

1 Introduction
For over 150 years, our knowledge of actinide science has been dictated by examinations of the dioxo-uranium(VI) ion UO22+. Interest in this species has and continues to be driven by a need to better understand its role in nuclear fuel reprocessing schemes and its environmental speciation [1]. Beyond these applications, this ion has played an important conceptual role in actinide science to demonstrate the importance of covalent interactions and f orbitals in An = O bonds [2]. Of the numerous studies performed on this species, amide and halide complexes have provided invaluable insight into the structure, bonding, and spectroscopic properties of this unique ion [3]. These derivatives have also proven useful as precursors for the synthesis of other dioxo uranium(VI) compounds which have been utilized to broaden our general knowledge about this ion [3]. Recently, we reported the synthesis of the isoelectronic imido analogue of the UO22+ ion, and investigated its structure, bonding, and reactivity to further highlight the importance of f orbitals and covalent interactions [4]. While these isoelectronic ions possess some similarities in terms of their structure and orbital involvement in the U = L bond, we have noted several distinct differences in both their coordination chemistry and reactivity. For example, simple phosphine adducts of the U(NR)22+ ion are known, however UO22+ congeners have yet to be reported [4b,d]. In our continuing studies of the U(NR)22+ ion, we sought to synthesize amide and halide bis(imido) uranium(VI) derivatives and make rational comparisons with the UO22+ ion to further expand our knowledge about high oxidation state uranium chemistry.
In this article, we report the synthesis of cis- and trans-bis(sulfonamide) derivatives of the U(NR)22+ ion and their reactions with TMSX (TMS = SiMe3; X = Cl,Br) and ArEH (E = O, Ar = 2-tBuC6H4; E = S, Ar = C6H5) to yield dihalide and dichalcogenate uranium(VI) complexes, respectively. Furthermore, we report theoretical studies that investigate trends in covalent interactions of the U-X bond of dihalide complexes and compare these values to analogous UO22+ dihalide derivatives. Surprisingly, we have found that these interactions are in remarkable contrast to previous studies that examined covalent interactions in the U-E bond (E = chalcogenate) of analogous bis(imido) uranium(VI) dichalcogenate complexes.
2 Results and discussion
To isolate high oxidation state uranyl(VI)-amide complexes, the electron-deficient bis(trimethylsilyl)amido ligand [N(SiMe3)2]− has been extensively used to stabilize the high valent U(VI) center [3]. Several studies have shown that these amide derivatives possess interesting structural, bonding, and spectroscopic features in addition to being useful synthetic precursors for other uranium(VI) complexes. Given this finding, we believed that this class of amido ligand might also be suitable to stabilize bis(imido) uranium(VI) amide complexes. Surprisingly, we have found that reactions between two equivalents of MN(SiMe3)2 (M = Na, K) and U(NlBu)2(I)2(L)x (L = OPPh3, x = 2 (1); Me2bpy, x = 1 (2); Me2bpy = 4,4’-dimethyl-2,2’-bipyridyl) in THF did not provide tractable uranium(VI) bis(amide) complexes. In contrast, we found that the coordination of electron-deficient sulfonamide donors [N(Me)(SO2Ar’)]− (Ar’ = 4-Me-C6H4) proceeds in a straightforward manner to yield desired bis(imido) uranium(VI) disulfonamide complexes. Complexes with the general formula U(NtBu)2[N(Me)(SO2Ar’)]2(L)x (L = OPPh3 (3); Me2bpy (4)) can be synthesized in near quantitative yield by the reactions of two equivalents of KN(Me)(SO2Ar’) and U(NtBu)2(I)2(L)x in THF. The identity of these complexes was confirmed by NMR spectroscopy, elemental analysis, and in some cases single crystal X-ray diffraction studies. The lH NMR of 4 is representative of these two complexes and features broad resonances for sulfonamide and bipyridyl ligands at room temperature. For example, broad bpy resonances are observed at 7.34 (overlapping with one of the resonances for the sulfonamide ligand), 8.53, and 10.82 ppm. A second broad resonance for the sulfonamide ligand is observed at 8.21 ppm. Cooling a CDCl3 solution of 4 to −45 °C results in the resolution of these broad resonances to well-refined multiplets that would be consistent with a uranium species that possesses Cl symmetry in solution. This is evidenced by the observation of well-defined unsymmetrical bypyridyl resonances at 7.27, 7.41, 8.46, 8.60, 10.77, 10.87 ppm and sulfonamide resonances at 7.20, 7.48, 8.05, and 8.37 ppm. In addition, two distinct singlets are observed at −0.024 and 0.068 ppm which suggest inequivalent tert-butyl imido ligands. In the case of complex 3, similar broad resonances attributable to OPPh3 and sulfonamide ligands were noted in the lH NMR spectrum at room temperature that could be well-resolved at −45 °C.
The solid-state molecular structure of 4 is shown in Fig. 1 and features a U–O interaction between the 0-atom from the SO2 moiety on the sulfonamide ligand and the U-center. Similar coordination of the sulfonamide oxygen atoms to early transition metals has been observed in group 4 sulfonamide complexes [5a,b]. As a result of this interaction, the uranium center assumes a pseudo-pentagonal bipyramidal geometry with trans-oriented imido ligands occupying the axial coordination sites (N3-U1-N4 = 167.0(3)°). Interestingly, the U-O bond length of 2.494(5) Å is quite long relative to the U-O bond distances found in U(NtBu)2(I)2(OPPh3)2 (ave = 2.338 Å) [4b]. This suggests a weak interaction between U and O atoms which we believe accounts for the observations seen by NMR spectroscopy at different temperatures. At room temperature, this weak U-O interaction is fluxional in nature and may exchange with other O-atoms present in the sulfonamide donors. However, at lower temperatures, this process becomes slower and enables a Cl symmetric species to be observed in solution that is consistent with the solid state structure shown in Fig. 1. Because there is a four-coordinate chiral S-center present in complex 4, the tert-butyl imido resonances are inequivalent as is observed in the low temperature proton NMR spectrum. The U-N(sulfonamide) bond distances in 4 (ave = 2.481(8) Å) are significantly longer than the average U-N(amide) bond distances found in [Na(THF)2][UO2(N(SiMe3)2)3] (ave 2.310(4) Å). Similar differences in M-N(sulfonamide) and M-N(amide) bond distances are also observed in related titanium sulfonamide transition metal complexes [5]. One further important note is that the coordination of these sulfonamide donors does not appear to perturb U-N(imido) and U-N(bpy) bond distances significantly as these values observed in 4 are comparable to other structurally characterized bis(imido) uranium(VI) complexes [4]. Although the solid state structure of the analogous complex 3 has yet to be determined, we believe that a similar coordination environment is present at the uranium center given that similar fluxional processes are observed in both complex 3 and 4 as judged by NMR spectroscopy.

Solid-state molecule structure of U(NtBu)2(η1-N(Me)(SO2Ar’)(η3-N(Me)(SO2Ar’)(Me2bpy)-CH2Cl2 (4) with thermal ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and angles (deg): U1-N1 = 2.569(7), U1-N2 = 2.535(7), U1-N3 = 1.864(8), U1-N4 = 1.822(8), U1-N5 = 2.502(7), U1-N6 = 2.459(8), U1-O1 = 2.494(5), U3-U1-N4 = 167.0(3).
Uranyl(VI)-amide complexes like UO2[N(SiMe3)2]2(THF)2 have been shown to undergo protonolysis reactions with substituted phenols to provide uranyl bis(aryloxide) derivatives [3c]. We examined if similar reactivity would be observed with bis(imido) uranium(VI) sulfonamide complexes 3 and 4 by adding two equivalents of ArOH (Ar = 2-tBuC6H4) to U(NtBu)2[N(Me)(SO2Ar’)]2(L)x in THF. In reactions with the OPPh3-derivative 3, the diphenolate uranium complex U(NtBu)2(O-2-tBuC6H4)2(OPPh3)2 (5) was recovered in near quantitative yield as identified by NMR spectroscopy. The lH and 13C{lH} NMR spectra of 5 are in agreement with previously published work [4e]. In the case of protonolysis reactions between two equivalents of ArOH and U(NtBu)2[N(Me)(SO2Ar’)]2(Me2bpy) (4), the reaction does not yield the expected cis-diphenolate uranium(VI) product. Instead, we recover a mixed valent uranium(V)-uranium(VI) complex that has been identified by NMR spectroscopy and X-ray diffraction experiments. This mixed valent species was also isolated from metathesis reactions between two equivalents of KOAr and U(NtBu)2(I)2(Me2bpy) [4e]. A full description of these complexes will be published in due course. Protonolysis reactions were also performed with two equivalents of PhSH and U(NtBu)2[N(Me)(SO2Ar’)]2(L)x in THF to determine if this general reaction would extend to uranium(VI)-bis(thiolate) chemistry. In both of these reactions, bis(arylthiolate) complexes with the general formula U(NtBu)2(SPh)2(L)x (L = OPPh3 (6); Me2bpy (7)) were recovered in quantitative yield and possessed spectroscopic properties similar to previously reported bis(imido) uranium(VI) dithiolate compounds [4e].
We were also intrigued about the potential to utilize the bis(sulfonamide) complexes 3 and 4 as synthons for bis(imido) uranium(VI) dihalide complexes. The lighter halide derivatives of the U(NR)22+ ion are desirable synthetic targets in this area of chemistry for their conceptual importance and their potential to be starting materials for the preparation of other bis(imido) uranium(VI) complexes. We had anticipated that the addition of TMSX (X = Cl, Br) to U(NtBu)2[N(Me)(SO2Ar’)]2(L)x derivatives might generate the desired halide complexes with the general formula U(NtBu)2(X)2(L)x. Indeed, reactions between two equivalents of TMSX and U(NtBu)2[N(Me)(SO2Ar’)]2(L)x in CH2C12 proceed to give complexes with the general formula U(NtBu)2(X)2(L)x (X = Cl, L = OPPh3 (8), L = Me2bpy (10); X = Br, L = OPPh3 (9), L = Me2bpy (11)) in quantitative yield. In the course of these studies, we also found that complexes 8–11 could also be prepared by a more direct route by the addition of two equivalents of AgX (X = Cl, Br) to U(NtBu)2(I)2(L)x (1, 2) in CH2Cl2.
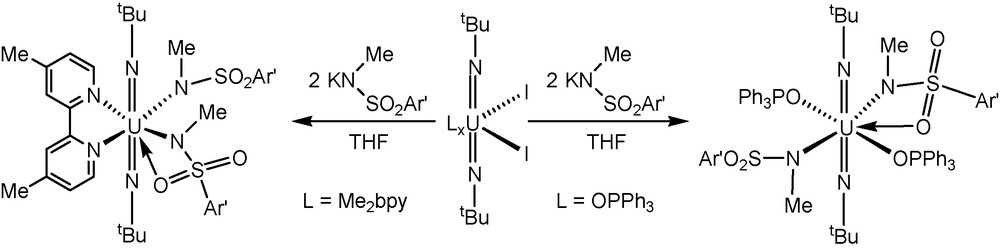
Synthesis of cis- and trans -bis(imido) uranium(VI) disulfonamides.
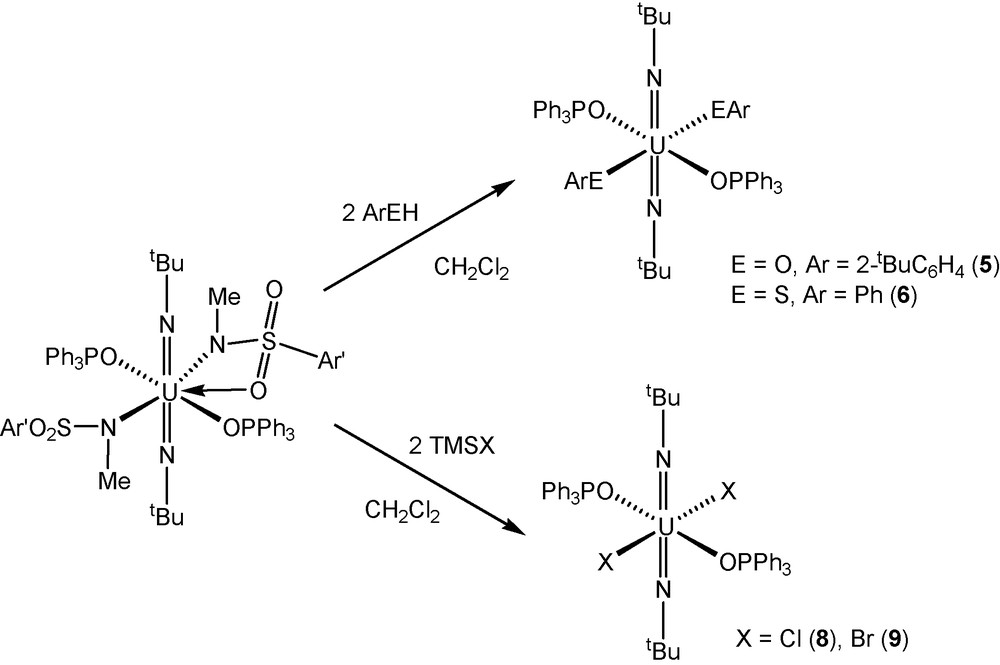
Synthesis of trans-bis(imido) uranium(VI) dihalides and dichalcogenates.
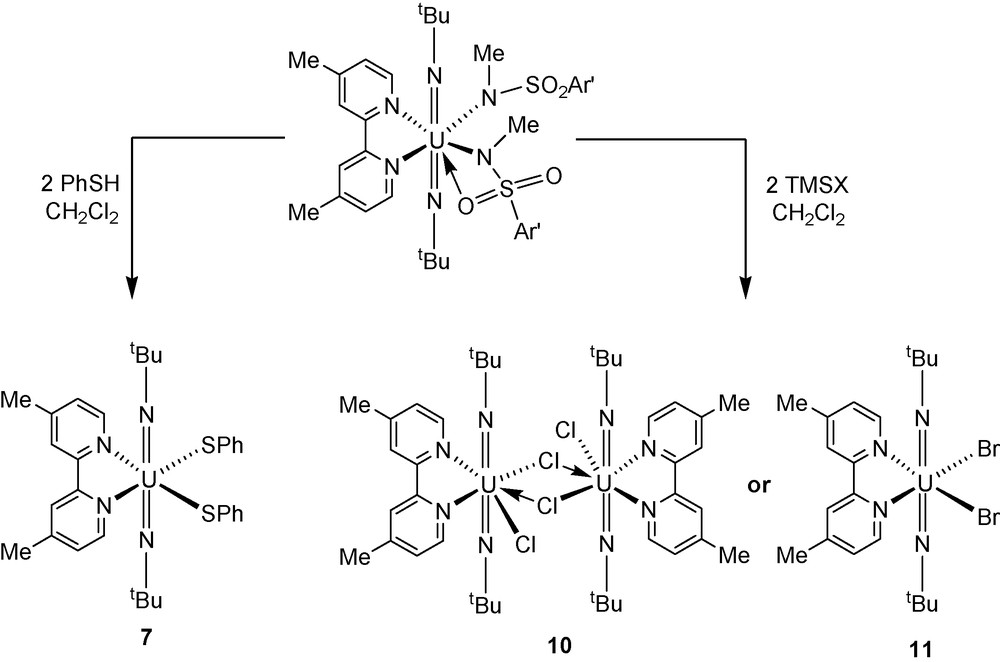
Synthesis of cis-bis(imido) uranium(VI) dihalides and dichalcogenates.
In the case of trans-oriented dihalide complexes U(NtBu)2(X)2(OPPh3)2 (Cl (8), Br(9)), X-ray quality crystals were grown from a layered CH2Cl2/hexanes solution; the solid state molecular structures of the dichloride and dibromide derivatives are shown in Figs. 2 and 3, respectively. Complexes 8 and 9 are isostructural and feature bis(imido), triphenylphosphine oxide and halide ligands arranged in an all trans-orientation. Furthermore, complexes 8 and 9 feature U-N(imido) bond lengths similar to the previously described bis(imido) uranium(VI) iodide complex U(NtBu)2(I)2(OPPh3)2 [4b]. In the case of U-X(halide) bond distances, the values found in dichloride derivative 8 (U-Cl = 2.7466(12) Å) and dibromide derivative 9 (U-Br = 2.8686(16) Å) are longer than those found in trans-UO2Cl2(OPPh3)2 (U-Cl = 2.645(5) Å) [6] and trans-UO2Br2(OAsPh3)2 (U-Brave = 2.839(2) Å) [7].
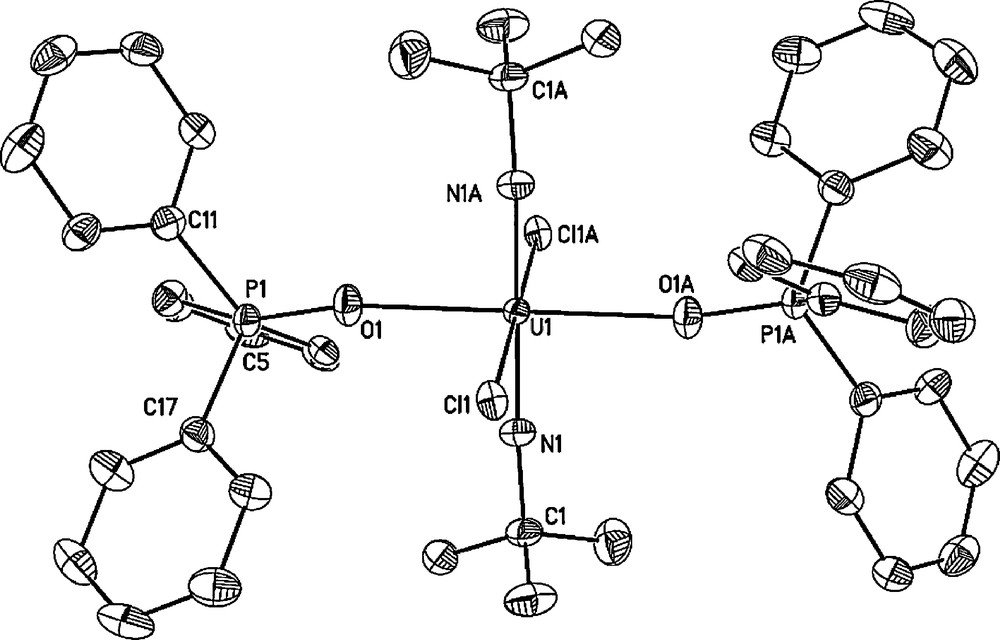
Solid-state molecule structure of trans-U(NtBu)2(Cl)2(OPPh3)2 (8) with thermal ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and angles (deg): U1-N1 = 1.848(4), U1-O1 = 2.344(3), U1-Cl1 = 2.7466(12), N1-U1-N1A = 180.00(16).
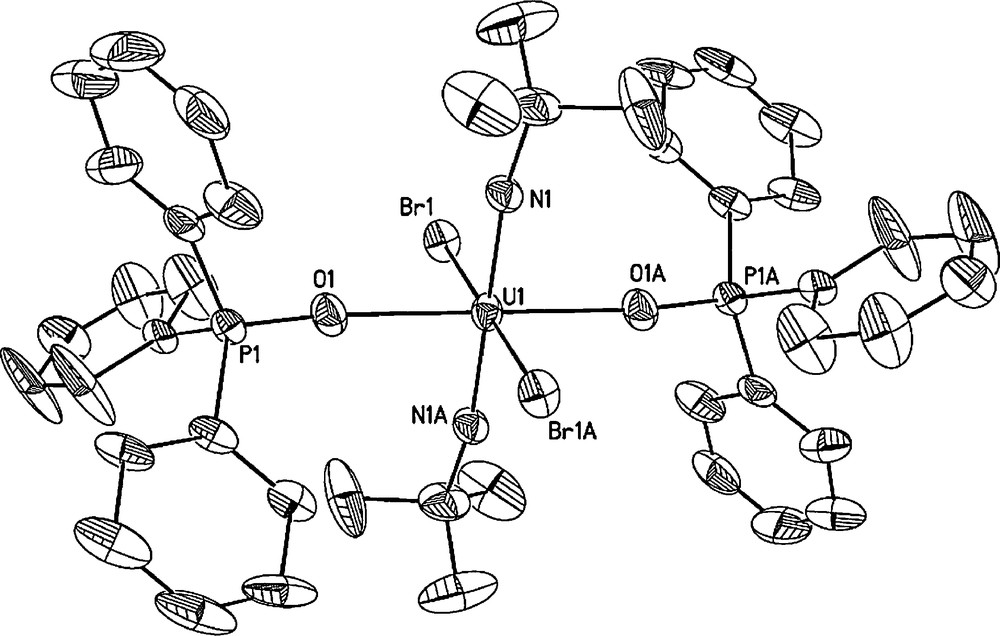
Solid-state molecule structure of trans-U(NtBu)2(Br)2(OPPh3)2 (9) with thermal ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and angles (deg): U1-N1 = 1.828(9), U1-O1 = 2.335(7), U1-Br1 = 2.8677(14), N1-U1-N1A = 180.0(3).
The behavior of complexes 8 and 9 in solution is quite different from what is observed in the solid state. The 1H and 31P{1H} NMR spectra of 8 are representative and feature two species in solution which we believe can be attributed to the cis and trans dihalide isomers. Similar solution cis-trans isomerization has been observed in the uranyl phosphine oxide and phosphinimine complexes UO2Cl2(EPCy3)2 (E = O, NH) [8]. To investigate this phenomenon, we examined the 1H and 31P{1H} NMR spectra in solvents that possessed different polarities. In solvents which possess a high dielectric constant, the cis-isomer is expected to be stabilized whereas the trans-isomer is expected to predominate in solvents which possess a lower dielectric constant. We chose CD2Cl2 (ɛ = 8.93 for CH2Cl2) and CDCl3 (ɛ = 4.81 for CHCl3) as the two solvents based on their different dielectric constants and found that the concentration of the cis-isomer increased with an increase in the concentration of CD2Cl2. For example, in CD2Cl2, the ratio of major/minor isomers of 8 as indicated by the 31P NMR spectrum and integration of tert-butyl imido resonance in the lH NMR spectrum was 0.74/0.26 whereas in CDCl3 the ratio was 0.86/0.14. These solution studies combined with results from single crystal diffraction experiments suggest that the dominant species in solution is the trans-isomer. Such a hypothesis is also consistent with the trans-diiodide complex U(NtBu)2(I)2(OPPh3)2 in which no cis-isomer is observed in solution or the solid state and with other trans-disposed actinyl dihalide complexes which have the general formula AnO2(X)2(L)2 (X = halide, L = Lewis base) [6,7,9].
In the case of cis-oriented dihalide complexes, we found that the nature of the solid state structure appears to be dependent upon the halide substituent. The dichloride derivative (10) appears as a dimeric species in the solid state with bridging chloride interactions between uranium centers. There are two independent molecules in the unit cell; the solid state molecular structure of one of them is shown in Fig. 3. Each uranium center appears to be in a distorted pentagonal bipyramidal geometry with trans-oriented imido ligands present in the axial positions (N1-U1-N2 = 172.3(3) Å). As expected, the terminal U-Cl bond distance (2.700(2) Å) is significantly shorter than bridging U-Cl bond lengths (2.8296(18) Å and 2.8399(19) Å) and similar to the U-Cl bond distance found in U(NtBu)2(Cl)(I)(tBu2bpy) (U-Cl = 2.7076(19) Å, tBu2bpy = 4,4’-di-tert-butyl-2,2’-bipyridyl) [4g]. The U-N(imido) and U-N(bpy) bond lengths are unremarkable and similar to previously described bis(imido) uranium(VI) complexes [4e].
In contrast to the dichloride species 10, the solid state structure of the dibromide derivative U(NtBu)2(Br)2(Me2bpy) (11) does not feature a dimeric interaction with bridging bromide ligands. Instead, a mononuclear species is observed which is presented in Fig. 4. Complex 11 appears to be isostructural with its diiodide analogue U(NtBu)2(I)2(Me2bpy) (2) [4e] and possesses a distorted octahedral geometry at the uranium center with trans-disposed bis(imido) ligands (N1-U1-N2 = 169.78(14)°). The U-N(imido) and U-N(bpy) bond lengths are unexceptional and similar to the diiodide analogue. Furthermore, the average U-Br bond distances of 2.8297(5) Å are similar to those in the trans-dibromide complex 9.

Solid-state molecule structure of [U(NtBu)2(Cl)(μ-Cl)(Me2bpy)]2-CH2Cl2 (10) with thermal ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and angles (deg): U1-N1 = 1.822(6), U1-N2 = 1.845(6), U1-N3 = 2.600(6), U1-N4 = 2.650(6), U1-Cl1 = 2.700(2), U1-Cl2 = 2.8296(18), U1-Cl2A = 2.8399(19), N1-U1-N2 = 172.3(3).
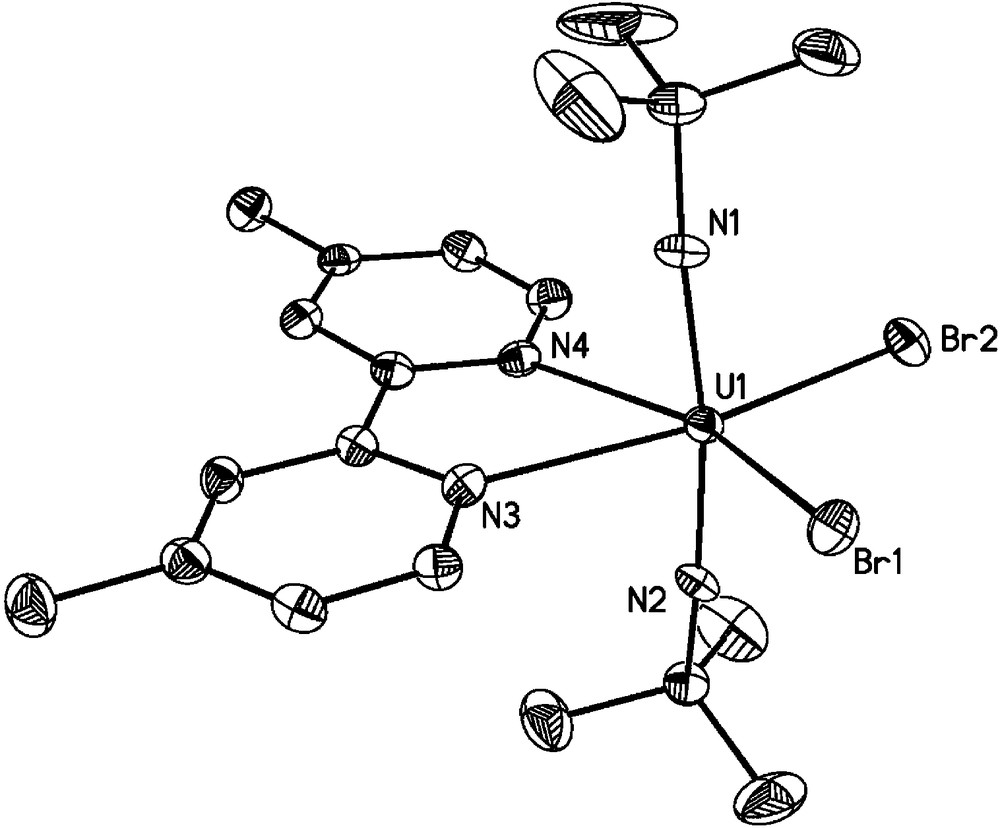
Solid-state molecule structure of U(NtBu)2(Br)2(Me2bpy) (11) with thermal ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and angles (deg): U1-N1 = 1.836(3), U1-N2 = 1.840(3), U1-N3 = 2.526(3), U1-N4 = 2.496(3), U1-Br1 = 2.8476(4), U1-Br2 = 2.8117(5), N1-U1-N2 = 169.78(14).
The dimeric dichloride species [U(NtBu)2(Cl)(μ-Cl)(Me2bpy)]2 (10) and cis-dibromide derivative U(NtBu)2(Br)2(Me2bpy) (11) also possess distinctive NMR spectroscopic differences. In the case of complex 10, broad 1H resonances are observed at room temperatures in the 1H NMR spectrum that can be resolved by cooling the CDCl3 solution to −30 °C. At low temperature, unsymmetrical bipyridyl aryl resonances are observed at 7.74, 7.82, 8.64, 8.60, 10.79 and 10.85 ppm that are consistent with a chloride bridged species in solution, similar to the solid state molecular structure. In contrast, the 1H NMR spectrum of the dibromide derivative 11 is not fluxional at room temperature and features a C2v symmetric species in solution with equivalent bipyridyl resonances at 7.76, 8.63, and 10.83 ppm and singlets at 2.71 and 0.04 ppm for the methyl bipyridyl and tert-butyl imido ligands, respectively.
Given the successful isolation of the lighter dihalide derivatives, we attempted to synthesize the trans- and cis-fluoride analogues with the general formula U(NtBu)2(F)2(L)x. Unfortunately, all attempts to synthesize these complexes have been unsuccessful. For example, the reactions between fluorinating reagents such as AgF, Et4NF, and Me3SnF and complexes 3 and 4 have failed to result in the formation of products with U-F bonds. While these results may be influenced by the hygroscopic nature of these fluorinating reagents, it is surprising that the preparation of these compounds is not as straightforward as chloride and bromide derivatives.
In our previous studies of the U(NR)22+ ion, we have effectively employed density functional theory (DFT) calculations to show remarkable differences in the U-N and U-O multiple bonds in U(NR)22+ and UO22+ ions, respectively [4a,4e]. Along these lines, we examined the overall charge on the uranium and halide centers as well as the U(=L)2 core to examine how covalent interactions in the U-X bond changes as the size of X becomes larger. To validate our calculations, we first compared the solid state structure of the dihalide derivatives of U(NR)22+ and UO22+ ions and found that the calculated geometries of the all trans U(NtBu)2(X)2(OPPh3)2 and UO2(X)2(OPPh3)2 series agree well with values described above and in the literature (within 0.1 Å, Supplemental Information for complete details). Although no molecular structures have been reported for UO2F2(OPPh3)2 and UO2Br2(OPPh3)2, geometrical parameters from similar compounds ([{UO2(μ-F)(O = PMePh2)3}2][BF4]2 [10] and UO2Br2(OAsPh3)2) [7] were used for comparison.
The charges extracted from the Natural Bond Orbital analysis (NBO) [11] on the uranium, halide atoms, and U(=L)2 core in both UO22+ and U(NR)22+ ions are shown in Table 1. In both ions, there is a steady decrease in the charge on the uranium center and a monotonic decrease in the negative charge of the halide atom as the size of X becomes larger. A steady decrease in positive charge of the UE2 unit is also observed upon descending the halide series. Such findings suggest that in both U(NR)22+ and UO22+ ions there is an increase in covalent interactions in the U-X bond as the size of X increases.
NBO charges for the Uranium center, the O=U=O/N=U=N unit, and the Halide atom in U(NtBu)2(X)2(OPPh3)2 and UO2(X)2(OPPh3)2 (X = F, Cl, Br, I).
| U(NtBu)2(X)2(OPPh3)2 | UO2(X)2(OPPh3)2 | |||||
| X | q(U) | -q(X) | q(UN2) | q(U) | -q(X) | q(UO2) |
| F | 1.702 | 0.641 | 0.421 | 1.853 | 0.537 | 0.699 |
| Cl | 1.473 | 0.632 | 0.208 | 1.619 | 0.496 | 0.528 |
| Br | 1.447 | 0.629 | 0.188 | 1.643 | 0.495 | 0.529 |
| I | 1.443 | 0.628 | 0.167 | 1.572 | 0.455 | 0.440 |
Calculations have also enabled a comparison in the covalency of the U-X bonds between U(NR)22+ and UO22+ ions. We have previously shown that the metal center and the U-N multiple bond in the U(NR)22+ ion possesses increased covalency compared to the UO22+ ion [4a]. It appears from the NBO charges described in Table 1 that similar results are observed at the uranium center in this study given that the magnitude of charges on both the uranium center and the UE2 core are systematically lower in the U(NR)22+ ions compared to UO22+ counterparts. Interestingly, the magnitude of the negative charge on the halide donor appears to be systematically lower in the UO22+ ion. Thus, it appears that the U-X bond in UO22+ complexes is more covalent in character than analogous bonds in U(NR)22+ ions. Such findings are in contrast with results from the study of the U-E (E = chalcogenate) bonds in the dichalcogenate series of U(NR)22+- and UO22+ ions where the U-E bond possessed a more covalent nature in the U(NR)22+ derivatives [4e]. At this time, the reason for this discrepancy in covalent interactions between U-X and U-E bonds is not clear but may simply arise from differences in the spatial and energetic overlap in the orbitals found in uranium and halide/chalcogenate atoms. Current efforts are focused on the use of X-ray absorption spectroscopy to further understand the differences in covalency in ligand interactions with U(NR)22+ and UO2 ions.
3 Conclusion
In summary, we have shown that bis(imido) uranium(VI) disulfonamide complexes can be synthesized and utilized as precursors for the generation of other cis- and trans-disubstituted uranium(VI) complexes. In the case of the dihalide series, we have further investigated covalent interactions in the U-X bond and made important comparisons to analogous UO22+ complexes. DFT calculations show that covalent interactions in the U-X bond of both UO22+ and U(NR)22+ ions increase as the size of the halide donor increases. Comparison of the trend in the NBO charges on the halide atoms in complexes 8, 9 and 1 with the analogous trend in the chalcogen atoms in the U(VI) chalcogenate complexes [4e] shows that much less negative charge is found on the heavier chalcogens than on the heavier halides, suggesting that the U-chalcogen interaction is more covalent in nature than the U-halogen interactions in this class of compounds. On-going efforts with theoretical and spectroscopic examinations are being performed to provide more insight into these interactions and understand the differences between the dihalide and dichalcogenate series.
4 Experimental
All reactions and subsequent manipulations were performed under anaerobic and anhydrous conditions either under high vacuum or an atmosphere of argon. Hexanes and THF were dried by passage over activated alumina and CH2Cl2 was purchased anhydrous and stored over activated 4Å molecular sieves for 24 h before use. CD2Cl2 was dried over activated 4Å molecular sieves for 24 h before use. U(NtBu)2(I)2(OPPh3)2 (1) and U(NtBu)2(I)2(Me2bpy) (2) were synthesized by published procedures [4b,e]. The potassium amide reagent K[N(Me)(SO2Ar’)] (Ar’ = 4-Me-C6H4) was prepared by a modification of the literature procedure [12] by adding 1.1 equivalent of HN(Me)(SO2Ar’) to a THF suspension of KH. The voluminous white solid that precipitated was isolated, washed several times with THF, and dried in vacuo. All other reagents were purchased from commercial suppliers and used as received. NMR spectra were recorded on a Bruker AVA300 spectrometer at room temperature unless otherwise stated. 1H, 13C{1H}, and 31P{1H} NMR spectra are referenced to external SiMe4 using the residual protio solvent peaks as internal standards (1H NMR experiments), the characteristic resonances of the solvent nuclei (13C NMR experiments), or using H3PO4 as an external standard (31P NMR experiments). Elemental analyses were performed at the UC Berkeley Microanalytical Facility, on a Perkin-Elmer Series II 2400 CHNS analyzer.
Computational Details. The B3LYP hybrid density functional was employed to optimize the equilibrium molecular structures of all the complexes studied [13]. The small-core Stuttgart RSC 1997 relativistic effective core potential (RECP) [14] was used to model the uranium center, with the associate basis set [6s/6p/5d/3f]. For the halide atoms we used the 6-311g** basis set that includes diffused and polarization functions. The remainder of the ligand atoms (H, C, P) was described with 6-31g*. The geometries of all the molecules were optimized without symmetry constraints. All the calculations reported in this paper were carried out with the Gaussian 03 code [15] and the charge analysis was carried out with the NBO 5.G package [11].
X-Ray Crystallographic Details. The crystal structures of compounds 4-CH2Cl2, 8, 9, 10-CH2Cl2, and 11 were determined as follows: The crystal was mounted in a nylon cryoloop from Paratone-N oil under an argon gas flow. The data were collected on a Bruker SMART APEX II charge-coupled-device diffractometer, with a KRYO-FLEX liquid nitrogen vapor cooling device. The instrument was equipped with a graphite monochromatized Mo Kα X-ray source (λ = 0.71073 Å), with MonoCap X-ray source optics. A hemisphere of data was collected using co scans, with 5s frame exposures and 0.3° frame widths. Data collection and initial indexing and cell refinement were handled using APEX II software [16]. Frame integration, including Lorentz-polarization corrections, and final cell parameter calculations were carried out using the SAINT+ software [17]. The data were corrected for absorption using the SADABS program [18]. Decay of reflection intensity was monitored via analysis of redundant frames. The structure was solved using direct methods and difference Fourier techniques. All hydrogen atom positions were idealized and rode on the atom they were attached to. The final refinement included anisotropic temperature factors on all non-hydrogen atoms. Structure solution, refinement, graphics, and creation of publication materials were performed using SHELXTL [19]. CCDC 756008–756012 contain the supplementary crystallographic data for this paper.
4.1 Typical procedure for the preparation of bis(imido) uranium(VI) bis(sulfonamides)
To a THF (5 mL) suspension of U(NtBu)2(I)2(OPPh3)2 (200 mg, 0.17 mmol) in a 20 mL scintillation vial was added a THF (2 mL) suspension of K[N(Me)(SO2Ar’)] (75 mg, 0.34 mmol). After stirring overnight, the orange suspension was filtered through Celite and the solvent reduced in vacuo until several milliliters remained. Hexanes were added to precipitate a bright orange powder which was recrystallized from CH2Cl2/hexanes.
4.1.1 U(NtBu)2[η1−(N(Me)(SO2Ar’)][(η3−(N(Me)(SO2Ar’)](OPPh3)2 (3)
Yield = 72%. 1H NMR (CD2Cl2, −45 °C): δ −0.012 (s, 9H, −C(CH3)3), 0.028 (s, 9H, −C(CH3)3), 1.95 (s, 3H, −NCH3), 2.14 (s, 3H, −ArCH3), 2.19 (s, 3H, −ArCH3), 2.26 (s, 3H, −NCH3), 7.16 (d, 3J{H,H}=7 Hz, 2H, −ArH), 7.36 (d, 3J{H,H}=7 Hz, 2H, −ArH), 7.62 (m, 18H, −ArHOPPh3), 8.00 (d, 3J{H,H}=7 Hz, 2H, −ArH), 8.39 (m, 18H, −ArHOPPh3), 8.48 (d, 3J{H,H}=7 Hz, 2H, −ArH). 13C{1H} NMR (CD2Cl2, −45 °C): 29.2 (−ArCH3), 29.5 (−ArCH3), 30.6 (−NCH3), 31.2 (−NCH3), 31.4 (−C(CH3)3), 34.2 (−C(CH3)3), 70.6 (−C(CH3)3), 70.9 (−C(CH3)3), 122.3 (−ArC), 122.8 (−ArC), 124.3 (−ArC), 128.1 (−ArC), 128.6 (−ArC), 129.0 (−ArC), 130.2 (−ArC), 130.5 (−ArC), 132.9 (−ArC), 139.3 (1J{C,P}=14Hz, −ArC), 145.0 (−ArC), 145.3 (−ArC). 31P{1H} NMR (CD2Cl2, −45 °C): 49.2 (s). Anal. Calcd for C60H68N4O6P2S2U: %C, 55.21; %H, 5.25; %N, 4.29. Found: %C, 55.29; %H, 5.33; %N, 4.32.
4.1.2 U(NtBu)2[η1−(N(Me)(SO2Ar’)][(η3−(N(Me)(SO2Ar’)](Me2bpy) (4)
Yield = 79%. lH NMR (CD2Cl2, −45 °C): 5 −0.024 (s, 9H, −C(CH3)3), 0.068 (s, 9H, −C(CH3)3), 2.05 (s, 3H, −NCH3), 2.10 (s, 3H, −ArCH3), 2.16 (s, 3H, −ArCH3), 2.23 (s, 3H, −NCH3), 2.71 (s, 3H, −CH3), 2.76 (s, 3H, −CH3), 7.20 (d, 3J{H,H} = 7 Hz, 2H, −ArH), 7.27 (d, 3J{H,H} = 5 Hz, 1H, −bpyH), 7.41 (d, 3J{H,H} = 5 Hz, 1H, −bpyH), 7.48 (d, 3J{H,H} = 7 Hz, 2H, −ArH), 8.05 (d, 3J{H,H} = 7 Hz, 2H, −ArH), 8.37 (d, 3J{H,H} = 7 Hz, 2H, −ArH), 8.46 (s, 1H, −bpyH), 8.60 (s, 1H, −bpyH), 10.77 (d, 3J{H,H} = 5 Hz, 1H, −bpyH), 10.87 (d, 3J{H,H} = 5 Hz, 1H, −bpyH). 13C{1H} NMR (CD2Cl2, −45 °C): 28.5 (−ArCH3), 29.2 (−ArCH3), 30.1 (−NCH3), 30.7 (−NCH3), 35.0 (−C(CH3)3), 35.2 (−C(CH3)3), 35.6 (−CH3), 35.9 (−CH3), 69.8 (−C(CH3)3), 70.3 (−C(CH3)3), 120.0 (−ArC), 120.7 (−ArC), 121.8 (−ArC), 122.1 (−ArC), 127.1 (−ArC), 127.3 (−ArC), 127.5 (−ArC), 127.9 (−ArC), 131.1 (−ArC), 131.4 (−ArC), 143.8 (−ArC), 144.6 (−ArC), 145.6 (−ArC), 146.8 (−ArC), 154.4 (−ArC), 154.9 (−ArC), 165.0 (−ArC), 166.1 (−ArC). One molecule of CH2C12 was present in the solid state lattice. Under vacuum, the crystalline material obtained readily loses this solvent. Anal. Calcd for C36H40N6O4S2U: %C, 46.85; %H, 4.37; %N, 9.11. Found: %C, 46.90; %H, 4.41; %N, 9.16.
4.2 Typical procedure for the preparation of bis(imido) uranium(VI) dichalcogenates
To a THF (3 mL) solution of complex 3 (200 mg, 0.15 mmol) in a 20-mL scintillation vial was added a THF (2 mL) solution of 2-tert-butylphenol (46 mg, 0.30 mmol). The solution darkened immediately from a bright orange to a dark red color during the addition of the phenol reagent. After stirring overnight, the solvent was reduced in vacuo until several milliliters remained. Hexanes were added to precipitate a dark red powder which was recrystallized from CH2Cl2/hexanes.
4.2.1 U(NtBu)2(O-2-tBuC6H4)2(OPPh3)2 (5)
Yield = 94%. The 1H and 13C NMR spectra are identical to previous reported complexes [4e].
4.2.2 U(NtBu)2(SPh)2(OPPh3)2 (6)
Yield = 91%. The 1H and 13C NMR spectra are identical to previous reported complexes [4e].
4.2.3 U(NtBu)2(SPh)2(Me2bpy) (7)
Yield = 97%. The 1H and 13C NMR spectra are identical to previous reported complexes [4e].
4.3 Typical procedure for the preparation of bis(imido) uranium (VI) dihalides
To a THF (3 mL) solution of complex 3 (200 mg, 0.15 mmol) in a 20-mL scintillation vial was added a THF (2 mL) solution of TMSCl (33.3 mg, 0.30 mmol). No colour change was moted as a result of the addition of this reagent. After stirring overnight, the orange suspension was filtered through Celite and the solvent reduced in vacuo until several milliliters remained. Hexanes were added to precipitate a bright orange powder which was recrystallized from CH2Cl2/hexanes.
4.3.1 U(NtBu)2(Cl)2(OPPh3)2 (8)
Yield = 91%. 1H NMR (CD2Cl2): trans-isomer δ −0.07 (s, 18H, −C(CH3)3), 7.62 (m, 18H, −ArH), 8.39 (m, 18H, −ArH). cis-isomer δ −0.10 (s, 18H, −C(CH3)3) and aryl resonances overlapping with trans-isomer. 13C{1H} NMR (CD2Cl2): trans-isomer δ 35.0 (−C(CH3)3), 71.2 (−C(CH3)3), 125.2 (−ArC), 127.6 (−ArC), 131.6 (−ArC, 138.9 (d, 1J{C,P} = 14 Hz, −ArC). 31P{1H} NMR (CD2Cl2, −45 °C): 44.9 (s, major), 44.6 (s, minor). Anal. Calcd for C44H48Cl2N2O2P2U: %C, 52.44; %H, 4.80; %N, 2.78. Found: %C, 52.42; %H, 4.85; %N, 2.84.
4.3.2 U(NtBu)2(Br)2(OPPh3)2 (9)
Yield = 87%. 1H NMR (CD2Cl2): trans-isomer δ −0.11 (s, 18H, −C(CH3)3), 7.64 (m, 18H, −ArH), 8.40 (m, 18H, −ArH). cis-isomer δ −0.16 (s, 18H, −C(CH3)3) and aryl resonances overlapping with trans-isomer. 13C{1H} NMR (CD2Cl2): trans-isomer δ 35.2 (−C(CH3)3), 71.8 (−C(CH3)3), 124.6 (−ArC), 128.8 (−ArC), 132.8 (−ArC), 138.2 (d, 1J{C,P} = 14 Hz, −ArC). 31P{1H} NMR (CD2Cl2, −45 °C): 45.1 (s, major), 44.8 (s, minor). Anal. Calcd for C44H48Br2N2O2P2U: %C, 48.19; %H, 4.41; %N, 2.56. Found: %C, 48.27; %H, 4.48; %N, 2.62.
4.3.3 [U(NtBu)2(Cl)(μ-Cl)(Me2bpy)]2 (10)
Yield = 90%. 1H NMR (CD2Cl2, −45 °C): δ 0.09 (s, 18H, −C(CH3)3), 2.72 (s, 3H, −CH3), 2.74 (s, 3H, −CH3), 7.74 (d, 3J{H,H}=5Hz, 1H, −bpyH), 7.82 (d, 3J{H,H}=5Hz, 1H, −bpyH), 8.64 (s, 1H, −bpyH), 8.60 (s, 1H, −bpyH), 10.79 (d, 3J{H,H}=5Hz, 1H, −bpyH), 10.85 (d, 3J{H,H}=5Hz, 1H, −bpyH). 13C{1H} NMR (CD2Cl2, −45 °C): δ 29.0 (−C(CH3)3), 35.6 (−CH3), 36.2 (−CH3), 71.8 (−C(CH3)3), 120.3 (−bpyC), 121.0 (−bpyC),126.8 (−bpyC), 127.2 (−bpyC), 147.5 (−bpyC), 148.3 (−bpyC), 153.6 (−bpyC), 154.8 (−bpyC), 164.7 (−bpyC), 165.1 (−bpyC). One molecule of CH2Cl2 was present in the solidstate lattice. Under vacuum, the crystalline material obtained readily loses this solvent.Anal. Calcd. For C40H60N8Cl4U2: %C, 37.80; %H, 4.76; %N, 8.82. Found: %C, 37.84; %H, 4.82; %N, 8.89.
4.3.4 U(NtBu)2(Br)2(Me2bpy) (11)
Yield = 84%. 1H NMR (CD2Cl2): δ 0.04 (s, 18H, −C(CH3)3), 2.71 (s, 6H, −CH3), 7.76 (d, 3J{H,H} = 5 Hz, 2H, −bpyH), 8.63 (s, 2H, −bpyH), 10.83 (d, 3J{H,H} = 5Hz, 2H, −bpyH).13C{1H} NMR (CD2Cl2): δ 28.5 (−C(CH3)3), 37.1 (−CH3), 70.5 (−C(CH3)3), 121.6 (−bpyC), 127.0 (−bpyC), 148.0 (−bpyC), 154.8 (−bpyC), 165.2 (−bpyC). Anal. Calcd. For C20H30N4Br2U: %C, 33.16; %H, 4.18; %N, 7.74. Found: %C, 33.22; %H, 4.23; %N, 7.82.
Acknowledgements
P.Y. and L.P.S. thank the Seaborg Institute for their postdoctoral fellowships. E.R.B. was partially supported by the Division of Chemical Sciences, Office of Basic Energy Sciences, US Department of Energy under the Heavy Element Chemistry program at LANL. We thank the Center for Integrated Nanotechnology at LANL for computing support. LANL is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the US DOE under contract DEAC52-06NA25396. CCDC 756008–756012 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/datarequest.cif.