

1 Introduction
Transesterification is an old and essential reaction in laboratory organic chemistry, allowing the transformation of one ester to another through interexchange of the alkoxy moieties [1–3]. Transesterification has also a long history in industry where the process costs are of crucial importance [4,5]. Recently, transesterification has received renewed attention due to its use in biodiesel production from animal or vegetable oils [6,7]. Another application of industrial importance concerns the preparation of high boiling point (meth)acrylic esters from readily available methyl and ethyl esters. Reports are scarce in the academic literature [8], the bulk of the state of the art being disclosed in patents. In this area, the preferred catalysts in industrial plants remain titanium alkoxides because of their low cost, selectivity and activity [9], even if alternative catalysts such as organotin derivatives have been proposed [10,11]. It must be mentioned that sulfonic acid resins, often used in esterification, seem to be ineffective in that case [12]. Industrial plants are running continuous homogenous processes where the equilibrated reaction is driven to completion by azeotropic distillation [13]. At the end of the process, titanates are hydrolysed and removed by filtration [14]. The catalyst is then degraded and appears difficult to recycle. Water-soluble cationic polyelectrolytes based on salts of aminoalkyl esters of (meth)acrylic acids attract considerable interest as a novel class of water-soluble polymers which have potential applications in wastewater treatment and sludge dewatering, in oil extraction and in paper production [15,16]. Amongst these esters, N,N-dimethylaminoethylacrylate (DMAEA) is the most widely used and the most industrially produced [17]. Various catalysts, such as potassium titanate [18] or organotin compounds [19], have been proposed for the synthesis of DMAEA by transesterification; however, the only catalysts used in industrial plants are soluble titanium alkoxides [20]. In a typical process, dimethylaminoethanol (DMAE) and ethyl acrylate (EA) in slight excess are heated with tetraethyltitanate (0.5 to 1 mol %) at 90–100 °C under slightly reduced pressure [21]. The EA-ethanol azeotropic mixture is distilled off to displace the equilibrium and the DMAEA produced is purified by distillation. The poorly soluble catalyst remains in the reactor and is difficult to remove and recycle. Several attempts to improve the recycling ability of titanium alkoxides have already been reported, as use of polymer supports by Lewis et al. [22,23]. The authors prepared solid-phase titanates by reacting chlorotri(isopropoxy)titanium with crosslinked polystyrene beads functionalised with acetyl acetone or polyethylene glycol ligands. In spite of a good transesterification activity, few details were given concerning the metal leaching and recycling ability. A more comprehensive work was performed using silica as support [24–26]. However, loss of activity during successive uses, attributed to leaching of metal in the homogeneous phase, has always been reported. This point can be related, for a large part, to the known high sensitivity of TiO bonds toward hydrolysis and alcoholysis, causing the leaching of the metal [27]. The present work is part of our efforts [28] to apply the heterogenization of homogeneous catalysts concept [29] to the industrial preparation of (meth)acrylic esters.
2 Experimental
2.1 Instruments
Specific surface areas were determined by N2 adsorption measurements performed on a Micromeritics ASAP 2010. The resulting data were subjected to the Brunauer, Emmett and Teller (BET) [30] and the Barrett, Joyner and Halenda (BJH) treatments [31].
Chromatography experiments were carried out using a Varian 3300 and the Star chromatography software. The capillary column was a DB5-like (5% phenyl groups). The DMAEA conversion was calculated from the peak area ratio of DMAEA relative to undecane, as obtained from GC analysis using a calibration curve. Elemental analyses on solids were executed by the Service central d’analyses, Vernaison, France. Results are expressed in weight percent (wt %). Titanium content in effluents was estimated using ICP-MS technique by Antellis Company, Toulouse, France. Results are expressed in mg of Ti kg–1 (ppm).
2.2 Chemicals
All the monomers, styrene (Aldrich), divinylbenzene (80%, mixture of isomers Aldrich), 4-acetoxystyrene (a gift from TOSH Corp, Japan) and other components involved in the polymerisation (azobisisobutyronitrile [AIBN, Acros], 2-ethylhexanol [Aldrich] and acacia gum (Aldrich) were used as received). Tetrabutoxytitanium (Aldrich) was also used without further purification. DMAE and EA (Aldrich) were distilled under reduced pressure from CaH2. All solvents were dried according to published procedures.
2.3 Typical preparation of polymer support (ACetoxyPolymer [ACP])
Poly(4-acetoxystyrene-co-styrene-co-divinylbenzene) beads were prepared by suspension polymerisation according a previously reported procedure [28]. The suspension polymerisation was performed in a 500 mL parallel-sided glass reactor already described [32]. In a typical experiment, the organic phase was composed of 4-acetoxystyrene (8.6 g, 53 mmol), styrene (8.3 g, 80 mmol), divinylbenzene (17.3 g, 133 mmol), AIBN (0.34 g) as the initiator and 2-ethylhexanol (22.6 g) as the porogen. The mixture was suspended in water (350 mL) containing a suspension stabiliser (acacia gum, 7.0 g) and stirred at 220 rpm. The polymerisation was performed at 80 °C for 15 hours, after what the resulting spherical particles were washed with water, then continuously extracted in a Soxhlet apparatus with ethanol (24 h), and finally dried under vacuum to give 33.5 g of white beads (ACP) with a diameter between 200 and 500 μm (yield 99%).
2.4 Conversion of ACP to poly(4–hydroxystyrene-co-styrene-co-divinylbenzene) beads (HP)
The ACP resin beads (31.0 g) were suspended in methanol (100 mL), then a sodium hydroxide aqueous solution (200 mL, 1 mol L–1) was added and the reaction medium mechanically stirred for 2 days at RT. Next, the beads were filtered and copiously washed successively with water and methanol, then continuously extracted in a Soxhlet apparatus with water (24 h), followed by THF (24 h), and finally chloroform (24 h). Drying under vacuum at 50 °C gave 29.0 g of white beads (HP), yield 100%.
2.5 Synthesis of supported catalyst TiP
The white opaque resin beads (HP, 29.0 g, 53 mmol of phenoxy group) were suspended in anhydrous chloroform (300 mL) in a round-bottomed flask under argon. Ti(OBu)4 (21.6 g, 1.2 eq Ti/OH) was injected and the reaction mixture was mechanically stirred for 2 days at room temperature, after what the resulting orange beads were continuously extracted for 2 days with chloroform using a Soxhlet apparatus. Finally, the TiP resin was dried under vacuum at 50 °C to give 33.1 g of orange-yellow beads, yield 94%, (titanium content: 0.71 mmol g–1).
2.6 Transesterification of EA with DMAE catalysed by TiP resin, batch reaction
In a typical procedure, a 50 mL, three-necked, round-bottomed flask connected to a condenser cooled at 0 °C and equipped with a mechanical stirrer, was charged with TiP resin (0.31 g, ∼0.3 mmol of Ti, ∼0.1 mol % /DMAE) suspended in a mixture of DMAE (3.0 g, 30 mmol), EA (6.0 g, 60 mmol), 4-methoxyphenol (0.037 g, 0.30 mmol) as polymerisation inhibitor and undecane (0.47 g, 3 mmol) as internal standard for GC analysis. The mixture was heated at reflux (∼115 °C) under a slow bubbling of dry air. Aliquots were periodically taken from the reaction mixture over a given period and diluted with diethyl ether (20 mL) before GC analysis for DMAEA conversion determination.
2.7 Transesterification of EA with DMAE catalysed by TiP resin, continuous reaction
A stainless steel HPLC-type column (7 mm ID, 300 mm useful length) was filled with TiP resin beads (5.1 g, 175 mg Ti) in a glove box and joined to a second column containing 3 Å molecular sieve (8.0 g). The system, protected from air moisture, was then connected to the pump by the desiccant column input, the reactor column being placed in the furnace heated at the given temperature. The reagents feedstock consisted of a mixture of DMAE (577.6 g, 6.48 mol), EA (1292.3 g, 12.91 mol), 4-methoxyphenol (31.0 g, 0.249 mol) and phenothiazine (64.0 g, 0.310 mol) as polymerisation inhibitors, and undecane (100.8 g, 0.645 mol) as internal standard for GC analysis. The columns were first flushed with a flow of dry DMAE until an acceptable water level in the exit flow was obtained (70 ppm). Then, the reagent feedstock was pumped through the system with a flow rate adjusted in order to obtain the expected average residential time. After cooling at −5 °C in order to avoid any loss of volatile products, the exit flow was fractionated every 8 hours and the DMAEA conversion estimated by GC.
2.8 Automated image analysis of polymer beads and size distribution
Optical microscopy images of dry beads were obtained with a 5 Mpixels digital camera to acquire greyscale photographs in TIFF format. Care to minimize aggregation was taken by coating the glass slide with a thin layer of oil. The intention was to obtain high-quality digital images with high contrast and resolution. Each image was taken from randomly chosen independent sample of each polymer beads batch to give a reliable representation of the size distribution (at least 100 beads were present in the image). The picture file was then transferred to Scion software (Scion, MD, http://www.scioncorp.com) for processing. The software identifies objects within the image by the difference in pixel intensity after thresholding. The beads were counted and their perimeter was calculated as were the size values. The results were then filtered to exclude any particle with a perimeter < 100 μm and the data were exported to Excel for plotting of size distribution histograms with the calculated best-fit distribution.
3 Results and discussion
Our previous studies on polymer-supported titanium catalysts [33] seemed to confirm the early observation that titanium phenoxide bonds Ph-OTi were much more resistant toward hydrolysis and alcohol exchange than alkyl titanate C-OTi bonds [34]. The grafting of a trialkoxytitanium moiety onto a polymeric network through a titanium-phenoxy bond led to the synthesis of an active catalyst in transesterification reactions exhibiting a low metal leaching [35].
3.1 Preparation of the catalyst support
In order to present some perspective of use in an industrial plant, the solid catalyst should possess the following properties. (i) The monomers used should be low cost and easily available compounds. (ii) The polymer support synthesis should use a well-established and straightforward procedure. (iii) The grafting of the titanate moiety should be simple and from a catalytic species already in use, and (iv) problems arising from metal loading should be strongly minimized. In order to fill these requirements, our choice focused on the suspension copolymerisation of styrene, divinylbenzene and 4-acetoxystyrene (Scheme 1). The two former compounds are universally used monomers, whereas the latter can be available in bulk quantities at relatively low cost with a high purity, due to its use as negative photoresist in photolithographic imaging technology – in the form of derivatives of poly(4-hydroxy)styrene [36]. 4-vinylphenol is conveniently prepared, either by a chemical route in a two-step sequence involving the Knoevenagel condensation of malonic acid and hydroxybenzaldehyde [37] or, alternatively, by an enzymatic decarboxylation of 4-hydroxycinnamic acid [38]. Furthermore, 4-acetoxystyrene behaves similarly to styrene in free radical polymerization and, therefore, its distribution in styrene copolymers is almost random [39].
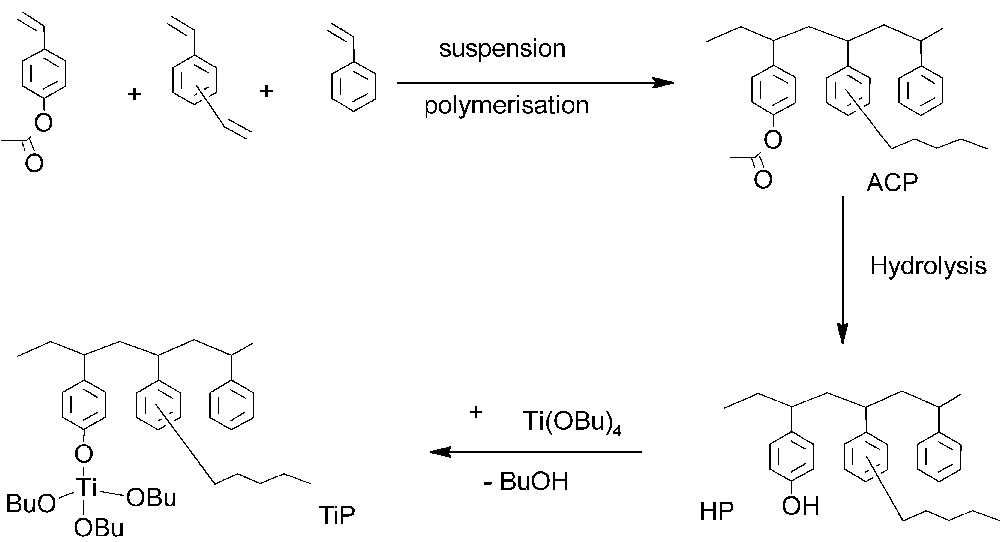
Synthesis of the supported catalyst.
3.2 Polymer beads and size distribution analysis
ACP beads samples chosen from 14 successive suspension polymerization batches were analyzed automatically for diameter size distribution analysis [40]. Fig. 1 is a representative example of automated particle analysis image. Histograms of the particles size analysis for batches prepared, determined by image analysis and by sieving for comparison are reported in Fig. 2. The two distribution graphs fit quite well. Average diameter value generally does not fit the maximum of the histogram, as the distribution does not follow a symmetrical Gaussian distribution.

Typical example of automated particles analysis.
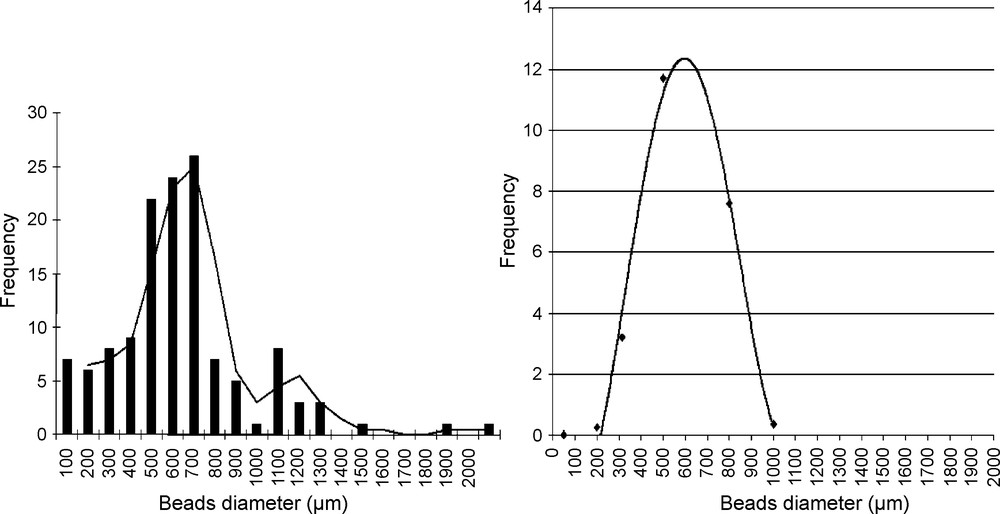
Distribution histogram of ACP bead diameters (a) by image analysis, (b) by sieving.
Organic titanates tend to associate in solution [41]. Electrospray mass spectroscopy suggests that tetra(n-butyl)titanate is trimeric, tetra(isopropyl)titanate being monomeric in nature [42]. They are also highly hydrolysable, giving access to much less active oligomers [41]. In order to insure the grafting of a stable, highly active monomeric titanate form, the polymer supports were designed with a high crosslinking level and a rather low catalyst loading. Furthermore, in the expectation to obtain a single-site heterogeneous catalyst [43], the morphology chosen was the so-called macroporous structure using a precipitant porogen (2-ethylhexanol, 40 wt %) in order to obtain a specific surface area value in the medium range [44].
Particles of suitable size (∼500 μm) for use in heterogeneous catalysis are readily prepared by suspension polymerisation in which the organic monomer phase containing dissolved free radical initiator is dispersed as droplets in a continuous aqueous phase [45,46]. The latter contains a water-soluble polymeric stabiliser, avoiding the coagulation of the droplets during the “sticky phase”. Styrene/divinylbenzene-based resins are synthesized on a large industrial scale by using a batch polymerization procedure carried out in steel, stirred-tank reactors [47]. Laboratory-scale resin synthesis is problematic and some optimization program is usually necessary for each functional comonomer. The use of a specially designed baffled parallel-sided reactor with a relatively flat base is recommended if reproducible high yields of material with a narrow diameter distribution are to be obtained [48].
Beads of poly(4-acetoxystyrene-co-styrene-co-divinylbenzene) (20/30/50 mol %) copolymer to be used as a catalyst support were thus obtained by suspension polymerisation (Scheme 1), after a careful optimisation of the experimental parameters (reactor design, stirring speed, organic/aqueous phase ratio), as previously reported [49].
We were thus able to obtain reproducible high yields (99%) of beads having a relatively narrow size distribution (Table 1) [28].
Characteristics of the ACP support beads.
| Specific surface area (BET) (m2 g–1) | 208 |
| Standard deviation σ | 35 |
| Pores size (BJH) (nm) | 11.1 |
| Standard deviation σ | 0.7 |
| Average particle diameter (μm) | 380 |
| Standard deviation σ | 12 |
3.3 Grafting of the catalytic species onto the polymer support
In order to generate strong, covalent titanium-phenoxy bonds between the supported catalyst and the polymer support, the simplest route is a two-step reaction involving alcohol interexchange between a soluble titanium tetraalkoxide and pendant phenolic moieties present on the support. Therefore, polymer-supported phenols were first generated by a based-catalysed hydrolysis reaction of acetoxy functional groups [50], followed by a treatment with tetrabutoxytitanium in chloroform at room temperature. The titanium loading of the supports were estimated to 0.8 ± 0.1 mmol g–1. Attempts to graft directly the trialkoxytitanium catalytic species on the support from the acetoxy moiety, according to a single-step reported procedure [51], resulted in a much less active supported catalyst.
3.4 Heterogeneous continuous catalytic experiments
The reaction studied was the transesterification of ethyl acrylate (EA) with dimethylaminoethanol (DMAE), giving dimethylaminoethylacrylate (DMAEA) and ethanol (EtOH) in the bulk (no solvent) with a molar ratio EA:DMAE (2:1) (Scheme 2). In these conditions, the experimental equilibrium constant value is about 0.5, giving a DMAEA conversion at equilibrium around 55%. This value is almost independent of the temperature.

Transesterification reaction.
The main objective of this work was the evaluation of the lifetime of the heterogeneous catalyst. The intrinsic activity of a supported catalyst is less crucial, given that it is always possible, in industrial plants, to increase the product conversion by a feedback loop. Therefore, we chose to work under equilibrium conditions (i.e., in a thermodynamically closed system) in order to obtain reproducible results. Some preliminary experiments were conducted in a 100 mL, mechanically stirred, glass batch reactor (not shown). A DMAEA conversion of 20%, (35% of the equilibrium value) was obtained reproducibly by heating the mixture at 115 °C for 8 hours, in the presence of ca. 0.4 mol % of supported catalyst. However, recycling catalyst experiments using this apparatus appeared tedious and poorly reproducible, mainly because of an almost unavoidable wet air intake during handling. Therefore, we developed a continuous flow reactor with an expanded catalytic bed allowing a drastic reduction in handling. The diagram of the reactor system is depicted in Scheme 3. The feed stock solution was continuously supplied to the bottom of a first vertical column containing 3 Å molecular sieve, followed by a second similar vertical column filled with the supported catalyst, this second column being placed in a furnace under controlled temperature.
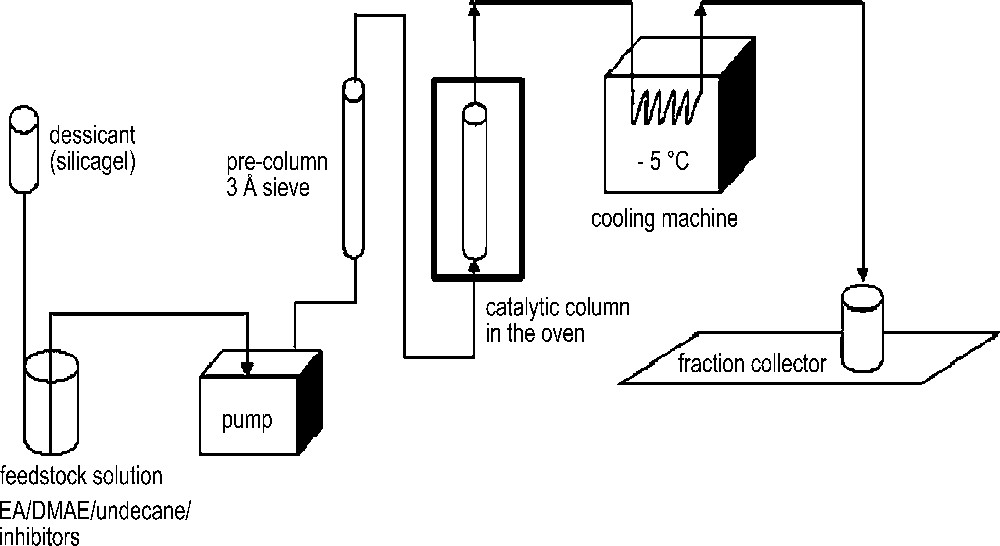
Continuous flow reactor device.
The residence time of the fluids in the expanded bed was regulated by changing the flow rate of the pump. As the reactive fluids flow upward, the catalyst resin floats upward and the expanded bed forms. The pressure inside the column remains below 1 MPa in any working conditions. The exit flow was cooled to −5 °C to avoid any loss of volatile compounds. Fractionation of the flow allowed an average daily DMAEA conversion estimation to be conducted by GC. The experimental conditions are summarised in Table 2.
Experimental conditions of the expanded bed flow reactor.
| Working temperature (°C) | 115 or 130 |
| Total column volume (cm3) | 12 |
| Beads weight (g) | 5.1 |
| Catalyst amount (mg of Ti) | 175 |
| Initial EA concentration (mol L–1) | 5.6 |
| Initial DMAE concentration (mol L–1) | 2.8 |
| Inner pressure (MPa) | 0.5 |
| Flow rate (mL h–1) | 1.5 or 0.48 |
The reactor was first conditioned by pumping pure DMAE for two days at a flow rate corresponding to an average residential time of the flow in the column of 8 h. The water level of the exit flow was then estimated to about 70 ppm (Phase 0, not reported). The feed solution was next pumped at that rate, the catalytic column being heated to 115 ± 0.5 °C, (Table 3 and Fig. 3, Phase 1). The conversion being low and unstable, the furnace temperature was increased to 130 ± 0.5 °C on Day 3 and maintained at that value until Day 10 (Table 3 and Fig. 3, Phase 2). At that time, the flow rate was decrease to obtain an average residence time of 24 h. A stable daily DMAEA conversion was thus obtained for 14 days (Table 3 and Fig. 3, Phase 3). At that moment (Day 26), the furnace temperature was lowered back to 115 ± 0.5 °C, the flow rate remaining constant and the reaction was conducted for 18 additional days (Table 3 and Fig. 3, Phase 4). The DMAEA conversion slowly decreased with time and the experiment was stopped on Day 42. We must highlight that this experiment can be seen as the equivalent of about 125 batches recycling.
Reaction working conditions and Catalyst Efficiency.
| Day number | Flow rate (mL h-1) | Column Temperature (°C) | TOF mmol of DMAEA·(g Ti)–1·day–1 | TON mmol of DMAEA·(g Ti)–1 | |
| Phase 1 | 1–2 | 1.5 | 115 | – | – |
| Phase 2 | 3–10 | 1.5 | 130 | 68 | 544 |
| Phase 3 | 11–24 | 0.48 | 130 | 46 | 644 |
| Phase 4 | 25–42 | 0.48 | 115 | 36 | 648 |
| TON total (mmol of DMAEA·(g Ti)–1) | 1996 |
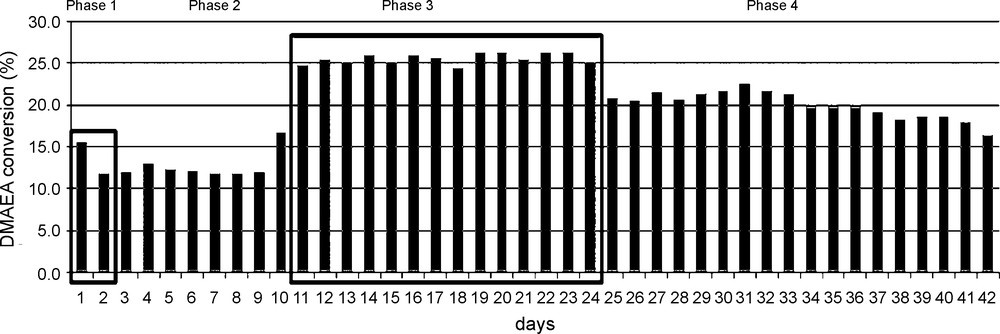
Daily DMAEA yield in continuous flow transesterification. Phase 1: T = 115 °C, flow rate = 1.5 mL h–1, Phase 2: T = 130 °C, flow rate = 1.5 mL h–1, Phase 3: T = 130 °C, flow rate = 0.48 mL h–1, Phase 4: T = 115 °C, flow rate = 0.48 mL h–1.
The efficiency of the catalysis according to the working conditions (reaction temperature and flow rate) can be estimated by calculation of the turn over frequency (TOF = mmol of DMAEA (g Ti)–1 day–1) for each experiment phase (phase 1 was omitted due to conversion instability). The results are reported in Table 3. From these values, it can be concluded that: (i) a high flow rate is favourable to the catalyst productivity; and (ii) the catalyst is more efficient at 130 °C than at 115 °C.
Loss of activity of the supported titanium catalyst with time could be attributed to three main causes: (i) hydrolysis of the titanium alkoxide groups, leading to inactive species; (ii) poisoning of the catalytic active sites; and (iii) leaching of the metal in solution due to alcoholysis of the Ti grafting bond.
We estimated the hydrolytic contribution to catalyst deactivation as marginal, considering the low water level observed in the reagents flow after drying with DMAE and insertion of a molecular sieve column in the device. Concerning the second point, it is well known in industry that the reaction conditions used for the titanium alkoxide catalysed transesterification of (meth)acrylic esters involve some polymerisation of those monomers [52]. In order to limit this very important problem, several radical inhibitors such as phenols derivatives, thiazines or nitroxyl compounds are added to the reagents mixture and, in addition, the reaction vessel is always aerated by a stream of oxygen [53,54]. Despite several attempts, oxygen aeration appeared to be not technically possible in our device, which strongly limited the efficiency of the added inhibitors. However, we did not observe in our experiments significant polymerisation of (meth)acrylates capable of reducing the grafted catalyst efficiency by encapsulation (polymerisation in the column is usually evidenced by a rapid increase of the fluid back pressure due to the reduction of the flow circulation). Thus, the metal leaching from the support remains the most probable cause of activity decrease.
3.5 Analysis of titanium leaching
In order to evaluate the catalyst leaching level during the reaction, the daily reactor production was collected and analyzed for titanium traces. Leaching during the first day was important and could be attributed to non-grafted catalyst incompletely washed from the support. Afterward, the average daily leaching during the first 32 days can be estimated to about 9.5 ppm, which corresponds to about 0.1% of the total amount of titanium grafted on the support engaged in the reaction. Finally, in 42 days of run, the total titanium leaching was estimated to about 12 mg of Ti that is, about 7% of the initial amount of grafted titanium involved.
4 Conclusions
We have prepared a polymer-supported titanium catalyst for application in a transesterification reaction of industrial interest with the idea to replace a homogeneous process. In order to assess the efficiency and stability of this catalyst, it was tested in a laboratory-scale continuous flow reactor under equilibrium conditions. The results obtained indicate a good stability to metal leaching with deactivation being attributed to the slow alcoholysis of the phenoxy-titanium bond in the long term. To our knowledge, this result is the best obtained, to date, concerning the heterogenization of a titanate for catalysis in transesterification reactions.
Acknowledgments
This research was strongly supported by the Arkema Company through Aid-in-Grant funding (to M-H. A). Mélanie Bousquet (US2B, Talence, France) is acknowledged for her gracious Karl-Fisher measurements.