3 cm−1 |
| a | 494 | 30,400 |
| b | 474 | 23,000 |
| c | 452 | 16,300 |
| d | 476 | 8530 |
| e | 463 | 14,815 |
| f | 482 | 23,200 |
| g | 466 | 14,500 |
| h | 477 | 28,500 |
| i | 484 | 7300 |
3 Antioxidant activities of compounds 4a–i
The synthesized compounds 4a–i were tested for their antioxidant and antibacterial activities. It is well known that free radicals play an important role in the inflammatory process [34]. Many non-steroidal anti-inflammatory drugs have been reported to act either as inhibitors of free radical production or as radical scavengers [35]. Consequently, compounds with antioxidant properties could be expected to offer protection in rheumatoid arthritis and inflammation and to lead to potentially effective drugs. The potential of the antioxidant activity is shown in Fig. 4 and Table 3.
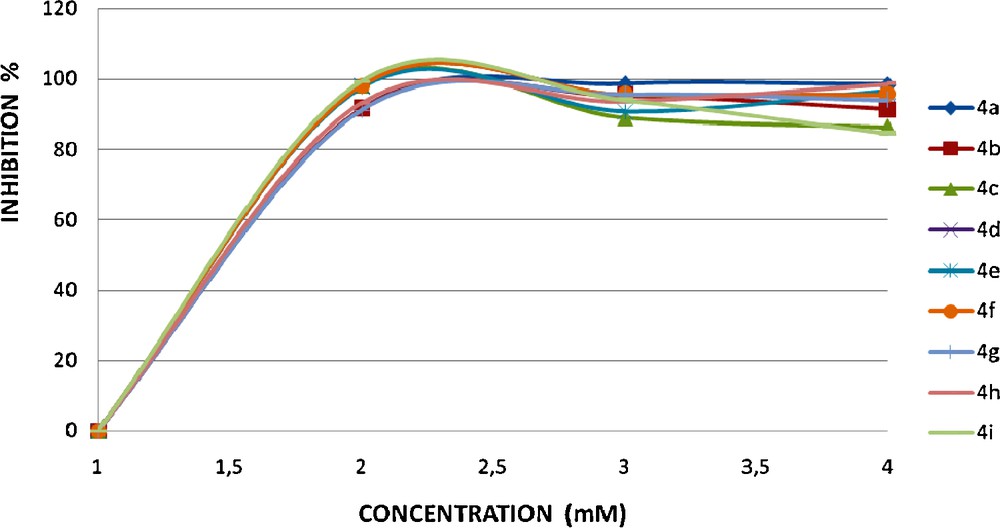
The DPPH radical scavenging capacity of the obtained coumarinic derivatives 4a-i.
The EC50 values exhibited by coumarinic chalcones 4 are summarized in the following table.
| Compounds 4a-i | EC50 (μM) |
| 4a | 2.39 |
| 4b | 2.34 |
| 4c | 3.03 |
| 4d | 2.24 |
| 4e | 2.10 |
| 4f | 2.16 |
| 4g | 2.42 |
| 4h | 2.29 |
| 4i | 2.18 |
Results of a single assay can give only a suggestion on the protective potential of tested compounds. Among the plethora of methods used for the evaluation of the antioxidant activity, the 1, 1-diphenyl-2-picrylhydrazyl (DPPH) test is very useful in the micromolar range demanding minutes to hours for α, β-unsaturated ketones of the coumarinic series samples. Where the structure of the electron donor is not known, this method can afford data on the reduction potential of the sample, and hence can be helpful in comparing the reduction potential of unknown materials. The competition of the compounds with DMSO for hydroxyl radicals has been used, whereas antioxidant capability as well as lipid peroxidation inhibitory activity was assayed by the inhibition of the oxidizing enzyme LOX [36]. The interaction of the examined compounds with the stable free radical DPPH was studied (Table 2). This interaction indicates their radical scavenging ability in an iron-free system using a stable free radical which can accept an electron or hydrogen atom.
The comparative study among the resulting test results for compounds shows that compounds 4a and 4d are potent antioxidant compounds while coumarinic chalcone 4c is inactive even at a concentration of 3.77 mM. Compounds 4e and 4i proved to be the most active compounds in this study. Comparing the activity of compounds 4f–h it was concluded that there is not a substantial difference when we change the nature of the substituent Ar.
4 Antibacterial activities of compounds 4a–i
New antimicrobials are needed because of the emergence of organisms that are resistant to available antimicrobials.
The new coumarinic fluorine chalcones derivatives 4a–i were also screened in vitro for their antimicrobial activity against a variety of bacterial strains.
The results of these studies are summarized in Table 4.
Antimicrobial activity spectrum of compounds 4a–i.
| Organism indicator | Inhibition zone (mm) | Compounds |
| Staphylococcus aureus (CIP 7625) | 30 | 4a |
| 28 | 4b | |
| 25 | 4c | |
| 32 | 4d | |
| 35 | 4e | |
| 26 | 4f | |
| 24 | 4g | |
| 31 | 4h | |
| 24–28 | Gentamycin | |
| Staphylococcus aureusa | 25 | 4a |
| 27 | 4b | |
| 26 | 4c | |
| 31 | 4d | |
| 34 | 4e | |
| 27 | 4f | |
| 25 | 4g | |
| 32 | 4h | |
| 39 | 4i | |
| 24 | Gentamycin | |
| Escherichia coli ATCC 25922 | 28 | 4a |
| 26 | 4b | |
| 34 | 4c | |
| 31 | 4d | |
| 30 | 4e | |
| 27 | 4f | |
| 25 | 4g | |
| 32 | 4h | |
| 22–26 | Gentamycin | |
| Klebsiella pneumonia CIP 104727 | 23 | 4a |
| 25 | 4b | |
| 30 | 4c | |
| 29 | 4d | |
| 27 | 4e | |
| 28 | 4f | |
| 26 | 4g | |
| 31 | 4h | |
| 32 | 4i | |
| 21 | Gentamycin | |
| Pseudomonas aeruginosa 27853 (CIP 76110) | 27 | 4a |
| 25 | 4b | |
| 33 | 4c | |
| 32 | 4d | |
| 30 | 4e | |
| 26 | 4f | |
| 27 | 4g | |
| 30 | 4h | |
| 31 | 4i | |
| 15,5–22,5 | Gentamycin |
a Methicillin-resistant clinical isolates.
It could be observed that all the tested compounds were active toward E. Coli,
The antibacterial data of the above compounds having pBrC6H4 group have increased antibacterial activity while 4h–i are found to be more biologically active than 4b–c [37].
The antibacterial test results depicted in Table 4 shows that the new compounds 4a–i show interesting antibacterial activities but moderate. Nevertheless, the obtained results clearly demonstrate that this kind of compounds could be an effective antibacterial agent and the activity is depending on the chemical composition of the coumarine derivative.
5 Conclusion
In the present study, as a first step, we have successfully developed a synthetic route for the preparation of novel coumarins derivatives. Their synthesis is easy, quit and convenient with satisfactory yield. The synthetic procedure is not sensitive to oxygen and water and it is easy to operate at room temperature. The flexibility of this methodology should provide access to many various coumarins chalcones. All these compounds are observed to exhibit excellent absorbance properties. The DPPH method has been widely applied for estimating antioxidant activity in recent years but its applications should be carried out bearing in mind the basis of this method. Nevertheless, it can be concluded that the antioxidant activity of the coumarine-synthesized derivatives could be related to their structure. Antioxidant activities of these compounds against the stable free radical DPPH show that these species are compounds that could help to increase the overall antioxidant capacity of an organism. Therefore, the design of this type of dual acting molecules should be further explored on the base of the structural features of these compounds. The adducts 4e and 4i present the highest activity from all the tested compounds.
6 Experimental section
6.1 General procedures
Flash chromatography was carried out on 0.04–0.063 mm (Merck) silica gel, thin layer chromatography was carried out on aluminium backed silica plates by Merck and plates were revealed using a UV 254 light. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Varian VXR 300 instrument at 293 °K in CDCl3 and DMSO-d6. Spectra were internally referenced to TMS. Peaks are reported in ppm downfield of TMS. Multiplicities are reported as singlet (s), doublet (d), triplet (t), quartet (q). Characterization was made some time by using DEPT experiment. The IR-spectra were recorded on a Philips Analytical PU 9800 spectrometer. The melting points of compounds were determined in open glass capillaries in a paraffin bath and are uncorrected. The absorption spectra were measured on a Beckmann K25 spectrophotometer.Absorbance spectra for all compounds were collected at room temperature with a Hewlett-Packard (Palo Alto, CA) model 8453 diode array UV–vis spectrophotometer. Absorbance measurements were taken using Eppendorf (Hamburg, Germany) UVette cuvettes (220–1600 nm). Elemental microanalysis was obtained by the microanalytical laboratory services, university of Cadiz in Spain.
In the experiment part, the 13C {1H} NMR should be replaced by 13C NMR. The 1H-NMR have been checked for the multiplicities for H-atoms.
6.2 Materials
DPPH was obtained from Sigma. All other chemicals were of analytical grade purity. The 4-hydroxycoumarin and aromatic aldehydes were purchased from Fluka, DMF was purified, dried and distillated over CaH2 prior to use.
6.3 3-Acetyl-4-hydroxycoumarin
To a solution of 4-hydroxy-2H-chromen-2-one (3 g, 1.86 mmol) in acetic acid (16 ml) was added phosphorus oxychloride (5.6 ml). The mixture was heated at reflux for 30 min. After cooling, the precipitate was collected and recrystallized from ethanol to give 3-acetyl-4-hydroxy-2H-chromen-2-one as white needles. Yield 2.7 g (90%); mp = 135 °C. IR spectrum, ν cm−1: 3185 (OH); 1705 (CO); 1700 (OCO lactone). 1H NMR spectrum (CDCl3). δ ppm: 2.72 (3H, s, CH3); 7.98 (1H, s, H-5); 7.95 (1H, dd, 3J7.8 = 8.35, 4J6.8 = 1.2, H-8); 7.1–7.4 (2H, m, H-6 + H-7); 17.69 (1H, s, OH). 13C NMR spectrum (CDCl3), δ ppm: 29.9 (CH3); 178.5 (CO); 159.8 (C-4); 154.6 (C-2); 101.26 (C-3); 115.0–136.0 (Carom). Mass spectrum, m/z (I, %): 204 [M]+ (100); 189 (74); 161 (43).
6.4 3-acetyl-4-difluoro boryloxycoumarin coumarin
To a solution of 3-acetyl-4-hydroxycoumarin (3 g, 1.86 mmol) in toluene (16 ml) was added BF2OEt2 (5.6 ml). The mixture was heated at reflux for 60 min. After cooling, the precipitate was collected and recrystallized from ethanol to give 3-acetyl-4-difluoro boryloxycoumarin coumarin as white needles. Yield 2.7 g (90%); mp = 135 °C. IR spectrum, ν cm−1: 1705 (CO); 1700 (OCO lactone). 1H NMR spectrum (CDCl3). δ ppm: 2.3 (3H, s, CH3); 7.02–7.27 (m, Harom). 13C{1H} NMR spectrum (CDCl3), δ ppm: 26.7 (CH3); 198.7 (CO); 170.0 (C-4); 159.5 (C-2); 102.3 (C-3); 121.5–126.8 (Carom).
6.5 General procedure for the preparation of the coumarinic chalcones 4a–i
3-acetyl-4-difluoro boryloxycoumarin coumarin (0.031 mol) and the substituted aromatic aldehyde (0.03 mol) were dissolved in 30 mL of chloroform. A catalytic amount of piperidine (0.02 mol) was added and the reaction mixture was refluxed for 1.5 h. The chloroform was removed under vacuum and the residue was washed with methanol.
6.6 3-((2E)-3-(phenyl)prop-2-enoyl)-2(H)-chromen-2-one (4a)
Solid (yield 79%); mp = 145 °C, there is a lactone carbonyl in IR spectrum, ν cm−1: 1622 (CO), 1577 (CC), 1028 (s) (sym) (C-O-C), 1727 (O-CO). 1H NMR: δ (ppm): 7.2–8.3 (m, 11H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 178.6 (CO); 179.1 (C4); 155.1 (C2); 179.1 (C4), 179.1 (C4), 101.8 (C3), 126.2–136.5 (Carom); 129.7 (Céthyl1), 147.8 (Céthyl2). C18H1204 calc. C 73.97, H 4.10, O 21.92; found. C 73.90, H 4.10, O 21.80.
6.7 3-((2E)-3-(4-fluorophenyl)prop-2-enoyl)-2(H)-chromen-2-one (4b)
Solid (yield 80%); mp = 178 °C. IR spectrum, ν cm−1: 1622 (CO), 1577 (CC), 1028 (s) (sym) (C-O-C); 1H NMR: δ (ppm): 7.2–8.2 (m, 10H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 160.5 (CO); 179.1 (C4); 155.1 (C2); 179.1 (C4), 179.1 (C4), 101.8 (C3), 115.6–136.5 (Carom); 129.7 (Céthyl1), 147.8 (Céthyl2). C18H1104 F calc. C 69.67, H 3.54, O 20.64; found. C 69.70, H 3.60, O 20.70.
6.8 3-((2E)-3-(4-methoxyphenyl)prop-2-enoyl)-2(H)-chromen-2-one (4c)
Solid (yield 70%); mp = 135 °C. IR spectrum, ν cm−1: 1622 (CO), 1577 (CC), 1028 (s) (sym) (C-O-C). 1H NMR: δ (ppm): 3.74 (s,OCH3), 7.1–8.3 (m, 10H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 181.1 (CO); 190.9 (C4); 162.4 (C2); 100.3 (C3); 113.6–157.6 (Carom); 128.3 (Céthyl1), 154.0 (Céthyl2). C19H1405 calc. C 70.80, H 4.34, O 24.84; found. C 70.70, H 4.40, O 24.80.
6.9 3-((2E)-3-(p-tolyl) prop-2-enoyl)-2(H)-chromen-2-one (4d)
Solid (yield 75%); mp = 140 °C. IR spectrum, ν cm−1: 1625 (CO), 1580 (CC), 1030 (s) (sym) (C-O-C). 1H NMR: δ (ppm) 2.84 (s, 3H, CH3); (7.28 (d, 1H, Héthy1), 7.60 (d, 1H, Héthy2), 7.2–8.2 (m, 8H, Ar-H). 13C{1H} NMR (ppm): 30.0 (OCH3); 178.4 (CO); 154.6 (C4); 135.6 (C2); 101.3–135.9 (Carom). C19H1404 calc. C 74.50, H 4.57, O 20.91; found. C 74.40, H 4.60, O 20.90.
6.10 3-[(2E)-3-(4-bromophenyl)prop-2-enoyl]-2(H)-chromen-2-one (4e)
Solid (yield 85%); mp = 182 °C. IR spectrum, ν cm−1: 1622 (CO), 1577 (CC), 1028 (s) (sym) (C-O-C). 1H NMR: δ (ppm): 7.2–8.0 (m, 10H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 178.7 (CO); 181.5 (C4); 160.2 (C2); 101.3 (C3), 122.6–154.7 (Carom); 130.0 (Céthyl1), 154.3 (Céthyl2). C18H1104Br calc. C 58.22, H 2.96, O 17.25; found. C 58.30, H 2.90, O 17.30.
6.11 3-((2E)-3-(4-Nitro-phenyl) prop-2-enoyl)-2(H)-chromen-2-one (4f)
Solid(yield 85%); mp = 182 °C. IR spectrum, ν cm−1: 1622 (CO), 1577 (CC), 1028 (s) (sym) (C-O-C); 1H NMR: δ (ppm): 6.8–8.3 (m, 10H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 171.3 (CO); 180.5 (C4); 163.3 (C2); 100.9 (C3), 122.6–152.5 (Carom); 129.6 (Céthyl1), 151.7 (Céthyl2). C18H1106 N calc. C 64.1, H 3.26, O 24.48, N 4.15; found. C 64.10, H 3.30, O 24.50, N 4.10.
6.12 3-((2E)-3-(3,4,5-Trimethoxy-phenyl)prop-2-enoyl)-2(H)-chromen-2-one (4g)
Solid (yield 83%); mp = 193 °C. IR spectrum, ν cm−1: 1716 (s) (CO), 1577 (CC), 1018 (s) (sym) (C-O-C); 1H NMR: δ (ppm): 3.74 (s, 3H, OCH3), 3.85 (s, 6H, OCH3), 7.4–8.1 (m, 8H, Ar-H). 13C{1H} NMR (ppm): 55.9 (OCH3), 60.1 (OCH3), 191.2 (CO); 180.7 (C4); 159.5 (C2); 100.6 (C3), 126.2–136.5 (Carom); 129.6 (Céthyl1), 153.3 (Céthyl2). C21H1807 calc. C 65.96, H 4.71, O 29.31; found. C 65.90, H 4.80, O 29.30.
6.13 3-[(2E)-3-(naphthyl) prop-2-enoyl]-2(H)-chromen-2-one (4h)
Solid (yield 80%); mp = 185 °C. IR spectrum, ν cm−1: 1718 (s) (CO), 1578 (CC), 1019 (s) (sym) (C-O-C); 1H NMR: δ (ppm): 7.2–9.1 (m, 13H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 180.1 (CO); 195.0 (C4); 155.1 (C2); 117.4 (C3), 123.5–143.9 (Carom); 129.3 (Céthyl1), 149.5 (Céthyl2). C22H1404 calc. C 77.20, H 4.10, O 18.71; found. C 77.10, H 4.10, O 18.70.
6.14 3-((2E)-3-(4-Dimethylamino-phenyl) prop-2-enoyl)-2(H)-chromen-2-one (4i)
Solid (yield 85%); mp = 192 °C. IR spectrum, ν cm−1: 1712 (s) (CO), 1550 (CC), 1028 (sym) (C-O-C); 1H NMR: δ (ppm): 3.2 (s, 6H, 2CH3), 6.8–8.4 (m, 10H, Ar-H + Héthyl). 13C{1H} NMR (ppm): 40.5 (CH3), 180.1 (CO); 190.1 (C4); 153.3 (C2); 112.2 (C3), 116.1–135.8 (Carom); 132.6 (Céthyl1), 149.9 (Céthyl2). C20H1704 N calc. C 71.64, H 5.1, O 19.10, N 4.17; found. C 71.60, H 5.10, O 19.30, N 4.20.
7 The experimental procedure for antioxidant evaluation
The DPPH radical's scavenging capacity of the obtained coumarinic derivatives was measured from the bleaching of purple colored ethanol solution of DPPH. The method described by Hatano et al. [32] was used. Half a millilitre of each sample concentration was mixed with an equal volume of DPPH ethanolic solution. After incubation for 30 minutes in darkness at 25 °C, absorbance was read at 520 nm wavelength. A mixture of 0.5 ml of DPPH solution and 0.5 ml of ethanol was taken as a blank (absorbance is equal to zero). Inhibitory concentration (IC50) values denoting the concentration (microgram of natural substance per milliliter of ethanol) required to scavenge 50% of DPPH radicals were calculated. All measurements were performed in triplicate. Results were expressed in inhibition percentage versus sample concentrations (mg/ml) at 30 mins.
8 The experimental procedure for the antibacterial evaluation
Bacteria used as indicator strains for the determination of the antimicrobial activity spectrum were grown in appropriate culture conditions, as indicated in Table 4.
Stock cultures were maintained at −80 °C in broth supplemented with 25% (v/v) glycerol.
The standard drug used for comparison is gentamycin (MIC + 15 ug/ml).
The tested compounds were dissolved in MeOH to obtain 0.5 mg/mL solution and was tested for antibacterial activity by disc diffusion assay as previously described [38]. An aliquot (10 μl) of was applied onto discs (6 mm diameter) on agar plates previously inoculated with the indicator strain suspension. Plates were incubated for 24 h at 28 °C, the optimal temperature of the tested microorganisms, and diameter of inhibition zone was evaluated.
Acknowledgments
This work was carried out with financial aid of both Tunisian Ministry of Higher Education and Scientific Research and Technology and the Spanish Agency of International Cooperation through projects (A/9549/07 and A/8302/07).