

1 Introduction
Luminescent lanthanide complexes have attracted much recent attention because of their use in a wide variety of bio-imaging applications such as biofluoroimmunoassays [1–3], as biosensors [4–9], or for two-photon bioprobes for scanning microscopy [10–15]. In most cases, these complexes consist of a lanthanide ion coordinated to a ligand that shields the metal ion from water coordination and bears a sensitizing chromophore which transfers its excitation energy to the luminescent lanthanide ion. The presence of the organic chromophores able to strongly absorb UV-visible light and to transfer this energy to the lanthanide ion overcomes the limitation of an intrinsically small molar absorption coefficient of the forbidden f-f transitions (ɛ about 1–10 L.Mol−1.cm−1). This so-called “antenna effect” results in the considerable increase of the brightness defined as the product of the luminescence quantum yield and the absorption coefficient, B = ɛ.Φ. In addition, the luminescence quantum yield and lifetime are highly sensitive to the inner coordination sphere and dramatically decrease when OH vibrators are coordinated. Furthermore, for in vivo applications, the ligands have to bind strongly the lanthanide ion in order to form kinetically stable complexes in a biological medium to avoid any release of toxic-free Ln(III) ions.
The unique properties of these ions include line-like emission, long luminescence lifetimes (in the μs-ms range), the sensitivity to the surrounding symmetry and the relative insensitivity of their emission to the presence of dioxygen (that is due to very fast energy transfer from the transferring excited state to the f-f emitting state) [16,17]. Despite extensive research, the optimization of the energy transfer for the sensitization of the lanthanide ion by exciting complexed chromophores is still not fully understood and remains a challenging task. In this respect, the europium ion is an interesting probe since Beeby et al. [18] and Werts et al. [19] have shown that the steady state luminescence spectrum and the luminescence lifetime of the europium ion can be used to evaluate the efficiency of the sensitization process and therefore understand the limitation residing in the studied systems.
We recently showed that the water soluble functionalized dipicolinate ligand can possess absorption transitions in the UV-visible (up to 400 nm) with high molar absorption coefficient and can efficiently sensitize Eu(III) [12,20], Yb(III) or Nd(III) [20] ions. In the mean time, these complexes present large two-photon cross-section values. This exaltation is induced by intraligand charge transfer (ILCT) state. Unfortunately, despite a stability good enough to make in vitro measurements, the previously presented chelators are not stable enough for in vivo applications. In the progress of our research, we looked for a new scaffold to improve the stability of the complexes. In this context, we decided to base our complexes on the DTPA [21,22] and DO3A [23,24] macrocyclic units well known to lead to the formation of complexes with enhanced stability in aqueous media.
Here, we report on the synthesis and complexation to Eu(III) of two new ligands featuring donor-phenylethynyl functionnalities that are based on DTPA bis-amide ([Eu(L1)]) and DO3A ([Na][Eu(L2)]) (Scheme 1). Those ligands possess electron donating oxygen or sulfur donor atoms, respectively, connected to a π-conjugated skeleton to induce a charge transfer transition [20,25] and additional triethyleneglycol (PEG) chain to insure the water solubility. The optical properties were studied and reveal that the DTPA bis-amide derivative ([Eu(L1)]) is much less bright than the DO3A derivative ([Na][Eu(L2)]) because of the presence of a coordinated water molecule that quenches the emission. On the other hand, [Na][Eu(L2)] reaches excellent photophysical properties almost as good as our benchmark compound [12]. However, the two-photon cross-section of this complex was measured to be only 4 GM limiting its potentialities for two-photon applications.
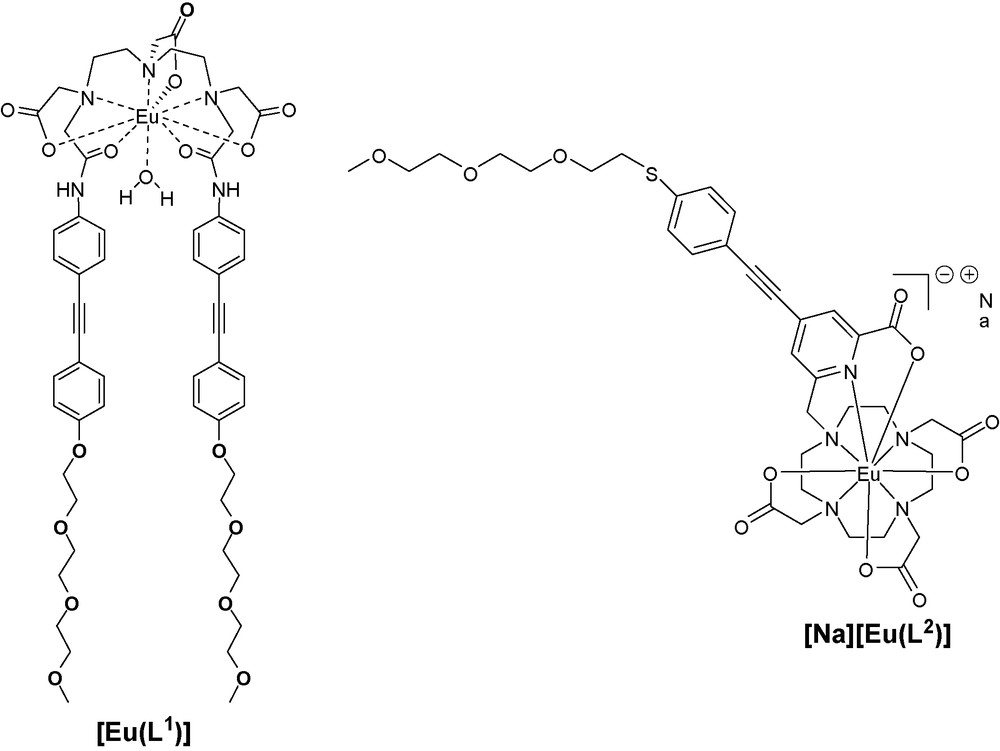
Structures of the investigated europium(III) complexes.
2 Results and discussion
2.1 Synthesis
The synthesis of the DTPA derivative (Scheme 2) was readily started with the opening of the bis-anhydride 1 by the addition of 4-iodoaniline giving the DTPA bis-amide derivative 2. After esterification of the three free carboxylic acid functions, the resulting tri-ester 3 was coupled to the acetylene derivative 4 [26] using a Pd-catalyzed Sonogashira cross-coupling reaction under standard conditions yielding 5 after column chromatography with an acceptable yield (66%). The final ligand L1 was obtained after deprotection of the ester moieties in basic methanol-THF solution (1 M sodium hydroxide). Complete analyzes are compiled in the experimental section. Mixing the ligand L1 with one equivalent of EuCl3·6H2O in aqueous solution followed by extraction with dichloromethane leads to the formation of analytically pure [Eu(L1)]·6H2O.

Synthetic pathway for the preparation of [Eu(L1)].
The DO3A derivative was prepared starting from the iododipicolinic diester 6 [27] (Scheme 3). The key step of this synthesis was the desymmetrisation of 6 into carboxylic acid-alcohol 7 thanks to a kinetically controlled monoreduction by sodium borohydride at room temperature in methanolic solution. After Sonogashira cross-coupling with 8 [26], the chromophore 9 was obtained in good yield (84%) after column chromatography. The alcohol function was then activated with mesyl chloride (10) and further attached to cyclen macrocycle giving the monosubstituted species 11. The remaining free amine functions of 11 were then alkylated by bromoacetate in basic medium and the final functionalized DO3A derivative 12 was purified using column chromatography on alumina oxide. After saponification of the four ester moieties, a one pot complexation to Eu(III) ion was carried out in a buffered solution at pH = 6.
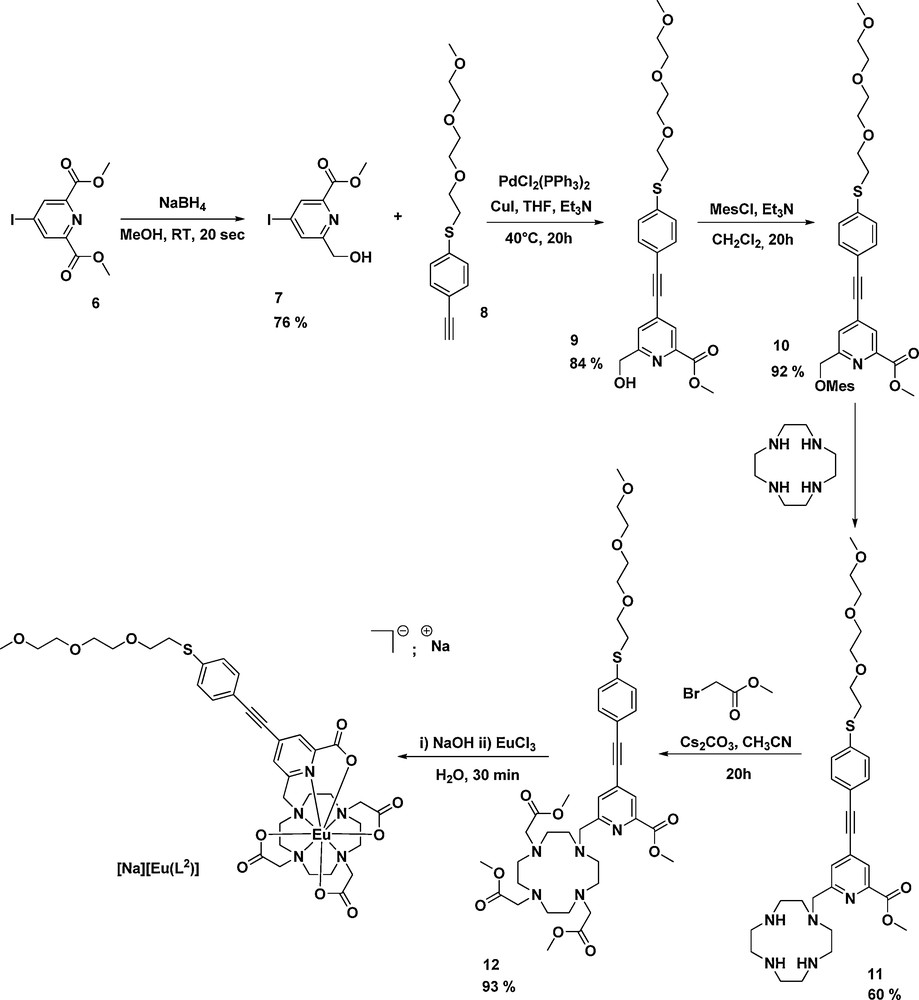
Synthetic pathway for the preparation of [Na][Eu(L2)].
2.2 Crystal structure of the Eu-DTPA model
Due to the presence of the long PEG chains, we were not able to prepare good quality crystals of [EuL1]. Therefore, white crystals of the model complex [Eu(2)] featuring iodophenyl pendant arm were obtained upon cooling down a hot methanol solution (see experimental section for the complex preparation). The complex crystallized as a methanol adduct with two methanol and one diethyl ether interstitial solvent molecules. At the molecular level (Fig. 1, top), the wrapping of the DTPA ligand around the central Eu(III) ion orientates the two iodophenyl in almost the same direction (the angle (I1N1),(I2N5) is estimated to be 12°). It is worth noting that the two phenyl rings are not parallel and form an angle of 32°, a distortion certainly related to the steric hindrance induced by the coordinated methanol molecule. The central europium ion is nona-coordinated by three nitrogen, the carboxylato- and two amido-oxygen atoms, the last position being occupied by an oxygen atom from a methanol molecule. The coordination polyhedron (Fig. 1, bottom) is a distorded tricapped trigonal prism with the Eu,O3,O9,N3 atoms lying in the equatorial plane (maximum standard deviation < 0.1 Å). Such a type of structure is relatively frequent for lanthanide DTPA complexes also dimer formation can be observed depending on the sterical hindrance afforded by the DTPA functionalization [28] (Table 1).
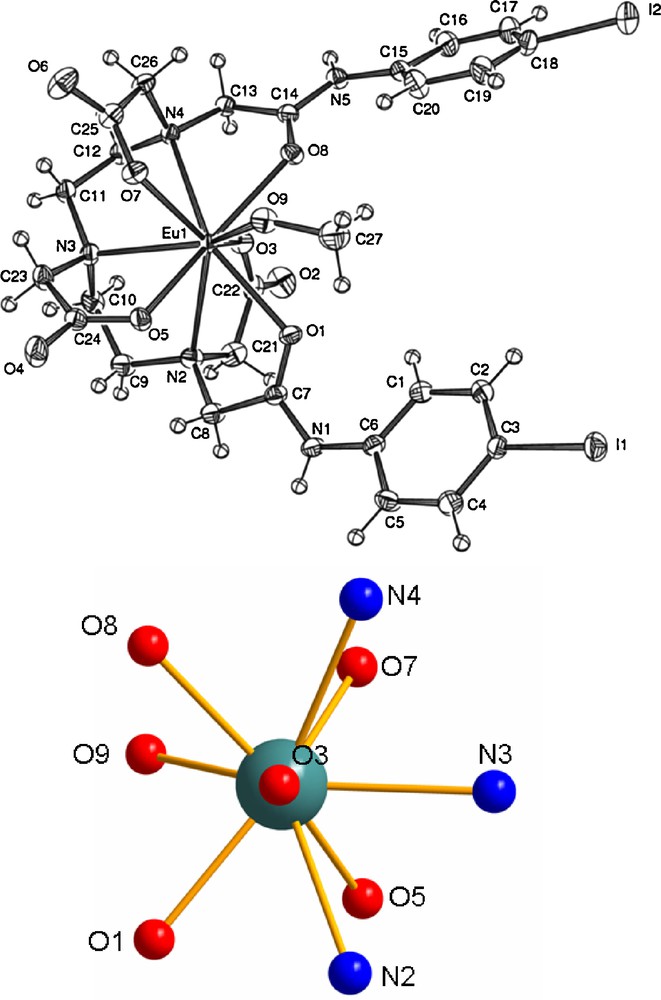
ORTEP drawing for [EuI2C27H33N5O9.CH3OH].3CH3OH.C4H10O. Interstitial solvent molecules were omitted for clarity (top). Coordination polyhedron around the Eu(III) ion (bottom). Selected bond lengths (Å): (amino) Eu-N2 = 2.755(3); Eu-N3 = 2.640(3); Eu-N4 = 2.673(3); (amido) Eu-O1 = 2.485(2); Eu-O8 = 2.481(2); (carboxylato) Eu-O7 = 2.394(2); Eu-O5 = 2.344(2); Eu-O3 = 2.331(3); (methanol) Eu-O9 = 2.483(3).
Crystal data and refinement parameters.
| Formula | C34H55EuI2N5O13 |
| f.w. (g.mol−1) | 1147.59 |
| Cryst. Syst. | Monoclinic |
| Space group | P21/n |
| a (Å) | 10.4117(2) |
| b (Å) | 14.7816(4) |
| c (Å) | 26.6463(6) |
| α (o) | 90.0 |
| β (o) | 99.2700(10) |
| γ (o) | 90.0 |
| V (Å3) | 4047.35(16) |
| Z | 4 |
| T (K) | 120 |
| λ(MoKα) (Å) | 0.71069 |
| D (g.cm−3) | 1.883 |
| μ (mm−1) | 3.143 |
| R(F)a, I > 1σ(Fo) | 0.037 |
| Rw(F2)b, I > 1σ(Fo) | 0.094 |
| S | 0.67 |
| Rint | 0.056 |
| θmax | 32.25 |
| h | −15 → 14 |
| k | −22 → 13 |
| l | −37 → 38 |
| Parameters | 496 |
| Measured reflections | 37666 |
| Independent reflections | 12950 |
| Reflections with I > 2.0σ(I) | 9882 |
| Δρmin | −2.095 e Å-1 |
| Δρmax | 2.217 e Å-1 |
a R(F) = Σ ||Fo| − |Fc||/Σ |Fo|.
b Rw(F) = Σ[w ((Fo2 − Fc2)2/Σ wFo4]1/2.
2.3 UV-visible absorption spectroscopy
The UV/visible absorption data for each of the Eu(III) complexes in water are depicted in Fig. 2 and all the data are summarized in Table 2. The two complexes possess their lowest absorption transition in the visible with maxima at 300 and 338 nm for [Eu(L1)] and [Na][Eu(L2)], respectively. This red shift observed for [Na][Eu(L2)] as compared to [Eu(L1)] will favor the former complex for two-photon applications since measurements are performed using Ti-sapphire laser sources between 700–900 nm (i.e. when doubled: 350–450 nm). Furthermore, the observed cut-off of the UV/visible absorption for [Eu(L1)] (at 350 nm) precludes any measurement of the two-photon excitation spectrum. In addition, the molar absorption coefficients of [Eu(L1)] and [Na][Eu(L2)] are 17,650 and 36,000 M−1cm−1, respectively. In this case, the molar absorption coefficient is in favor of [Na][Eu(L2)] that will give an advantageous brightness for this complex.

UV/visible absorption spectra in water of [Eu(L1)] (__) and [Na][Eu(L2)] (--).
2.4 Luminescence of Eu(III) complexes and determination of the sensitization parameters
The steady state emission spectra, the luminescence quantum yields and luminescence lifetimes of the Eu(III) complexes were measured in water (Table 2). The relevant radiative and non-radiative parameters were also determined following the works of Werts et al. [19] and Beeby et al. [18], and a complete summary of these data are reported in Table 3. As can be seen from Fig. 3, the spectra of the two Eu(III) complexes are very similar with intense J = 2 and J = 4 transitions. In both spectra, all transitions are split in numerous transitions revealing the low symmetry of those complexes [29]. In the two cases, the J = 0 seems to be a single transition which is typical of the presence of only one species in solution (full-width-at-half-maximum of the J = 0 transitions are 750 and 450 cm−1 for [Eu(L1)] and [Na][Eu(L2)], respectively). This observation is confirmed by the presence of a mono-exponential decay for both samples. The luminescence quantum yields measured in water are very different about 2% for [Eu(L1)] up to 28% for [Na][Eu(L2)] with luminescence lifetimes of 0.59 and 1.39 ms, respectively. Such differences in terms of luminescence quantum yields and lifetimes suggest the presence of water molecules in the inner sphere of [Eu(L1)] while the complex [Na][Eu(L2)] most likely presents a saturated coordination sphere. In order to determine the number of water molecules in the inner sphere of [Eu(L1)], the luminescence lifetime was also measured in deuterated water, yielding a value of 1.87 ms, which is far higher than that in water. The q number can therefore be obtained (Table 3) using the improved phenomenological Horrocks equation [30], revealing a q = 1.2 (substantially one water molecule) in agreement with the crystal structure of the model complex [Eu(2)] containing one solvent molecule. Similar water molecule coordination has already been observed in related complexes [31].
Calculated luminescence parameters τR, kr, Σknr, ηEu, and ηsens for the two investigated Eu(III) complexes using the experimental quantities ϕEu, τEu and [I(0,1)/Itot] in water.
| Complex | ϕEu | τEu/ms | τR/ms | ηEu | ηsens | [I(0,1)/Itot] | kr/s−1 | Σknr/s−1 |
| [Eu(L1)]a | 0.019 | 0.59 | 4.58 | 0.129 | 0.147 | 0.148 | 218 | 1477 |
| [Na][Eu(L2)] | 0.284 | 1.39 | 4.24 | 0.328 | 0.866 | 0.137 | 236 | 483 |
| [Eu(L1)]a,b | 0.078 | 0.98 | 3.86 | 0.254 | 0.305 | 0.152 | 259 | 761 |
a Not taking the J = 3 into account because of the overlap with the 2λ of the excitation.
b Measured in dichloromethane solution.
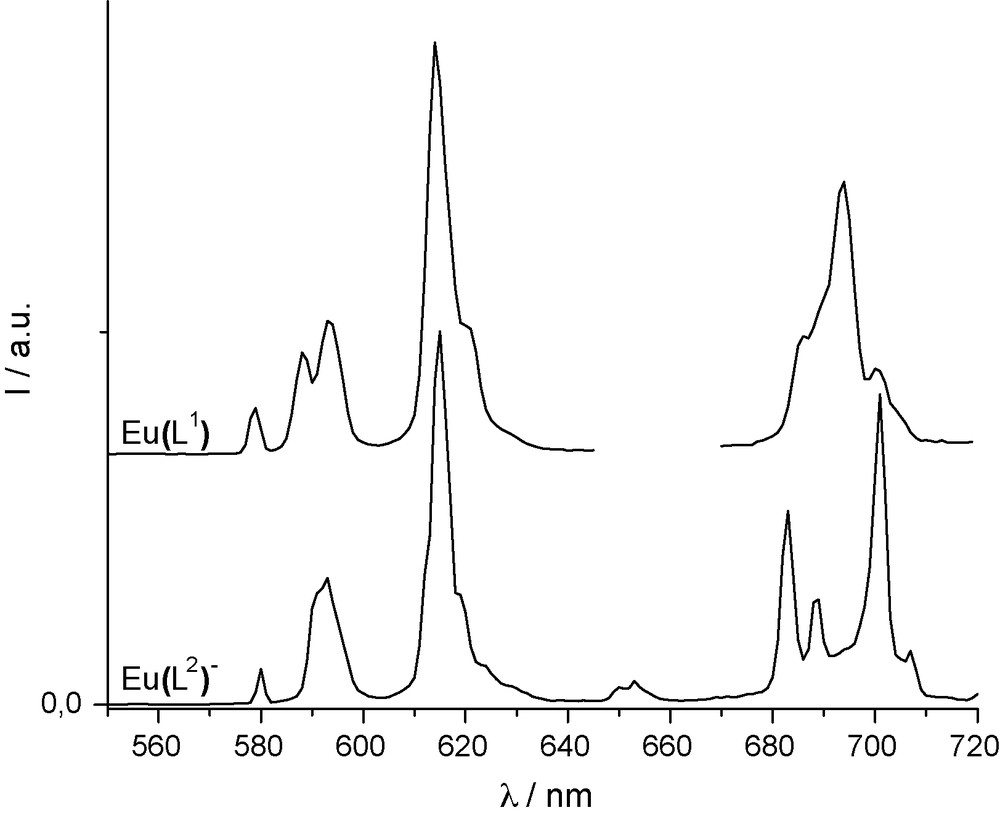
Normalized luminescence spectra (on J = 2) of [Na][Eu(L2)] (bottom, λex = 335 nm) and [Eu(L1)] (top, λex = 325 nm) in water.
To understand the limitation and advantage of such ligands, the radiative and non-radiative parameters were determined. As can be seen in Table 3, the symmetry of both complexes are almost equivalent giving identical ratio of intensities between the J = 1 and the whole spectrum. Consequently, both complexes exhibit identical radiative lifetimes (τr) and radiative decay rate (kr) [31]. Here, it has to be highlighted that both complexes possess a coordination number of 9 giving the possibility of a similar symmetry around the metal (in the case of [Eu(L1)], the ninth site being occupied by the water molecule). Therefore, for [Eu(L1)], the large decrease of the luminescence lifetime induces a drop of the europium centered efficiency (ηEu = 0.129) compared to [Na][Eu(L2)] (ηEu = 0.328) that is followed by a large difference in the non-radiative decay rate (483 s−1 vs. 1477 s−1 for [Na][Eu(L2)] and [Eu(L1)], respectively). As can be seen in the Table 3, all parameters related to the europium centre (τEu and ηEu) are very low in the case of [Eu(L1)]. This can be explained by the inner sphere water molecule bound to the metal cation (also increasing the non-radiative decay rate (Σknr)). It is worth noting that the sensitization efficiency (ηsens), that is independent of the presence of inner sphere water molecule, is low for [Eu(L1)] (0.147) while it is almost quantitative for [Na][Eu(L2)] (0.866). In the dichloromethane solution, this sensitization efficiency remains rather low (ηsens = 0.305) proving the inefficiency of L1 ligand to induce Eu(III) luminescence.
2.5 Two-photon absorption properties
Since [Eu(L1)] has its UV/visible absorption cut-off at 350 nm and is poorly emissive (ϕ = 0.019), no measurement of the two-photon excitation spectrum was performed. On the other hand, the two-photon absorption cross-sections (σ2PA) of [Na][Eu(L2)] have been determined in water using the two-photon excited fluorescence technique in the 700–900 nm wavelength range with a femtosecond Ti-Sapphire pulsed laser source, according to the experimental protocol described by Xu and Webb (coumarin-307 as a standard) [32]. The two-photon excitation spectrum matches the wavelength-doubled scale single-photon one (Fig. 4a). This similarity indicates that the low lying excited state in energy is one- and two-photon allowed and confirms that the europium luminescence is induced by a two-photon antenna effect with a similar photophysical sensitization process (the same excited states are involved in the one- or two-photon sensitization process). As can be seen in Fig. 4, the σ2PA found for [Na][Eu(L2)] in water is rather small compared to the value reported for the europium (III) dipicolinate derivatives [12,33] revealing a maximum of 4 GM. This can be partially rationalized by the presence of only one chromophore while three of such chromophores were present in the previously cited studies.

a: two-photon excitation spectrum of [Na][Eu(L2)] in water solution (■, upper abscissa). Superimposed on this plot is the single-photon absorption spectrum in wavelength doubled scale (__, lower abscissa); b: two-photon excitation spectrum of [Na][Eu(L2)] in water solution (■).
3 Conclusion
In conclusion, this article reported the first attempts at the design of europium complexes featuring high stability in biological media and excellent one- or two-photon brightness, which are the key requirements for a luminescent bioprobe for biphotonic microscopy. To that end, well-known macrocycles, namely DTPA and DO3A, were functionalized by already described two-photon antenna chromophores. In the case of DTPA-bis amide derivative [Eu(L1)], the presence of one water molecule in the first coordination sphere results in a strong quenching of the europium luminescence. On the other hand, the functionalized DO3A complex Na[Eu(L2)] presents an excellent one-photon brightness, but the presence of only one antenna chromophore reduces dramatically its two-photon absorption efficiency. New macrocycles functionalized by several two-photon antenna chromophores are currently being prepared in our group towards the design of this new generation of Ln-based bioprobes.
4 Experimental
4.1 Crystal structure analysis
The samples were studied on a Oxford Diffraction Xcalibur Saphir 3 diffractometer with graphite monochromatized MoKα radiation. Cell parameters were obtained with Denzo and Scalepack with 10 frames (psi rotation: 1° per frame). The structure was solved with SIR-97 [34], which reveals the non hydrogen atoms of structure. After anisotropic refinement, many hydrogen atoms may be found with a Fourier Difference. The whole structure was refined with SHEL-XL97 [35] by the full-matrix least-square techniques (use of F magnitude; x, y, z, αij for C, N and O atoms, x, y, z in riding mode for H atoms). ORTEP views were made with PLATON 98 [36]. All calculations were performed on a Silicon Graphics Indy computer. Crystallographic datum for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, CCDC 601476.
4.2 UV/visible absorption measurements
UV/visible absorption measurements were recorded on a JASCO V550 absorption spectrometer. The spectra were corrected using a solution of water as a reference.
4.3 Luminescence measurements
The luminescence spectra were measured using a Horiba-Jobin Yvon Fluorolog-3 spectrofluorimeter (see the preceding article for details). Phosphorescence lifetimes (> 30 μs) were obtained by pulsed excitation using an FL-1040 UP xenon lamp. Luminescence decay curves were fitted by least-squares analysis using Origin. Fluorescence quantum yields, Q, were measured in a diluted solution with an optical density lower than 0.1 using the following equation: Qx/Qr = [Ar(λ)/Ax(λ)][nx2/nr2][Dx/Dr], where A is the absorbance at the excitation wavelength (λ), n is the refractive index, and D is the integrated luminescence intensity. The “r” and “x” stand for reference and sample. Here, references are Ru(bpy)3 2Cl complexes in a water solution (Qr = 0.028) [37] and [Na]3[EuL1G3] (Qr = 0.157) [12] for each complex.
4.4 Two-photon excited luminescence measurements
The TPA cross-section spectra were obtained by up-conversion fluorescence using a Ti:sapphire femtosecond laser in the range of 700–900 nm. The excitation beam (5 mm diameter) is focalized with a lens (focal length 10 cm) at the middle of the fluorescence cell (10 mm). The fluorescence, collected at 90° to the excitation beam, was focused into an optical fiber (diameter 600 μm) connected to an Ocean Optics S2000 spectrometer. The incident beam intensity was adjusted to 50 mW in order to ensure an intensity-squared dependence of the fluorescence over the whole spectral range. The detector integration time was fixed to 1 s. Calibration of the spectra was performed by comparison with the published 700–900 nm Coumarin-307 two-photon absorption spectrum60 (Coumarin-307 quantum yield 0.56 in ethanol). The measurements were done at room temperature in dichloromethane and at a concentration of 10−4 M.
4.5 General procedure
4 and 8 were prepared following the published procedure [33]; all other starting materials were commercially available. All solvents for synthesis were analytic grade. For the spectroscopy, spectroscopic grade solvents were used. NMR spectra (1H and 13C) were recorded at room temperature on a BRUKER AC 200 operating at 200.13 and 50.32 MHz for 1H and 13C, respectively. Data are listed in parts per million (ppm) and are reported relative to tetramethylsilane (1H and 13C); residual solvent peaks of the deuterated solvents were used as an internal standard. Low resolution mass spectrometry was carried out on an Agilent 1100 Series LC/MSD apparatus. High-resolution mass spectrometry measurements and elemental analyses were performed at the Service Central d’Analyse du CNRS (Vernaison, France).
4.6 Synthesis
1. In a 250 mL round bottom flask, a suspension of DTPA (49 g, 125 mmol) in acetic anhydride (53 mL, 560 mmol) and dry pyridine (62 mL, 770 mmol) was heated at 65 °C, while stirring for 24 h. After cooling to room temperature, the precipitate formed was filtered off and then washed with diethyl ether. After drying under vacuum, pure DTPA-bis(anhydride) 4 was obtained (43 g, 120 mmol, 96%). 13C-NMR ([d6]-DMSO): 50.78, 51.72, 52.71, 54.73, 165.88, 171.82 ppm.
2. In a 50-ml round bottom flask, iodoaniline was solubilized in DMF (20 mL). Pyridine was added to the solution followed by 1. The solution was stirred for 1–2 h at room temperature. The DMF was removed by adding toluene, and the solid obtained was cleaned twice with dichloromethane (2 × 150 mL). Yield 85%. 1H-NMR (200.13 MHz, [d6]-DMSO): 10.12 (d, 2H), 7.55 (d, 3J = 8.8 Hz, 4 H), 7.45 (d, 3J = 8.8 Hz, 4H), 3.43 (m, 10H), 2.95 (m, 4H), 2.90 (m, 4H).
3. In a 250-ml round bottom flask, 2 was suspended in anhydrous MeOH (150 mL), 2 mL of concentrated H2SO4 was added and the solution was refluxed for 16 h. After a cooling period, the acid was neutralised by adding NaHCO3. The solution was filtered and the solvent was evaporated. Yield 95%. 1H-NMR (200.13 MHz, CDCl3): 9.77 (bs, 2H, NH), 7.57 (d, 3J = 6.8 Hz, 4H), 7.40 (d, 3J = 6.8 Hz, 4H), 3.68 (m, 9H), 3.48 (m, 10H), 2.71 (m, 8H); 13C-NMR (50.332 MHz, CDCl3): 177.06, 175.80, 174.23, 172.30, 141.64, 141.44, 126.05, 125.95, 90.86, 50.78, 50.36, 51.63, 51.21, 52.91, 52.49, 52.06, 55.00; m/z 838 (MH+ requires 838), 860 (MNa+ requires 860).
5. In a 100-mL schlenk flask, 3 (0.25 g, 0.30 mmol) was dissolved in a mixture of THF and triethylamine (15 mL/15 mL). The solution was thoroughly degassed by argon bubbling for 20 min and 4 [33] (0.75 mmol, 2.5 equiv.), copper iodide (0.2 equiv.) and (Ph3P)PdCl2 (0.l equiv.) were added under a flux of argon. The reaction was stirred at 40 °C for 14 h. The compound was purified by column chromatography on silica using gradient ethyl acetate/methanol (9/1 to 7/3) as eluent. This yields a brown oil (0.22 g, 66%). 1H-NMR (200.13 MHz, CDCl3): δ = 9.82 (bs, 2H), 7.59 (d, 3J = 8.6 Hz, 4H), 7.44 (m, 8H), 6.88 (d, 3J = 8.7 Hz, 4H), 4.12 (t, 3J = 5.6 Hz, 4H), 3.84 (t, 3J = 5.5 Hz, 4H), 3.75 (s, 9H), 3.40–3.72 (m, 26H), 3.54 (m, 2H), 3.36 (s, 6H), 2.74 (m, 8H); EI-MS: [M + H]+: 1110, [M + Na]+: 1132.
L1. In a 100-mL round bottom flask, 5 (0.236 g, 0.230 mmol) was dissolved in 30 mL of methanol and stirred at room temperature. Subsequently, 10 mL of a 1 M aqueous sodium hydroxide solution is added and the reaction is stirred at room temperature for 2 h. The solution is extended to 50 mL by adding water and the methanol is removed under vacuum. The aqueous solution is extracted with ethyl acetate (2 × 50 mL), then acidified to pH = 2 by adding 1 M HCl aqueous solution, and extracted with dichloromethane (3 × 50 mL). The dichloromethane solutions are regrouped and dried over sodium sulfate and the dichloromethane was evaporated yielding the pure 8 as a light yellow solid (0.22 g, 93%). 1H-NMR (200.13 MHz, CD3OD): δ = 7.56 (d, 3J = 8.0 Hz, 4H), 7.33 (m, 8H), 6.82 (d, 3J = 8.1 Hz, 4H), 4.06 (m, 4H), 3.89 (m, 4H), 3.31–3.83 (m, 59H); 13C-NMR (50.332 MHz, CD3OD): δ = 171.4, 169.1, 168.1, 1158.9, 137.5, 132.7, 131.6, 115.5, 114.6, 88.7, 87.4, 71.6, 70.4, 70.1, 69.4, 67.4, 59.7; 54.9, 52.7, 50.8; m/z (ESI) 1068 (MH+ requires 1068), 1090 (MNa+ requires 1090); elemental analysis: Calcd for C56H69N5O16·6H2O: C, 57.18; H, 6.94; N, 5.95; found: C, 56.85; H, 6.94; N, 5.66.
[Eu(L1)]. In a 100-mL round bottom flask, L1 (1 eq) and sodium carbonate (5 eq) are solubilized in water (50 mL). Europium(III) chloride hexahydrate (1 eq) is solubilized in water (10 mL) and added to the solution. The reaction flask is stirred for 1 h at room temperature before being extracted with dichloromethane (4 × 100 mL). The organic phases are combined, dried over sodium sulfate, and evaporated under vacuum giving a slightly yellow solid (0.22 g, 93%). m/z (ESI) 1218 (MH+-H2O requires 1218), 1239 (MNa+ requires 1239); elemental analysis: Calcd for C56H66N5O15Eu·6H2O: C, 50.75; H, 5.93; N, 5.28; found: C, 50.46; H, 5.50; N, 5.15.
[Eu(2)]. In a 100-mL round bottom flask, 5 (651 mg, 0.819 mmol) was dissolved in basic water (30 mL with K2CO3 (302 mg, 2.19 mmol)). Then, EuCl3·6H2O (305 mg, 0.832 mmol) was added and the solution was stirred for 3 h at room temperature. The precipitate was filtered and the white solid was cleaned with cold water and with ether. (764 mg, 98.8%) (M = 944.3 g.mol−1) M(HPLC-MS) = 945.2 [M + H]+, 966.0[M + Na]+.
7. To a stirred mixture of insoluble dimethyl 4-iodopyridine-2,6-dicarboxylate 6 (2 g, 6.23 mmol, 1 eq) and MeOH (40 mL) in a 250-mL round bottom flask, is added NaBH4 (473 mg, 12.5 mmol, 2 eq) at room temperature in one portion. The colorless solution becomes red while H2 is released. As soon as 6 is solubilized (approximately 20 s), the mixture is quenched by addition of saturated NaHCO3 solution (40 mL). The MeOH is evaporated under vacuum, and the aqueous solution is extracted with ethyl acetate (3 × 100 mL). The combined organic extracts are dried (Na2SO4), and the solvent evaporated under vacuum. The crude material is purified by chromatography on silica (CH2Cl2/Acetone 9/1) to afford the product as a white crystalline powder. Depending on the activity of the NaBH4 and the length of the reaction, the yields average from 50 to 76%. 1H NMR (200.13 MHz, CDCl3): 8.37 (s, 1H), 7.95 (s, 1H), 4.81 (d, 3J = 4.9 Hz, 2H), 3.99 (s, 3H), 3.09 (t, 3J = 4.9 Hz, 1H); 13C-NMR (50.332 MHz, CDCl3): 164.4, 161.4, 147.2, 133.2, 132.9, 106.8, 64.2, 53.2; LRMS m/z: 294 (MH+ requires 494), 316 (MNa+ requires 316).
9. To a degassed solution of 7 (0.865 g, 2.95 mmol, 1 eq) and (4-ethynylphenyl)(2-(2-(2-methoxyethoxy)ethoxy)ethyl)sulfane 8[33] (0.858 g, 3.25 mmol, 1.1 eq) in THF (20 mL) and Et3N (10 mL) under argon, is added copper iodide (0.112 g, 0.59 mmol, 0.2 eq) and Pd(PPh3)2Cl2 (0.207 g, 0.295 mmol, 0.1 eq). The dark brown mixture is heated in the dark at 40 °C while stirring for 20 h. After cooling to room temperature, the black precipitate is filtered and triturated with Et2O (2 × 40 mL). The remaining organic phase is washed with saturated aqueous ammonium chloride solution (2 × 50 mL), and brine (50 mL). The organic layer is dried (Na2SO4) and evaporated under vacuum. The crude residue is purified by flash chromatography (CH2Cl2/MeOH 49/1) yielding a light yellow oil (1.267 g, 84%). 1H NMR (200.13 MHz, CDCl3): 7.90 (d, 4J = 1.2 Hz, 1H), 7.56 (d, 4J = 1.2 Hz, 1H), 7.30 (d, 3J = 8.5 Hz, 2H), 7.17 (d, 3J = 8.5 Hz, 2H), 4.75 (d, 3J = 4.3 Hz, 2H), 4.29 (t, 3J = 4.4 Hz, 1H), 3.84 (s, 3H), 3.61–3.38 (m, 10H), 3.24 (s, 3H), 3.04 (t, 3J = 6.8 Hz, 2H); 13C-NMR (50.332 MHz, CDCl3): 165.1, 161.5, 147.0, 139.2, 133.3, 132.2, 127.6, 125.4, 125.3, 118.5, 95.0, 86.6, 71.8, 70.51, 70.47, 70.40, 69.6, 64.6, 58.9, 52.9, 32.1; LRMS m/z: 446 (MH+ requires 446), 468 (MNa+ requires 468).
10. To a stirred solution of 9 (1.10 g, 2.56 mmol, 1 eq) and triethylamine (0.777 g, 7.68 mmol, 3 eq) in CH2Cl2 (100 mL) in a 250-mL round bottom flask, is added methanesulfonyl chloride (0.440 g, 3.84 mmol, 1.3 eq) drop by drop at room temperature. After 30 min, the reaction is quenched by addition of saturated NaHCO3 solution (100 mL). The organic phases is extracted, dried over Na2SO4, and evaporated. The crude material is purified by chromatography on silica (CH2Cl2/MeOH 97/3) to afford the product as a bright yellow oil (1.19 g, 92%). 1H NMR (200.13 MHz, CDCl3): 8.04 (d, 4J = 1.2 Hz, 1H), 7.62 (d, 4J = 1.2 Hz, 1H), 7.37 (d, 3J = 8.5 Hz, 2H), 7.22 (d, 3J = 8.5 Hz, 2H), 5.32 (s, 2H), 3.91 (s, 3H), 3.66–3.42 (m, 10H), 3.28 (s, 3H), 3.13–3.04 (m, 5H); 13C-NMR (50.332 MHz, CDCl3): 164.7, 154.7, 147.9, 139.6, 134.1, 132.3, 127.6, 126.6, 126.4, 118.2, 96.1, 86.1, 71.9, 70.56, 70.55, 70.53, 70.46, 69.6, 59.0, 53.1, 38.0, 32.1; LRMS m/z: 524 (MH+ requires 524), 546 (MNa+ requires 546), 562 (MK+ requires 562).
11. To a stirred solution of 1,4,7,10-tetraazacyclododecane (1.92 g, 11.2 mmol, 4 eq) in CHCl3 (100 mL) in a 250-mL round bottom flask, is added a solution of 10 (1.46 g, 2.8 mmol, 1 eq) in CHCl3 (100 mL) drop by drop over 3 h at room temperature. After 20 h of stirring, the mixture is evaporated and the crude material is purified twice by chromatography on aluminum oxide (CH2Cl2/MeOH 97/3) to afford the product as a bright yellow oil (1.00 g, 60%). 1H NMR (200.13 MHz, CDCl3): 7.93 (d, 4J = 1.1 Hz, 1H), 7.47 (d, 43J = 1.1 Hz, 1H), 7.33 (d, 3J = 8.5 Hz, 2H), 7.20 (d, 3J = 8.5 Hz, 2H), 5.31 (b, 3H), 3.90–3.84 (m, 5H), 3.64–3.40 (m, 10H), 3.26 (s, 3H), 3.06 (t, 3J = 6.8 Hz, 2H), 2.65-2.90 (m, 16H); 13C-NMR (50.332 MHz, CDCl3): 165.2, 160.0, 147.5, 139.2, 133.3, 132.2, 127.7, 127.4, 125.7, 118.5, 95.1, 86.4, 71.8, 70.51, 70.50, 70.4, 69.6, 59.5, 59.0, 53.0, 51.8, 47.2, 45.7, 45.3, 32.1; LRMS m/z: 600 (MH+ requires 600), 622 (MNa+ requires 622).
12. To a stirred solution of 11 (0.852 g, 1.42 mmol, 1 eq) and Cs2CO3 (1.48 g, 4.55 mmol, 3.2 eq) in CH3CN (100 mL) in a 250-mL round bottom flask, is added methyl 2-bromoacetate (0.782 g, 5.11 mmol, 3.6 eq). After stirring for 20 h at room temperature, CH2Cl2 (100 mL) is added and the solution is filtered. After evaporation of the solvents, the crude material is purified by chromatography on aluminum oxide (CH2Cl2: MeOH 39/1) to afford the product as a bright yellow oil (1.08 g, 93%). 1H NMR (500 MHz, CDCl3): 7.92 (s, 1H), 7.44 (s, 1H), 7.33 (d, 3J = 8.4 Hz, 2H), 7.19 (d, 3J = 8.4 Hz, 2H), 3.87 (s, 3H), 3.67 (s, 3H), 3.59 (t, 3J = 6.8 Hz, 2H), 3.52-3.46 (m, 12H), 3.44–3.36 (m, 2H), 3.24 (s, 3H), 3.06 (t, J = 6.8 Hz, 2 H), 3.2–2.2 (m, 24H); 13C-NMR (50.332 MHz, CDCl3): 173.5, 172.4, 165.0, 159.2, 147.7, 139.5, 134.0, 132.2, 129.0, 127.5, 125.3, 118.0, 96.2, 85.9, 71.7, 70.31, 70.29, 70.26, 69.4, 59.1, 58.8, 55.2, 54.9, 53.0, 52.2, 51.9, 50.2, 32.0; λmax (H2O)/nm 333 (ɛ/dm3 mol−1 cm−1 27400); LRMS m/z: 816 (MH+ requires 816), 838 (MNa+ requires 838); HRMS m/z: 816.3848 (MH+ cald for C40H58N5O11S requires 816.3854).
[Na][Eu(L2)]. To a stirred solution of 12 (122 mg, 0.15 mmol, 1 eq) in H2O (10 mL) in a 50-mL round bottom flask, is added NaOH (36 mg, 0.9 mmol, 6 eq). After stirring for 1 h at 50 °C, the solution is cooled down to room temperature and acidified to pH = 6 by addition of 0.1 M HCl solution. At that stage, a solution of EuCl3·6H2O (66 mg, 0.18 mmol, 1.2 eq) in H2O (10 mL) is slowly added while the pH of the reaction is kept at pH = 6 by addition of 0.1 M NaOH solution. Once the pH of the reaction is stable, the reaction is stirred for an additional 20 h at 75 °C. λmax (H2O)/nm 334 (ɛ/dm3 mol−1 cm−1 36100); LRMS m/z: 910 (MHH+ requires 910), 932 (MHNa+ requires 932), 948 (MHK+ requires 948), 954 (MNaNa+ requires 954); HRMS m/z: 954.1859 (MNaNa+ cald for C36H45EuN5Na2O11S requires 954.1844).
Acknowledgments
The authors thank the ANR LnOnL NT05-3_42676 for financial support and for grants to AD and MA. We are also grateful to ENS Lyon for AP's grant.