

1 Introduction
In an effort to expand the range of rare earth carbene complexes which utilise a carbene that is not stable in the absence of a metal [1], we have recently reported a range of yttrium derivatives of the bis(iminophosphorano)methane H2C(PPh2NSiMe3)2 (H2BIPM) [2], exemplified by I-IV, Fig. 1, [3] and have reported preliminary studies of their reactivity towards unsaturated substrates [3c] and in salt elimination reactions to generate the first unsupported yttrium-gallium bond [3d].

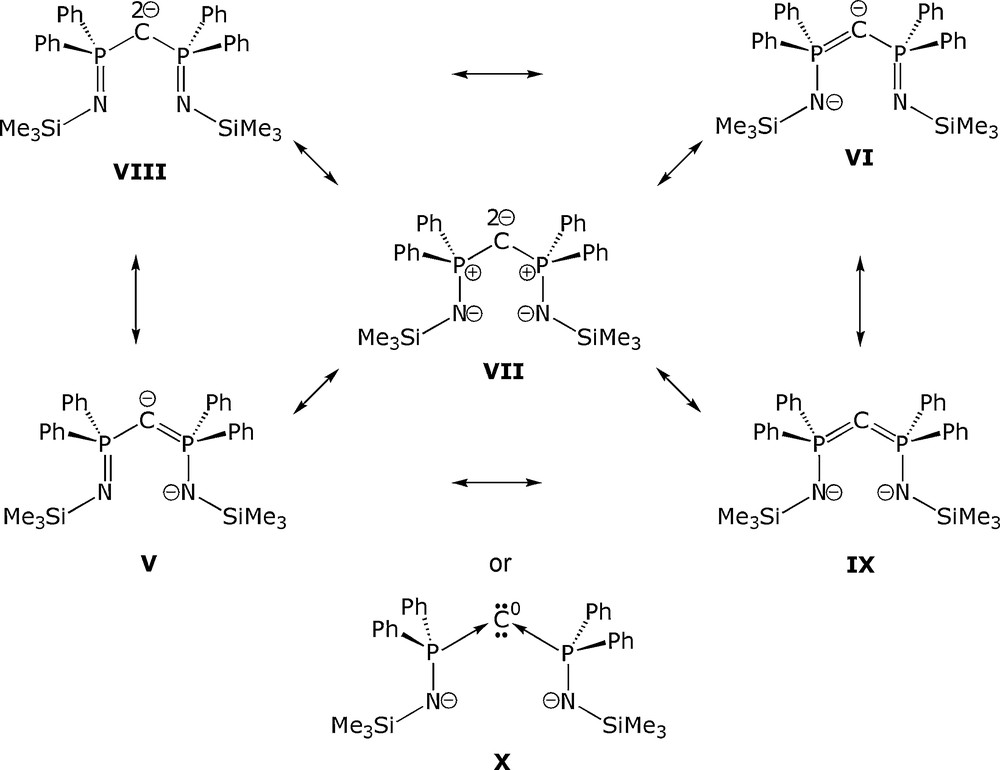
However, the electronic structure of BIPM2− is complicated by the fact that several resonance forms are valid [2–4] as exemplified by Fig. 2. Thus, BIPM2− can be considered to adopt resonance structures analogous to: N-heterocyclic carbenes (V and VI); a dipolar form with a geminal carbon dianion and two anionic amides (VII); a geminal carbon dianion and two imino groups (VIII); and a carbodiphosphorane with two anionic amides (IX). The later resonance form is particularly interesting because it is predicated on the assignment of a P(V) oxidation state, but an alternative and credible [5] assignment is that of two amido-functionalised P(III) centres datively coordinated to a zero-valent carbon centre (X), Fig. 2. Whilst DFT calculations have delivered insight into the electronic structure of the BIPM2− dianion and suggest the N−-P+-C2−-P+-N− dipolar resonance form is prevalent [3,4], the possibility of a captodative carbodiphosphorane assignment [5a] cannot be dismissed [3b].
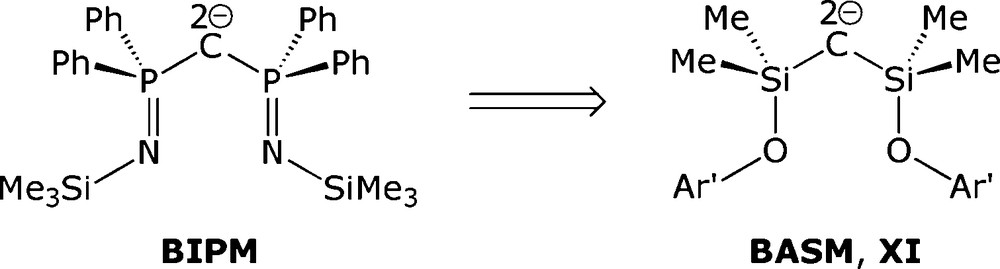
Consequently, we turned our attention towards an analogous silicon system because this would remove the P(III)/P(V) assignment problem since an unambiguous oxidation state of IV would be clear-cut for silicon. In order to retain a valence isoelectronic ligand framework which would still be dianionic following double deprotonation, RO rather than RN groups would be required. We therefore targeted a bis(aryloxysilyl)methane (H2BASM) exemplified by XI, Fig. 3. Herein, we report the synthesis of the sterically demanding bis(aryloxysilyl)methane (Ar′OSiMe2)2CH2 (2, Ar′ = 2,6-diisopropylphenyl) and its reactions with alkali metal and yttrium alkyls which reveal facile Si-O bond cleavage and Si-C bond formation reactions.
2 Results and discussion
2.1 Synthesis of 2
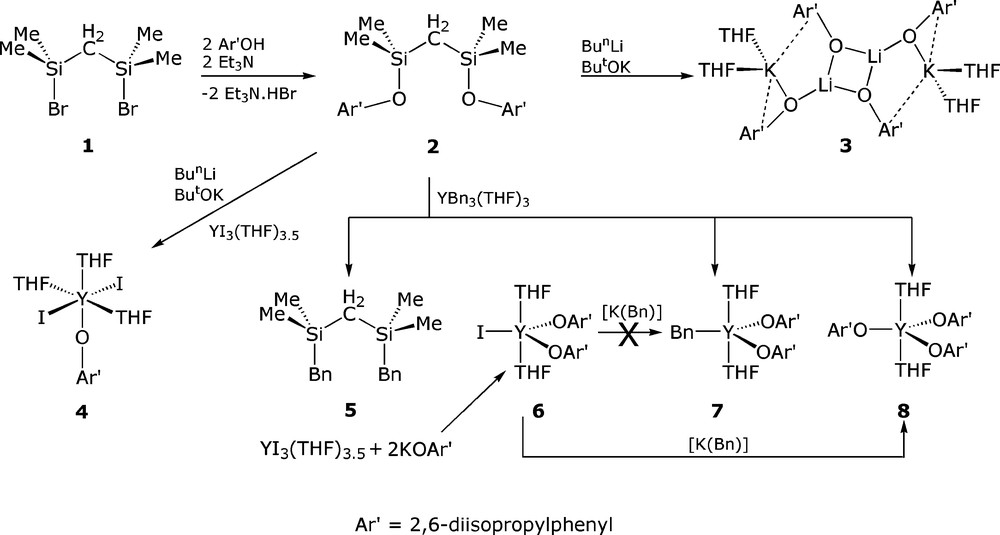
We selected (BrSiMe2)2CH2 (1) as a start point for the synthesis of ligands of type XI as its synthesis was reported previously by Barrau and Satge [6]. Accordingly, treatment of ClSiMe2CH2Cl with PhMgBr afforded PhSiMe2CH2Cl, and conversion to the corresponding Grignard and reaction with Me2SiHCl gave PhSiMe2CH2SiHMe2, which delivered 1 in good yield following treatment with bromine and vacuum distillation. We noted that the use of 1.1 equivalents of PhMgBr was necessary to ensure excellent (∼85 %) yields in the first step. Subsequent reaction of 1 with two equivalents of Ar′OH (Ar′ = 2,6-diisopropylphenyl) in the presence of the auxiliary base triethylamine afforded (Ar′OSiMe2)2CH2 (2) as a colourless oil in 85 % yield following separation from the Et3N.HBr by-product and work-up, Fig.4. The NMR spectra of 2 is straightforward as would be expected, and the CH2 resonates as a singlet at 0.23 ppm in the 1H NMR spectrum and a single resonance in the 29Si NMR spectrum is observed at 15.21 ppm.
2.2 Solid state structure of 2
Colourless single crystals of 2 were grown from hexane at −80 °C; the molecular structure is illustrated in Fig. 5 and selected bond lengths are compiled in Table 1. The structure is unremarkable; the Si-O and O-Caryl bond distances average 1.6676(12) and 1.3837(18) Å, respectively, which, for example, compares favourably to observed Si-O and O-Caryl bond lengths of 1.677 and 1.366 Å, respectively, in (But)3SiCH(SiMe3)SiMe2OC6F4-4-Me [7].
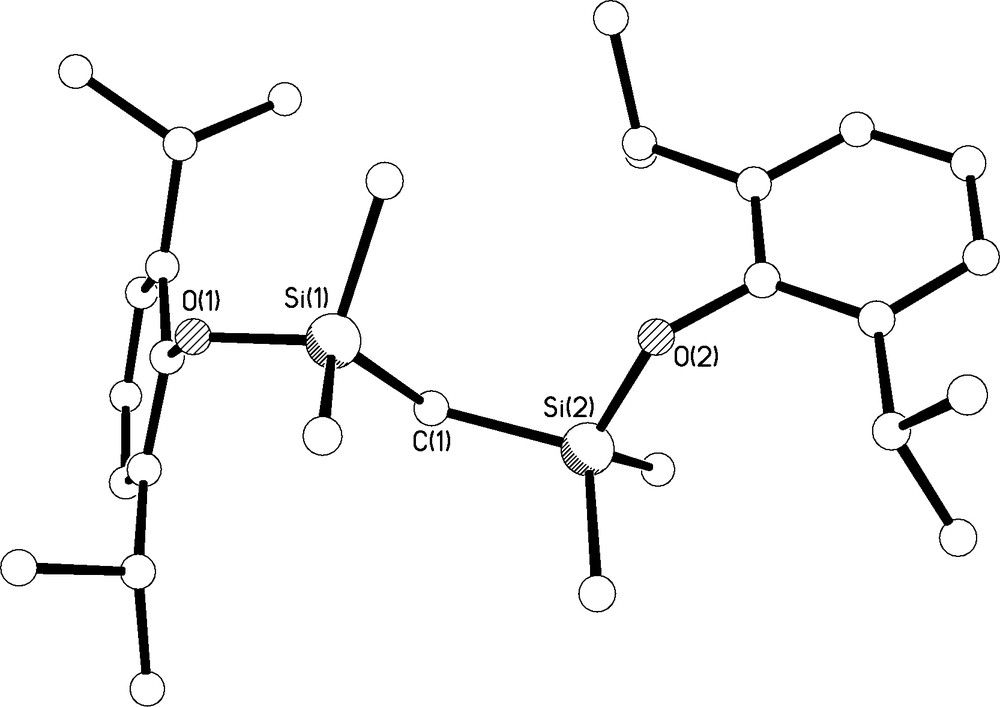
Molecular structure of 2 with selective labelling. Hydrogen atoms omitted for clarity.
Selected bond lengths for 2, 3.C4H8O, 4, 7.C6H5CH3, and 8.
| 2 | |||
| C(1)-Si(1) | 1.8655(16) | C(1)-Si(2) | 1.8559(16) |
| Si(1)-O(1) | 1.6717(12) | Si(2)-O(2) | 1.6635(12) |
| O(1)-C(4) | 1.3825(19) | O(2)-C(18) | 1.3849(18) |
| 3.C4H8O | |||
| Li(1)-O(1) | 1.888(11) | Li(1)-O(1A) | 1.840(10) |
| Li(1)-O(2) | 1.803(11) | K(1)-O(2) | 2.642(5) |
| K(1)-O(3) | 2.707(5) | K(1)-O(4) | 2.664(5) |
| K(1)⋯C(1) | 3.159(6) | K(1)⋯C(2) | 3.185(7) |
| K(1)⋯C(3) | 3.258(8) | K(1)⋯C(4) | 3.310(10) |
| K(1)⋯C(5) | 3.329(10) | K(1)⋯C(6) | 3.254(7) |
| K(1)⋯C(13) | 3.323(6) | ||
| 4 | |||
| Y(1)-O(1) | 2.046(3) | Y(1)-I(1) | 2.9868(4) |
| Y(1)-I(1A) | 2.9868(4) | Y(1)-O(2) | 2.393(4) |
| Y(1)-O(3) | 2.319(3) | Y(1)-O(3A) | 2.319(3) |
| 7.C6H5CH3 | |||
| Y(1)-C(1) | 2.416(5) | Y(1)⋯C(2) | 2.935(4) |
| Y(1)-O(1) | 2.099(2) | Y(1)-O(2) | 2.099(2) |
| Y(1)-O(3) | 2.323(3) | Y(1)-O(4) | 2.332(3) |
| 8 | |||
| Y(1)-O(1) | 2.086(2) | Y(1)-O(2) | 2.069(3) |
| Y(1)-O(3) | 2.085(2) | Y(1)-O(4) | 2.351(3) |
| Y(1)-O(5) | 2.344(3) |
2.3 Synthesis of 3
Encouraged by the fact that Lappert disclosed that (Me3Si)2CH2 could be deprotonated by [Li(Bun)] in the presence of [K(OBut)] to give the corresponding potassium alkyl [8], we initially sought to access alkali metal derivatives of 2 to employ in subsequent salt elimination chemistry, since mono-functionalised bis(organosilyl)methanides have been successfully prepared and utilised in tris-alkyl rare earth chemistry [9]. However, treatment of 2 with [Li(Bun)] in the presence of [K(OBut)] does not afford the corresponding potassium alkyl. Rather, the superbasic mixture is apparently too aggressive and instead promotes Si-O bond cleavage. Although the exact fate of 2 is not clear it seems likely that the silicon centres in 2 are functionalised with a mixture of butyl and tert-butoxy groups as colourless crystals of the complex shown to be [{Li(OAr′)}2{K(OAr′)}2(THF)4] (3) were isolated from THF as the sole isolable product in 50 % yield, Fig. 4. Although this fragmentation is unfortunate, ligand fragmentation was not entirely unexpected as Lappert showed that although alkali metal complexes of {CH(SiMe3)(SiMe2OMe)}− could be obtained in crystalline form, the corresponding bis-methoxy CH2(SiMe2OMe)2 compound fragmented on lithiation to give the expected complex [Li{CH(SiMe2OMe)2}]∞ as the minor product (4 %) and the tris-methoxy compound [Li{C(SiMe2OMe)3}]2 as the major product (64 %) [10]. Germane to these observations are that the fragmentation of methoxy-functionalised tris(triorganosilyl)methanide complexes by electropositive metals has been observed previously [11] and the cleavage of Si-O linkages in vacuum joint grease has ample precedent [12]. The room temperature 1H and 13C NMR spectra of 3 in THF exhibits only one Ar′ environment and a variable temperature study between +25 °C and −80 °C showed little variation and no de-coalescence which indicates facile and fast exchange of the aryloxide groups in solution compared to the NMR time-scale.
Synthesis of 1–8. Complexes 6 and 8 have been prepared by rational routes.
2.4 Solid state structure of 3
The molecular structure of 3 is shown in Fig. 6 and selected bond lengths are listed in Table 1. Complex 3 crystallises as a tetrametallic, di-lithium di-potassium aggregate. Although the crystallographic data were poor the identity and connectivity of 3 is clear-cut. The complex is constructed around a centrosymmetric Li2O2 core which resides over an inversion centre and each three-coordinate lithium centre bridges to another aryloxide which in turn bridges to a potassium centre. The coordination sphere of each potassium centre is completed by the oxygen atoms of two molecules of THF, an η6-interaction with the phenyl ring of the aryloxide associated with the Li2O2 core and an η1-interaction with the second Ar′ ligand. The construction of 3 is entirely in line with the hard nature of lithium compared to potassium; the lithium centres, although only three-coordinate, maximise their contacts to the hard aryloxide ligands whereas the softer potassium centres engage mainly with the soft π-systems of the aryloxide ligands. The Li-O, K-Oaryl, and K-OTHF bond lengths are unremarkable [13]. The η6-K-Caryl bond distances span the range 3.169(5)-3.303(7) Å (av. 3.238 Å) and compare favourably to the 3.1–3.9 Å range of reported η6-K-Caryl bond distances in the literature [14]. However, the η1-K-Caryl distance of 3.359(5) Å is rather longer than the η6-K-Caryl range.
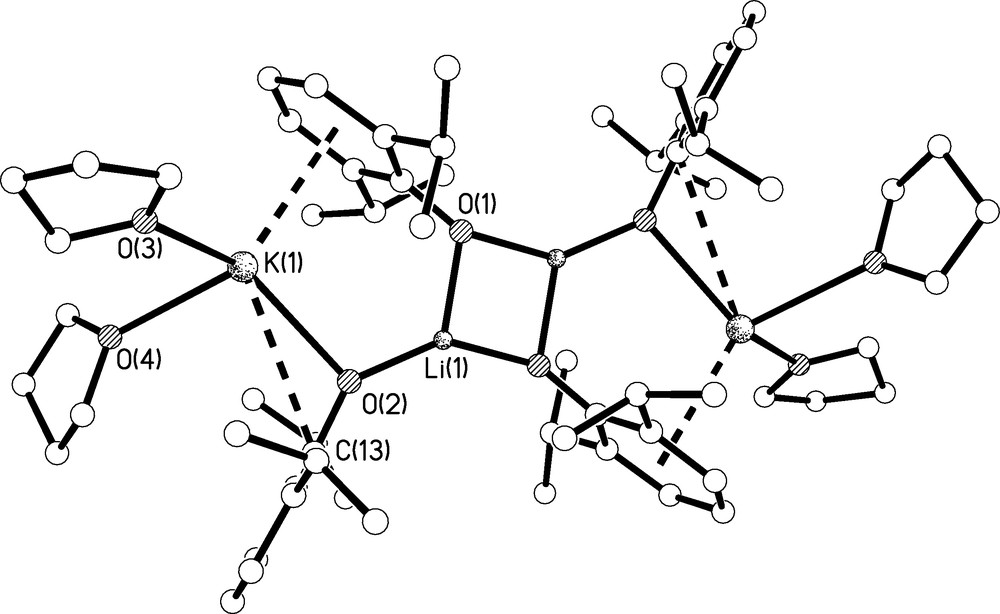
Molecular structure of 3.C4H8O with selective labelling of symmetry unique atoms. Hydrogen atoms omitted for clarity. Multi-hapto interactions are shown as dashed lines.
2.5 Synthesis of 4
Before the true identity of the product of the reaction between 2 and [Li(Bun)]/[K(OBut)] was known, we treated an in situ mixture of 2 and [Li(Bun)]/[K(OBut)] with [Y(I)3(THF)3.5]. Colourless crystals were isolated in 47 % yield and subsequently identified as the complex [Y(OAr′)(I)2(THF)3] (4), Fig. 8. The 1H and 13C NMR spectra exhibited resonances consistent with a mono-aryloxide complex which is noteworthy because monomeric, mono-aryloxide-di-halide rare earth complexes are few in number, and usually result from the use of aryloxides that are considerably more sterically demanding than diisopropylphenyl as otherwise dimers are isolated [15]. In order to confirm the structure, colourless single crystals of 4 were grown from a solution in toluene.
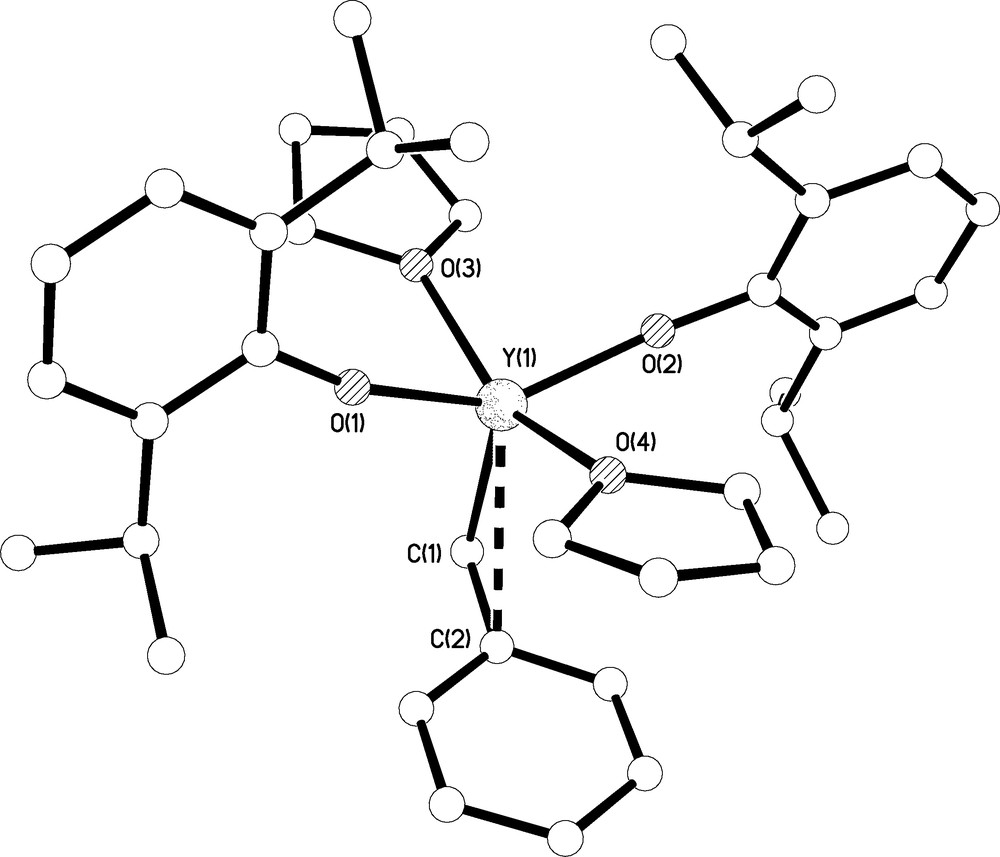
Molecular structure of 7.C6H5CH3 with selective labelling. Hydrogen atoms omitted for clarity. Multi-hapto interactions are shown as dashed lines.
2.6 Solid state structure of 4
The molecular structure of 4 is illustrated in Fig. 7 and selected bond lengths are given in Table 1. Complex 4 crystallises on a crystallographic two-fold rotation axis which intersects the O(1)-Y(1)-O(2) axis, which is therefore rigorously linear by definition. The complex is approximately octahedral at yttrium which is coordinated to the oxygen atom of the aryloxide, two iodides, and three THF molecules. Surprisingly, only two Y-OAr′ containing complexes have been crystallographically authenticated to date, namely [Y(OAr′){Al(μ-Me)(Me)2OAr′}2] by Anwander [16] and [Y{C6H3-2,6-(C6H4-2-OMe)2}(OAr′)2(THF)] by Rabe [17]. Complex 4 is thus the first mono-OAr′ complex of yttrium and is notable for its kinetic stability. The Y(1)-O(1) bond length of 2.046(3) Å compares with bond distances of 2.023 and 2.100/2.122 Å, respectively for the two aforementioned aryloxide complexes [16,17]. The Y(1)-I bond lengths are comparable to those observed in [Y(I)3(THF)3.5] [18] and the Y(1)-THF bond lengths are unremarkable.
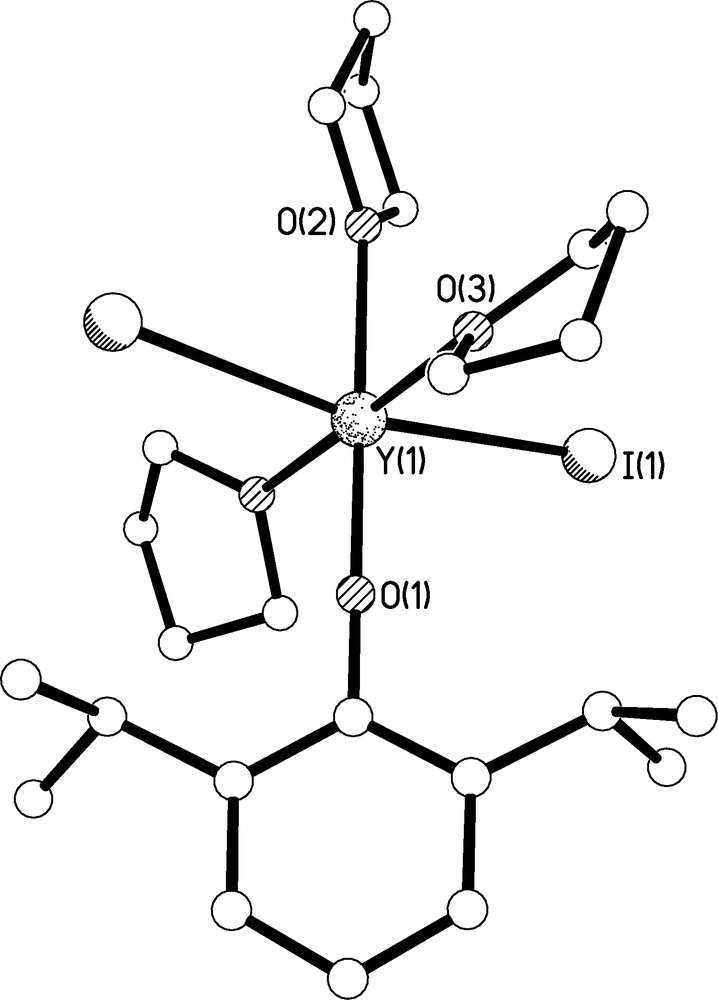
Molecular structure of 4 with selective labelling of symmetry unique atoms. Hydrogen atoms omitted for clarity.
2.7 Synthesis of 5–8
Since a superbasic mixture of [Li(Bun)]/[K(OBut)] was apparently too aggressive for 2, we switched our attention to [Y(Bn)3(THF)3] (Bn = CH2C6H5) [4c]. Since [Y(Bn)3(THF)3] and 2 do not react in toluene which is apparently due to the low solubility of the tribenzyl in arene solvents, we tested the reaction of [Y(Bn)3(THF)3] and 2 in THF. However, facile Si-O bond cleavage was observed again; although the resonance associated with the CH2 group was essentially unchanged in the 1H NMR spectrum, the 29Si NMR spectrum revealed a resonance at 1.17 ppm showing the formation of (BnSiMe2)2CH2 (5). The assertion of benzyl-aryloxide exchange was supported by the isolation of two new compounds by fractional crystallisation, namely [Y(OAr′)2(Bn)(THF)] (7) and [Y(OAr′)3(THF)2] (8). However, for 7 only enough material to obtain an X-ray diffraction data set was isolated. We therefore prepared [Y(OAr′)2(I)(THF)2] (6) from the reaction between [Y(I)3(THF)3.5] and two equivalents of [K(OAr′)] then treated it with one equivalent of [K(Bn)]. However, this route produced 8 as the sole isolable product, presumably from Schlenk-type ligand exchange. Unfortunately, X-ray diffraction quality crystals of 6 have not been obtained. Complex 7 was prepared rationally from the reaction of [Y(Bn)3(THF)3] and two equivalents of HOAr′ and was isolated as colourless crystals in 9 % yield. This low yield may be explained by the fact that solution NMR spectra of 7 are essentially identical to that of 8, which is presumably the result of facile Schlenk-type equilibria in solution. Complex 8 can also be prepared from the reaction of one equivalent of [K(OAr′)] with 6 or three equivalents of [K(OAr′)] with [Y(I)3(THF)3.5]. Surprisingly, although complex 4 appears to be kinetically stable in solution with respect to ligand exchange, dissolution of pure 6, as evidenced by NMR and CHN, in d6-benzene results in the establishment of a Schlenk-type equilibria over 12 h and the observation of resonances attributable to a small amount (∼10 %) of 8 in addition to the resonances for 6. Such an equilibrium could explain why 7 is not available from 6.
2.8 Solid state structure of 7.C6H5CH3
A very small crop of crystals of 7.C6H5CH3 suitable for X-ray diffraction were grown from toluene; the molecular structure is shown in Fig. 8 and selected bond lengths are complied in Table 1. Complex 7.C6H5CH3 crystallises as a monomer with an approximately square based pyramidal geometry at yttrium such that the benzyl carbon occupies the axial site. One molecule of toluene co-crystallises in the lattice. In addition to the Y(1)-C(1) bond distance of 2.416(5) Å, which compares with Y-CBn bond lengths spanning the range 2.452(3)-2.463(3) Å in [Y(Bn)3(THF)3] [3c], a Y(1)⋯C(2)ipso distance of 2.935(4) Å is suggestive of an η2-bonding mode of benzyl which is congruent with the compressed Y(1)-C(1)-C(2) angle of 95.9(3)° and Y⋯Cipso contact distance of 2.921(4) Å in II [3c]. The Y(1)-O(1) and Y(1)-O(2) bond lengths of 2.099(2) and 2.099(2) Å are significantly longer than the Y-OAr′ bond distance in 4 reflecting the more sterically crowded environment at yttrium in 7.C6H5CH3 compared to 4. The steric crowding may also be reflected in the fact the two Y-O-Cipso bond angles at O(1) and O(2) deviate from linear to 166.8(2) and 169.4(2)°, respectively, which contrasts to the linear Y-O-Cipso bond angle in 4.
2.9 Solid state structure of 8
Crystals of 8 suitable for X-ray crystallography were obtained from toluene; the molecular structure is illustrated in Fig. 9 and selected bond lengths are given in Table 1. Complex 8 crystallises as a monomer with approximately trigonal bipyramidal geometry at yttrium where the two coordinated THF molecules occupy the axial sites leaving the three aryloxides to occupy the more spacious equatorial positions. Overall, the structure is very similar to the lanthanum, praseodymium, samarium, gadolinium, dysprosium, erbium, and lutetium congeners [19,20]. Interestingly, the cerium analogue has also been isolated as a tris-THF adduct [21], the dimeric solvent free analogues have been characterised for lanthanum, neodymium, samarium, and dysprosium [19a,d,e], and monomeric mono-THF and THF-free analogues are known for dysprosium [19a]. The Y(1)-O(1), Y(1)-O(2), and Y(1)-O(3) bond lengths of 2.086(2), 2.069(3), and 2.085(2) Å compare well to the observed Y-OAr′ bond distances in 6. The two long and one shorter Y-O bond lengths match a trend in Y-O-Cipso bond angles; the angles at O(1), O(2), and O(3) of 174.7(3), 153.7(2), and 174.6(3)° are counter intuitive because conventional logic would suggest a shorter and stronger Y-O bond would be anticipated from a more linear Y-O-Cipso arrangement. However, we suggest the bond distances are not indicative of bond strength but rather of the sterically crowded environment at yttrium which forces the polar Y-O-Cipso bond angles to deviate from linear since the energy profile for bending in such ionic bonds would be expected to be rather flat.
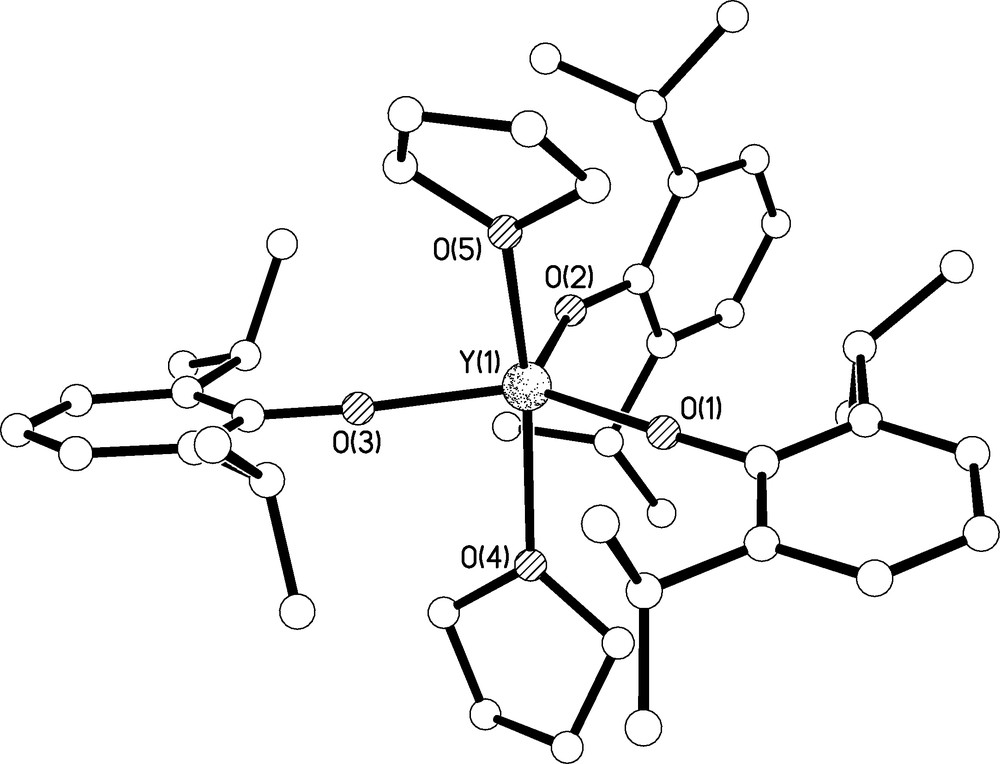
Molecular structure of 8 with selective labelling. Hydrogen atoms omitted for clarity.
3 Summary and conclusions
In summary, we have prepared a new bis(aryloxysilyl)methane (Ar′OSiMe2)2CH2 (2) and have tested it in reactions with electropositive metal alkyls. In the presence of [Li(Bun)/K(OBut)] 2 undergoes Si-O bond cleavage to give [{Li(OAr′)}2{K(OAr′)}2(THF)4] (3) and addition of [Y(I)3(THF)3.5] results in the isolation of [Y(OAr′)(I)2(THF)3] (4). The reaction of 2 with [Y(Bn)3(THF)3] in THF results in Si-O bond cleavage and Si-C bond formation to give (BnSiMe2)2CH2 (5), [Y(OAr′)2(Bn)(THF)] (7), and [Y(OAr′)3(THF)2] (8). The complex [Y(OAr′)2(I)(THF)2] (6), was prepared by a rational route, but attempts to prepare 7 from it resulted in the isolation of 8 and pure 6 was found to slowly establish a Schlenk-type equilibrium with 8 in benzene. Complex 7 was prepared rationally from [Y(Bn)3(THF)3] and two equivalents of HOAr′ but was found to convert to 8 in solution by Schlenk-type equilibria. We are currently investigating the behaviour of 2 towards [Li(But)] in pentane and are investigating the synthesis of the bis(aryloxysilyl)bromomethane (Ar′OSiMe2)2CHBr. The latter would enable low temperature lithium-halide exchange to take place and this would represent a milder route to the corresponding alkali metal bis(aryloxysilyl)methanides that would be less prone to ligand fragmentation reactions.
4 Experimental
4.1 General
All manipulations were carried out using standard Schlenk techniques, or an MBraun UniLab glovebox, under an atmosphere of dry nitrogen. Solvents were dried by passage through activated alumina towers and degassed before use. All solvents were stored over potassium mirrors (with the exception of THF which was stored over activated 4 Å molecular sieves). Deuterated solvents were distilled from potassium or CaH2, degassed by three freeze-pump-thaw cycles and stored under nitrogen. The compounds (BrSiMe2)2CH2 [6], [Y(I)3(THF)3.5] [18], [Y(Bn)3(THF)3] [3c], [K(OAr′)] [13], and [K(Bn)] [22] were prepared according to published procedures. Triethylamine was distilled from calcium hydride. Ar′OH was stored over 4 Å sieves. All other chemicals were purchased from Aldrich and were used as supplied.
1H, 13C, and 29Si NMR spectra were recorded on a Bruker 400 spectrometer operating at 400.2, 100.6, and 79.5 MHz, respectively; chemical shifts are quoted in ppm and are relative to TMS (1H, 13C, and 29Si). FTIR spectra were recorded on a Bruker Tensor 27 spectrometer. Elemental microanalyses were carried out by Mr Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK.
4.2 Preparation of (Ar′OSiMe2)2CH2 (2)
Ar′OH (10.36 ml, 55.2 mmol) in diethyl ether (30 ml) was added to a pre-cooled (−78 °C) mixture of (BrSiMe2)2CH2 (8.00 g, 27.6 mmol) and NEt3 (7.89 ml, 55.2 mmol) in diethyl ether (50 ml). The mixture was allowed to warm slowly to room temperature with stirring over 18 hours. The mixture was filtered and volatiles were removed in vacuo to afford 2 as a colourless oil. Yield: 11.21 g, 84 %. Crystals suitable for a X-ray crystallography were grown from hexane (10 ml) at −80 °C. Anal Calcd for C29H48O2Si2: C, 71.90; H, 9.92. Found: C, 72.00; H, 10.00. 1H NMR (d-chloroform, 298 K): δ 0.23 (s, 2H, CH2Si2), 0.46 (s, 12H, Si(CH3)2), 1.32 (d, 24H, JHH = 6.90 Hz, CH(CH3)2), 3.37 (sept, 4H, JHH = 6.90 Hz, CH(CH3)2), 7.09 (t, 2H, JHH = 7.40 Hz, p-Ar-CH) and 7.19 (d, 4H, JHH = 7.50 Hz, m-Ar-CH). 13C{1H} NMR (d-chloroform, 298 K): δ 3.30 (Si(CH3)2), 6.53 (CH2Si2), 23.59 (CH(CH3)2), 27.10 (CH(CH3)2), 122.13 (p-Ar-C), 123.53 (m-Ar-C), 139.13 (o-Ar-C) and 149.58 (ipso-Ar-C). 29Si NMR (d-chloroform, 298K): δ 15.21. IR (Nujol): 1589 (w), 1362 (m), 1330 (m), 1260 (s), 1196 (m), 1098 (m), 1047 (br, s), 918 (s), 814 (br, s), 756 (m).
4.3 Preparation of [{Li(OAr′)}2{K(OAr′)}2(THF)4] (3)
[Li(Bun)] (2 ml, 2.5 M in hexane) was added dropwise to a pre-cooled (−78 °C) mixture of 2 (2.42 g, 5 mmol) and [K(OBut)] (0.56 g, 5 mmol) in THF (20 ml). The mixture was allowed to slowly warm to room temperature with stirring over 18 hours. Volatiles were removed in vacuo and the resulting white solid was washed with hexane (20 ml) to afford 3. Crude yield: 2.05 g, 75 %. Crystals suitable for X-ray crystallography were grown from THF (5 ml) at −25 °C. Yield 1.36 g, 50 %. Anal Calcd for C64H100K2Li2O8: C, 70.53; H, 9.25. Found: C, 70.49; H, 9.31. 1H NMR (d8-THF, 298 K): δ 1.04 (d, 48H, JHH = 6.80 Hz, CH(CH3)2), 3.44 (sept, 8H, JHH = 6.80 Hz, CH(CH3)2), 6.07 (t, 4H, JHH = 7.60 Hz, p-Ar-CH) and 6.63 (d, 8H, JHH = 7.60 Hz, m-Ar-CH). 13C{1H} NMR (d8-THF, 298 K): δ 24.14 (CH(CH3)2), 27.23 (CH(CH3)2), 111.14 (p-Ar-C), 122.54 (m-Ar-C), 135.77 (o-Ar-C) and 164.47 (ipso-Ar-C). 7Li NMR (d8-THF, 298 K): δ −0.83. IR (Nujol): 1583 (m), 1345 (s), 1281 (m), 1263 (m), 1155 (w), 1137 (w), 1105 (w), 1042 (w), 886 (m), 849 (m), 802 (w), 763 (s), 752 (s).
4.4 Preparation of [Y(OAr′)(I)2(THF)3] (4)
THF (30 ml) was added to a pre-cooled (−78 °C) mixture of [Y(I)3(THF)3.5] (2.83 g, 3.92 mmol) and in situ formed 3 (2.05 g, 1.88 mmol). The mixture was allowed to slowly warm to room temperature with stirring over 24 h. The solution was filtered and volatiles were removed in vacuo and the resulting off-white solid was recrystallised from toluene (4 ml) to afford 4 as colourless crystals. Yield: 1.36 g, 47 %. Anal Calcd for C24H41I2O4Y: C, 40.02; H, 5.74. Found: C, 39.98; H, 5.67. 1H NMR (d6-benzene, 298 K): δ 1.45 (m, 12H, OCH2CH2), 1.58 (d, 12H, CH(CH3)2 JHH = 6.00 Hz), 4.08 (m, 12H, OCH2CH2), 4.52 (br, 2H, CH(CH3)2), 7.10 (t, 1H, JHH = 7.60 Hz, p-Ar-CH) and 7.35 (d, 2H, JHH = 7.60 Hz, m-Ar-CH). 13C{1H} NMR (d6-benzene, 298 K): δ 24.96 (br, CH(CH3)2 and OCH2CH2), 26.38 (CH(CH3)2), 71.25 (OCH2CH2), 118.62 (p-Ar-C), 123.36 (m-Ar-C), 137.63 (o-Ar-C) and 157.58 (ipso-Ar-C). IR (Nujol): 1588 (m), 1336 (s), 1267 (s), 1208 (m), 1173 (m), 1100 (m), 1039 (m), 1008 (s), 920 (m), 890 (m), 854 (s), 751 (m), 723 (m).
4.5 Preparation of [Y(OAr′)2(I)(THF)2] (6)
THF (30 ml) was added to a pre-cooled (−78 °C) mixture of [Y(I)3(THF)3.5] (1.89 g, 2.61 mmol) and [K(OAr′)] (1.13 g, 5.22 mmol). The mixture was allowed to slowly warm to room temperature with stirring over 24 h. The solution was filtered and volatiles were removed in vacuo and the resulting off-white solid was recrystallised from toluene (4 ml) to afford 6 as colourless crystals. Yield: 0.94 g, 51 %. Anal Calcd for C43H66IO5Y: C, 58.77; H, 7.57. Found: C, 58.66; H, 7.67. 1H NMR (d6-benzene, 298 K): δ 1.22 (m, 8H, OCH2CH2), 1.54 (d, 24H, JHH = 6.40 Hz, CH(CH3)2), 3.91 (m, 8H, OCH2CH2), 4.00 (br, 4H, CH(CH3)2), 7.09 (t, 2H, JHH = 7.60 Hz, p-Ar-CH) and 7.34 (d, 4H, JHH = 7.60 Hz, m-Ar-CH). 13C{1H} NMR (d6-benzene, 298 K): δ 23.96 (CH(CH3)2), 24.75 (OCH2CH2), 27.41 (CH(CH3)2), 71.93 (OCH2CH2), 118.49 (p-Ar-C), 123.11, 125.46 (m-Ar-C), 136.13 (o-Ar-C) and 157.68 (br, ipso-Ar-C). IR (Nujol): 1586 (w), 1334 (m), 1261 (s), 1209 (m), 1145 (m), 1042 (m), 1013 (m), 935 (m), 889 (m), 859 (m), 802 (m), 753 (m).
4.6 Preparation of [Y(OAr′)2(Bn)(THF)2] (7)
A solution of HOAr′ (0.75 ml, 4 mmol) in THF (10 ml) was added to a solution of [Y(Bn)3(THF)3] (1.24 g, 2 mmol) in THF (10 ml) dropwise at −78 °C. The solution was allowed to warm to room temperature and then stirred for 24 hours. Volatiles were removed in vacuo to yield a brown oil. The brown oil was washed with hexane (10 ml) to yield a white solid. Yield: 0.1 g, 9 %. Anal. calcd for C39H57O4Y: C, 69.03; H, 8.41. Found: C, 68.88; H, 8.34. IR (Nujol): 1459 (s), 1377 (s), 1331 (s), 1262 (w), 1209 (s), 1016 (w), 863 (s), 799 (s) 753 (s) and 692 (s).
4.7 Preparation of [Y(OAr′)3(THF)2] (8)
THF (10 ml) was added to a pre-cooled (−78 °C) mixture of 6 (0.76 g, 0.86 mmol) and [K(Bn)] (0.11 g, 0.86 mmol). The mixture was allowed to slowly warm to room temperature with stirring over 18 h. The solution was filtered and volatiles were removed in vacuo and the resulting off-white solid was recrystallised from toluene (2 ml) to afford 8 as colourless crystals. Yield: 0.26 g, 59 %. Anal Calcd for C44H67O5Y: C, 69.09; H, 8.83. Found: C, 69.01; H, 8.94. 1H NMR (d6-benzene, 298 K): δ 1.22 (m, 8H, OCH2CH2), 1.45 (d, 24H, JHH = 6.80 Hz, CH(CH3)2), 3.66 (sept, 4H, JHH = 6.80 Hz, CH(CH3)2), 3.91 (m, 8H, OCH2CH2), 7.08 (t, 2H, JHH = 7.40 Hz, p-Ar-CH) and 7.33 (d, 4H, JHH = 7.60 Hz, m-Ar-CH). 13C{1H} NMR (d6-benzene, 298 K): δ 23.63 (CH(CH3)2), 25.00 (OCH2CH2), 27.41 (CH(CH3)2), 71.59 (OCH2CH2), 117.49 (p-Ar-C), 122.94, (m-Ar-C), 135.93 (o-Ar-C) and 157.92 (d, 2JCY = 5.03 Hz, ipso-Ar-C). IR (Nujol): 1586 (m), 1332 (s), 1209 (m), 1041 (m), 1016 (m), 888 (m), 863 (s), 753 (m).
4.8 X-ray Crystallography
Crystal data for compounds 2, 3.C4H8O, 4, 7.C6H5CH3 and 8 are given in Table 2, and further details of the structure determinations are in the Supporting Information. Bond lengths are listed in Table 1. Crystals were examined on a Bruker AXS SMART APEX CCD area detector diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). Intensities were integrated from a sphere of data recorded on narrow (0.3°) frames by ω rotation. Cell parameters were refined from the observed positions of all strong reflections in each data set. Semi-empirical absorption corrections based on symmetry-equivalent and repeat reflections were applied. The structures were solved variously by direct or Patterson methods and were refined by full-matrix least-squares on all unique F2 values, with anisotropic displacement parameters for all non-hydrogen atoms, and with constrained riding hydrogen geometries; Uiso(H) was set at 1.2 (1.5 for methyl groups) times Ueq of the parent atom. The largest features in final difference syntheses were close to heavy atoms and were of no chemical significance. The Flack parameters for 8 refined to −0.017(5) confirming that the correct absolute structure had been adopted. The data for 3.C4H8O were persistently weak and of poor quality, despite several attempts at recrystallisation under different conditions, resulting in a high R1 but the chemical connectivity is unambiguous. Disorder of coordinated THF in 7.C6H5CH3 could not be modelled. The data for 8 were weak and/or absent at high angle but the structure is unambiguous. Programs were Bruker AXS SMART (control) and SAINT (integration) [23], and SHELXTL was employed for structure solution and refinement and for molecular graphics [24]. Disordered lattice THF in 3 which could not be modelled was treated with the Platon SQUEEZE procedure [25].
Crystallographic data for 2–4, 7.C6H5CH3, and 8.
| 2 | 3.C4H8O | 4 | 7.C6H5CH3 | 8 | |
| Formula | C29H48O2Si2 | C68H108K2Li2O9 | C24H41I2O4Y | C46H65O4Y | C44H67O5Y |
| Fw | 484.85 | 1161.62 | 736.28 | 770.89 | 764.89 |
| Cryst size, mm | 0.37 × 0.19 × 0.10 | 0.30 × 0.18 × 0.13 | 0.40 × 0.40 × 0.21 | 0.32 × 0.30 × 0.21 | 0.47 × 0.39 × 0.29 |
| Cryst syst | Triclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | P-1 | P21/c | Pbcn | P21/c | P21 |
| a, Å | 9.6735(7) | 10.705(2) | 9.2246(10) | 13.8410(7) | 9.6919(5) |
| b, Å | 12.2705(8) | 17.677(4) | 17.924(2) | 17.6892(10) | 19.3180(10) |
| c, Å | 13.8619(9) | 21.243(4) | 17.868(2) | 18.6316(10) | 12.1546(6) |
| α,° | 70.965(1) | ||||
| β,° | 74.690(1) | 120.060(8) | 107.996(2) | 109.519(2) | |
| γ,° | 87.871(1) | ||||
| V, Å3 | 1498.05(18) | 3479.2(12) | 2954.4(6) | 4338.5(4) | 2144.90(19) |
| Z | 2 | 2 | 4 | 4 | 2 |
| ρcalcd, g cm−3 | 1.075 | 1.109 | 1.655 | 1.180 | 1.184 |
| μ, mm−1 | 0.140 | 0.187 | 4.088 | 1.383 | 1.400 |
| No. of reflections measd | 13385 | 24847 | 16698 | 22477 | 7053 |
| No. of unique reflns, Rint | 6764, 0.0200 | 6145, 0.0984 | 3377, 0.0499 | 7614, 0.0317 | 4609, 0.0260 |
| No. of reflns with F2 > 2σ(F2) | 6060 | 3750 | 2414 | 5820 | 4291 |
| Transmn coeff range | 0.90–0.98 | 0.79–0.98 | 0.26–0.46 | 0.63–0.73 | 0.54–0.67 |
| R, Rwa (F2 > 2σ) | 0.0508, 0.1181 | 0.1260, 0.2640 | 0.0376, 0.0821 | 0.0417, 0.0989 | 0.0302, 0.0694 |
| R, Rwa (all data) | 0.0570, 0.1221 | 0.1667, 0.2787 | 0.0662, 0.0920 | 0.0620, 0.1087 | 0.0322, 0.0698 |
| Sa | 1.108 | 1.127 | 1.037 | 1.035 | 0.890 |
| Parameters | 310 | 352 | 145 | 468 | 463 |
| Max., min. diff map, e Å−3 | 0.493, −0.188 | 0.373, −0.493 | 1.062, −0.436 | 0.656, −0.306 | 0.380, −0.210 |
Acknowledgements
We thank the Royal Society, the UK EPSRC, and the University of Nottingham for supporting this work.
Annexe A Supplementary material
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC-745767-745771). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request.cif.