

1 Introduction
The organometallic and coordination chemistry of low-coordinate lanthanide(III) complexes has greatly expanded in recent years and has generated a range of unusual f-block complexes. For example, complexes containing bonds between the lanthanide and main group metals including gallium, [Nd(L′){Ga(NArCH)2}(N″)(THF)] (N″ = N(SiMe3)2, Ar = Dipp = 2,6-(Pri2C6H3)) [1] and aluminium [Ln(Cp∗)2–Al(Cp∗)] (Cp∗ = C5Me5) [2] have been isolated; the complexes [(N2)(LnZ2)2] are formed from the reductive activation of dinitrogen by LnN″3; molecular complexes of Nd(II), Tm(II), and Dy(II) have been isolated for the first time [3]; and a variety of reactions to activate inert substrates such as hydrocarbyl C–H and carbon dioxide CO bonds have been reported [4].
Over the last decade, the organometallic chemistry of lanthanide complexes has been expanded to include a variety of N-heterocyclic carbene complexes, in which the carbene functions as a σ-donor to the electropositive metal [5]. While the donor carbene complexes generally were not amenable to reductive activation chemistry [6], the σ-donor ligands did allow access to heteroleptic iodide complexes that were relatively inert to ligand scrambling [7]. This has allowed us to explore some metathetical routes to other low-coordinate lanthanide complexes.
The use of sterically encumbered ligands to make low-coordinate complexes is also of relevance to the continued search for isolable examples of terminal alkylidene and imido complexes of lanthanides [8].
One major class of bulky group that can protect a reaction site at a metal comprises substituted terphenyls based on aryls, such as the Dipp- and Trip-functionalised systems Ar′ = 2,6-Dipp2C6H3 and Ar∗ = 2,6-Trip2C6H3 (Dipp = 2,6-Pri2C6H3, Trip = 2,4,6-Pri3C6H2) [9].
Herein, we report the reactivity of the heteroleptic Nd(III) iodide [Nd(L)(I)(N″)] recently reported by us [5a], with primary arylamides, including the new potassium arylamide salt [KNHAr∗], and the reactivity of these amido complexes towards nucleophiles and bases.
2 Results and discussion
2.1 Preparation of 2
Reaction of the terphenyl-substituted primary amine Ar∗NH2 [Ar∗ = 2,6-(2,4,6-Pri3C6H2)2C6H3] with potassium hydride proceeds smoothly in THF to afford the corresponding potassium primary amide [KNHAr∗] 2, Eq. (1).
| Ar∗NH2 + KH → [Ar∗N(H)K] (2) + H2 | (1) |
Following filtration and removal of solvent, compound 2 is isolated from hexane as an air- and moisture-sensitive yellow powder in good yield. The 1H and 13C{1H} NMR spectra are as expected; the most salient features are the broad singlet at 2.25 ppm in the 1H NMR spectrum (cf. 2.06 ppm for the corresponding lithium salt reported by Power [10]), characteristic of an N–H group and the absence of any solvent resonances. The formulation of 2 as solvent free is corroborated by elemental analysis. We were unable to grow solvent free crystals of 2, but yellow crystals of [2(OEt2)2] were readily obtained from a cold, saturated solution of 2 in diethyl ether. The diethyl ether in [2(OEt2)2] is bound very loosely as evidenced by rapid degradation, on exposure to dynamic vacuum, of crystals of [2(OEt2)2] to a yellow powder which affords NMR spectra identical to that of 2, i.e. solvent free.
2.2 X-ray crystal structure of [2(OEt2)2]
The molecular structure of [2(OEt2)2] is illustrated in Fig. 1 and selected bond lengths and angles are presented in Table 1. Compound [2(OEt2)2] crystallises as a monomer; the potassium centre is pseudo tetrahedral, bonded to the anionic nitrogen, the neutral oxygen donors of two diethyl ether solvent molecules, and by an η6 interaction to one of the Trip arene rings of the terphenyl group. The K(1)–N(1) bond length of 2.611(3) Å represents a rare example of a terminal primary amide–potassium bond and is short in comparison to other K–N bonds [from a survey of the Cambridge Structural Database version 1.9], which are usually bridging, reflecting the formally low coordination number of potassium and the weak bonding nature of the donor groups. The K⋯Caryl distances span the range 3.098(3)–3.463(3) Å, which is towards the lower end of the range of K⋯Caryl distances [11].
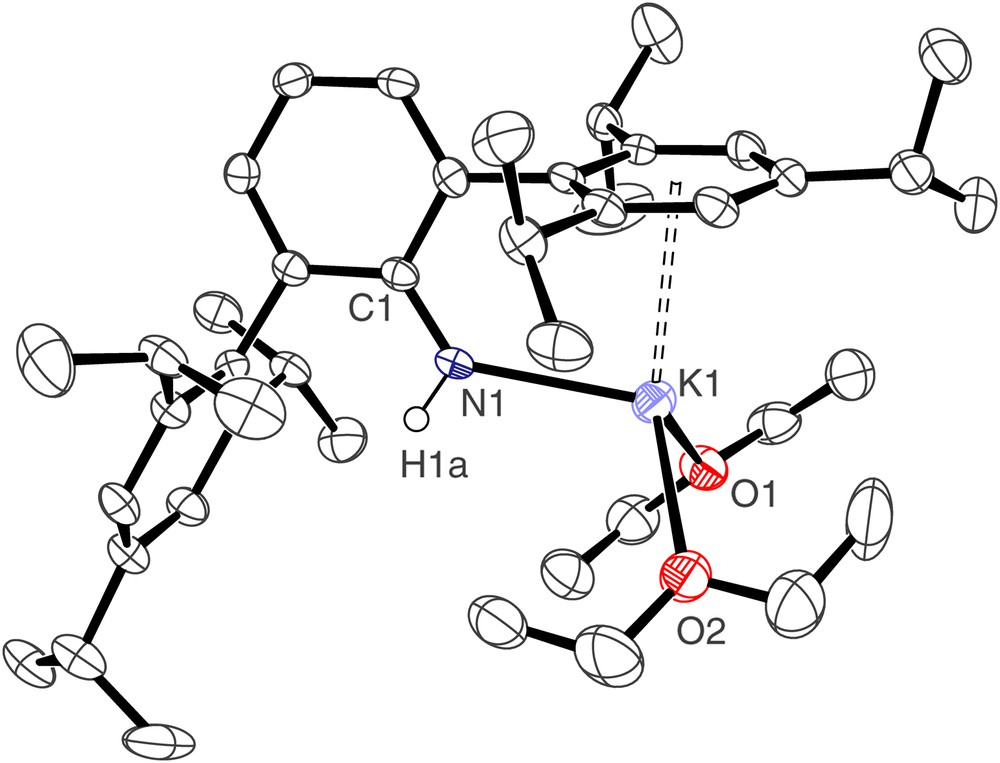
Molecular structure of [2(OEt2)2]; carbon-bound hydrogen atoms omitted for clarity.
Selected bond lengths (Å) and angles (°) for 2(OEt2)2, 3, and 4·1⅓OEt2
| 2(OEt2)2 | |||
| K(1)–N(1) | 2.611(3) | K(1)–O(1) | 2.719(3) |
| K(1)–O(2) | 2.715(3) | K(1)–C(7) | 3.115(3) |
| K(1)–C(8) | 3.098(3) | K(1)–C(9) | 3.269(3) |
| K(1)–C(10) | 3.463(3) | K(1)–C(11) | 3.442(3) |
| K(1)–C(12) | 3.296(3) | ||
| N(1)–K(1)–O(2) | 106.13(9) | N(1)–K(1)–O(1) | 109.15(9) |
| O(2)–K(1)–O(1) | 103.70(8) | N(1)–K(1)–C(8) | 69.50(9) |
| O(2)–K(1)–C(8) | 105.85(9) | O(1)–K(1)–C(8) | 149.48(9) |
| N(1)–K(1)–C(7) | 58.22(8) | O(2)–K(1)–C(7) | 130.32(9) |
| O(1)–K(1)–C(7) | 125.89(8) | C(8)–K(1)–C(7) | 26.26(8) |
| N(1)–K(1)–C(9) | 94.33(9) | O(2)–K(1)–C(9) | 99.04(9) |
| O(1)–K(1)–C(9) | 140.79(9) | C(8)–K(1)–C(9) | 24.83(8) |
| C(7)–K(1)–C(9) | 44.36(9) | N(1)–K(1)–C(12) | 75.94(8) |
| O(2)–K(1)–C(12) | 148.60(9) | O(1)–K(1)–C(12) | 105.02(8) |
| C(8)–K(1)–C(12) | 44.48(8) | C(7)–K(1)–C(12) | 25.27(8) |
| C(9)–K(1)–C(12) | 49.92(8) | N(1)–K(1)–C(11) | 99.15(8) |
| O(2)–K(1)–C(11) | 134.76(9) | O(1)–K(1)–C(11) | 102.47(8) |
| C(8)–K(1)–C(11) | 49.91(9) | C(7)–K(1)–C(11) | 43.15(8) |
| C(9)–K(1)–C(11) | 41.36(8) | C(12)–K(1)–C(11) | 23.82(8) |
| N(1)–K(1)–C(10) | 108.37(8) | O(2)–K(1)–C(10) | 111.76(9) |
| O(1)–K(1)–C(10) | 117.14(9) | C(8)–K(1)–C(10) | 43.07(9) |
| C(7)–K(1)–C(10) | 50.68(8) | C(9)–K(1)–C(10) | 23.66(8) |
| C(12)–K(1)–C(10) | 42.16(8) | C(11)–K(1)–C(10) | 23.17(8) |
| Compound 3 | |||
| Nd(1)–N(2) | 2.252(3) | Nd(1)–N(5) | 2.342(3) |
| Nd(1)–N(4) | 2.363(3) | Nd(1)–C(2) | 2.612(4) |
| N(1)–C(2) | 1.360(5) | N(3)–C(2) | 1.372(5) |
| N(2)–Nd(1)–N(5) | 121.92(12) | N(2)–Nd(1)–N(4) | 11.14(12) |
| N(5)–Nd(1)–N(4) | 108.84(11) | N(2)–Nd(1)–C(2) | 79.17(12) |
| N(5)–Nd(1)–C(2) | 105.69(12) | N(4)–Nd(1)–C(2) | 122.76(12) |
| Nd(1)–N(4)–Si(2) | 119.40(16) | N(1)–C(2)–N(3) | 102.9(3) |
| Nd(1)–N(4)–Si(3) | 116.38(16) | ||
| 4·1⅓OEt2 | |||
| Nd(1)–N(2) | 2.289(5) | Nd(1)–N(5) | 2.332(5) |
| Nd(1)–N(4) | 2.365(5) | Nd(1)–C(2) | 2.603(6) |
| N(1)–C(2) | 1.355(7) | N(3)–C(2) | 1.362(7) |
| Nd(1A)–N(2A) | 2.270(5) | Nd(1A)–N(5A) | 2.326(5) |
| Nd(1A)–N(4A) | 2.366(5) | Nd(1A)–C(2A) | 2.602(6) |
| N(1A)–C(2A) | 1.366(7) | N(3A)–C(2A) | 1.362(7) |
| Nd(1B)–N(2B) | 2.269(5) | Nd(1B)–N(5B) | 2.338(4) |
| Nd(1B)–N(4B) | 2.361(5) | Nd(1B)–C(2B) | 2.603(7) |
| N(1B)–C(2B) | 1.357(7) | N(3B)–C(2B) | 1.364(7) |
| N(2)–Nd(1)–N(5) | 97.97(17) | N(2)–Nd(1)–N(4) | 110.55(17) |
| N(5)–Nd(1)–N(4) | 133.43(17) | N(2)–Nd(1)–C(2) | 84.07(17) |
| N(5)–Nd(1)–C(2) | 110.61(17) | N(4)–Nd(1)–C(2) | 108.36(18) |
| N(1)–C(2)–N(3) | 103.0(5) | N(2A)–Nd(1A)–N(5A) | 100.55(18) |
| N(2A)–Nd(1A)–N(4A) | 111.3(2) | N(5A)–Nd(1A)–N(4A) | 129.60(18) |
| N(2A)–Nd(1A)–C(2A) | 81.64(19) | N(5A)–Nd(1A)–C(2A) | 104.55(19) |
| N(4A)–Nd(1A)–C(2A) | 117.60(19) | N(3A)–C(2A)–Nd(1A) | 120.3(4) |
| N(2B)–Nd(1B)–N(5B) | 95.57(16) | N(2B)–Nd(1B)–N(4B) | 112.06(17) |
| N(5B)–Nd(1B)–N(4B) | 133.17(17) | N(2B)–Nd(1B)–C(2B) | 84.37(18) |
| N(5B)–Nd(1B)–C(2B) | 106.59(17) | N(4B)–Nd(1B)–C(2B) | 112.89(18) |
| N(1B)–C(2B)–N(3B) | 103.2(5) |
2.3 Synthesis of 3 and 4
Treatment of 1 with 1 equiv of [Ar′N(H)K] [12] in cold THF affords compound 3 as blue crystals, which, after workup and recrystallisation from tert-butyl methyl ether, are isolated in low yield, due to the high solubility of 3, Scheme 1. The chemical shifts in the 1H NMR spectrum fall in the range +65 to −15 ppm due to the paramagnetic neodymium(III) centre, but all resonances are observed. Similarly, treatment of 1 with 1 equiv of 2 in cold THF affords compound 4 as blue crystals in high yield after workup and recrystallisation from diethyl ether, Scheme 1. Both 3 and 4 are highly air- and moisture-sensitive. Crystals of 4 were shown to be the etherate 4·1⅓OEt2 by X-ray crystallography (see below); exposure of crystals of 4 to dynamic vacuum results in loss of lattice solvent and collapse of the crystals into a blue glass. The 1H NMR spectrum of 4 is resolved at room temperature over the range +54 → −20 ppm and is broader than that observed for 3, perhaps reflecting the sterically more demanding environment around the neodymium(III) centre in 4 compared to 3. Interestingly, solutions of 3 and 4 are dichroic; dilute solutions (or samples viewed through a short path length) appear pale blue whilst more concentrated (or longer path length) solutions appear dark red.
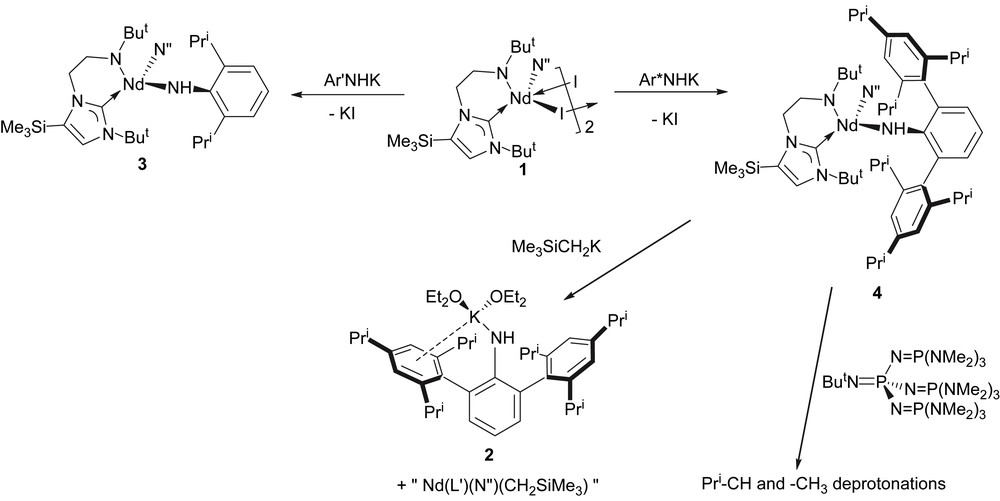
Synthesis of 2 and 4, and reactions of 4.
2.4 X-ray crystal structure of 3
The molecular structure of 3 is depicted in Fig. 2 and selected bond lengths and angles are in Table 1. The four-coordinate neodymium centre adopts a distorted tetrahedral geometry, bonding to the neutral carbene centre of the NHC and the anionic nitrogen centres of the Dipp amide, N″ ligand, and amido tether of the NHC. The Nd(1)–C(2) bond length of 2.612(4) Å is towards the lower end of Nd–CNHC bonds which we have previously reported [7a] and statistically invariant compared to the Nd–CNHC bond length of 2.609(3) Å in [Nd(L)(N″)2], which also features a four-coordinate, distorted tetrahedral neodymium(III) centre [7a]. The Nd(1)–N(2), Nd(1)–N(5), and Nd(1)–N(4) bond lengths of 2.252(3), 2.342(3), and 2.363(3) Å, respectively, are unexceptional, and follow the anticipated trend of Ndialkyl < Nanilide < Nsilyl. The Nd(1)⋯Si(3) distance of 3.475 Å is suggestive of a close β-Si–C contact, and is corroborated by a smaller metal–amide–silicon angle at the amido nitrogen to Si(3) of 116.38(16)° than that to Si(2) which is 119.40(16)°. Similarly, the complex [Nd(L′)(N″)2(THF)] contains a close Nd–Si(N) distance of 3.353 Å to one silylamide group [7a], while the structure of [NdN″3] also contains an Nd–Si distance of 3.398 Å [13].
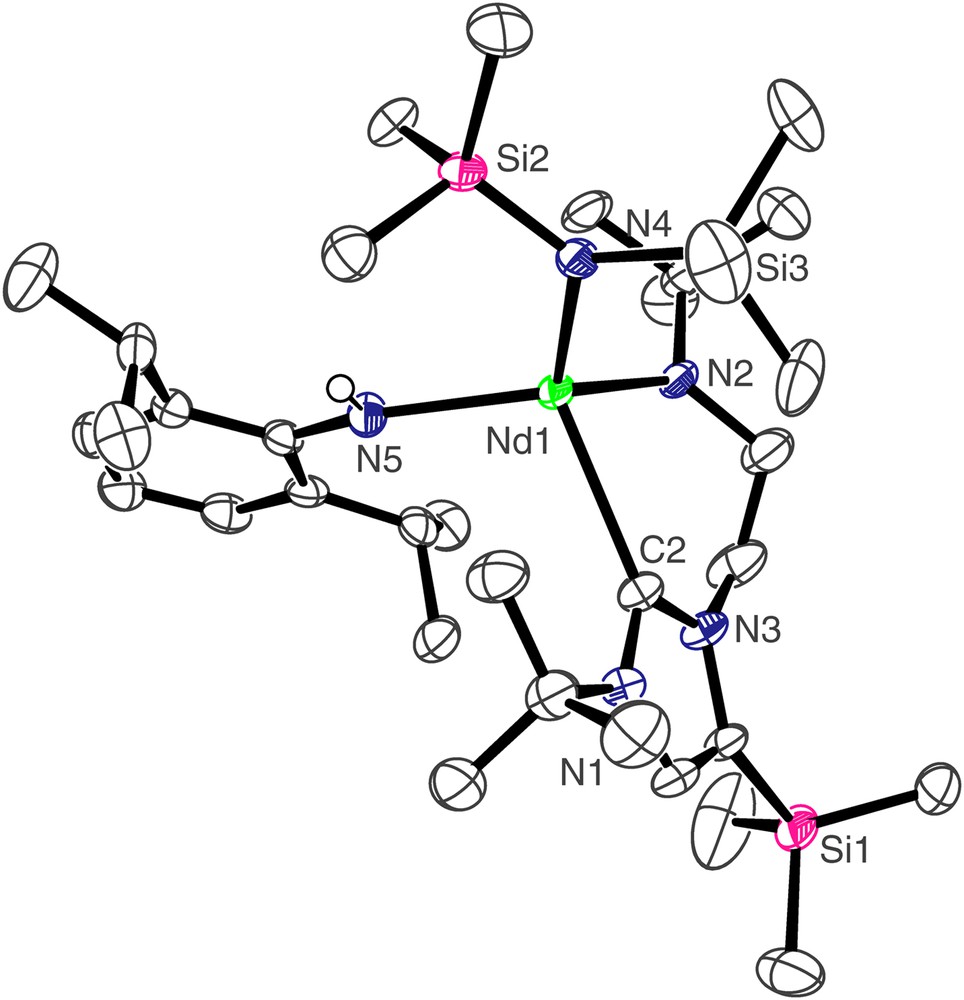
Molecular structure of 3; carbon-bound hydrogen atoms omitted for clarity.
2.5 X-ray crystal structure of 4
The molecular structure of compound 4 is shown in Fig. 3, and selected bond lengths and angles are in Table 1. Three molecules of 4 crystallise in the asymmetric unit but they differ from each other in only minor details so the structure of only one is discussed. As for 3, the neodymium centre in 4 adopts a distorted tetrahedral geometry, bonding to the neutral carbene centre of the NHC and the anionic nitrogen centres of the terphenyl amide, N″ ligand, and amido tether of the NHC. The Nd(1)–C(2) bond length of 2.603(6) Å is, statistically, essentially the same as in 3. However, the Nd–C bond is much more distorted in 4 than it is in 3 as a consequence of the much greater steric demands of the terphenyl group compared to the Dipp group. We have previously described pitch and yaw, Fig. 4 [14], to quantify such deviations and the magnitude of the bends away from the ideal trigonal planar M–CN2 moiety for 3 and 4 are 6.5° and 4.3° and 18.0° and 0.9°, respectively. Whereas the Nd(1)–N(4) and Nd(1)–N(5) bond lengths of 2.365(5) and 2.332(5) Å, respectively, in 4 are statistically invariant to the equivalent bonds in 3, the Nd(1)–N(2) bond length of 2.289(5) Å is slightly longer than the equivalent bond length in 3, reflecting the greater steric congestion at neodymium in 4 compared to 3. As in 3, an Nd(1)⋯Si(3) distance of 3.419 Å in 4 is suggestive of a β-Si–C contact due to the low coordination number of neodymium.
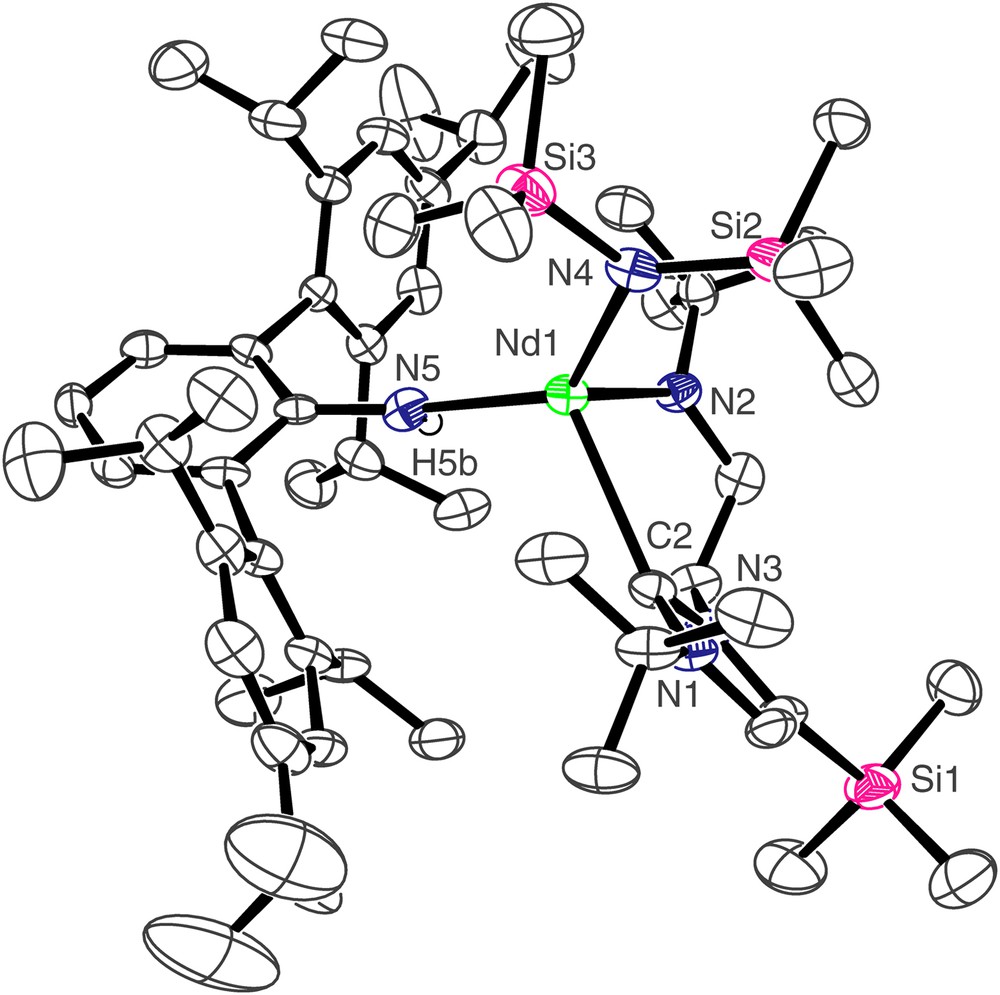
Molecular structure of 4; carbon-bound hydrogen atoms omitted for clarity.
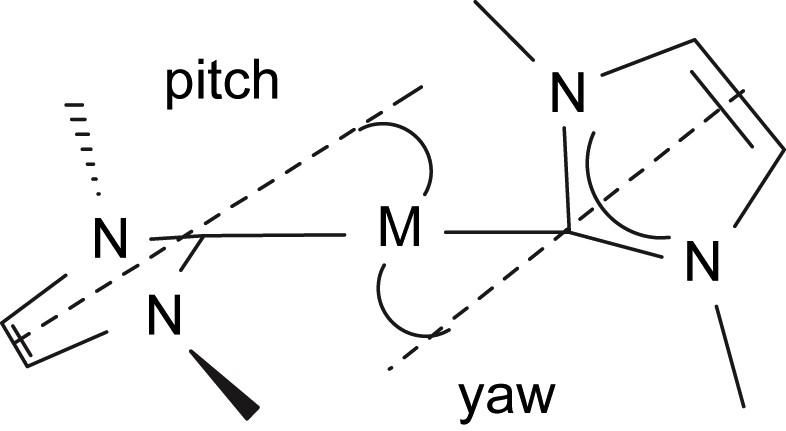
Illustration of pitch and yaw in metal–NHC complexes.
2.6 Reactivity of 3 and 4 towards alkali metal alkyls
In an effort to deprotonate the primary amide in 3 and 4 a second time to afford anionic imido species, 3 and 4 were treated with alkali metal alkyls MR (M = Li or K, R = Me or CH2SiMe3). In the case of 3, intractable, oily products were obtained which we were unable to unambiguously identify due to the paramagnetic nature of neodymium(III). However, treatment of 4 with alkali metal alkyls led to isolable products; in each case, the corresponding alkali metal terphenyl amide [Ar∗N(H)M] (M = Li or K) was eliminated affording heteroleptic species of the general form [Nd(L′)(N″)(R)], which doubtless undergo Schlenk-type equilibria. The identities of [Ar∗N(H)Li] [15] and 2 were confirmed by comparison of their NMR spectra with authentic samples, and by a unit cell determination of the latter crystallised as 2(OEt2)2 (see above).
2.7 Reactivity of 4 towards the phosphazene superbase ButNP{NP(NMe2)3}3
Since the primary aryl amide ligands had proven to be vulnerable to ligand exchange in the presence of strong nucleophiles such as alkyls, apparently a process thermodynamically more favourable compared to double deprotonation of the primary amide centre, the reaction of 4 with the bulky phosphazene superbase ButNP{NP(NMe2)3}3 was attempted, Scheme 1, since it is a base with great thermodynamic strength but very poor nucleophilicity [16]. Treatment of 4 with 1 equiv of ButNP{NP(NMe2)3}3 afforded an intractable brown oil which could not be recrystallised despite exhaustive attempts. In order to gain insight as to the nature of the reaction product(s) an aliquot was quenched with D2O and a 2H NMR spectrum obtained. The resulting spectrum exhibited broad 2H resonances at 1.82, 3.14, 4.16, and 6.91 ppm, characteristic of ortho- and para-methyl and methine deprotonations of the iso-propyl groups as well as minor deprotonation at the aryl para position of the terphenyl ligand. Due to the bulk of the phosphazene superbase and the steric congestion around the amide–proton linkage we suggest that deprotonation at the C–H sites must be kinetically more favourable than removal of the amide proton, despite the latter being surely more acidic than the former grouping (for analogous reactivity of a phosphazene superbase with a scandium amide supported by a β-diketiminate ligand system see Ref. [17]).
3 Conclusions
The potassium terphenyl primary amide [Ar∗N(H)K] (2) is readily prepared and isolated in high yield. The reaction of [Nd(L′)(N″)(μ-I)] (1) with 1 equiv of [Ar′N(H)K] or 2 affords 3 and 4 cleanly. Both complexes exhibit four-coordinate, tetrahedral Nd(III) centres supplemented by β-Si–C interactions. Treatment of 3 or 4 with alkali metal alkyls results in ligand exchange to give alkali metal primary amides and heteroleptic species of the generic form [Nd(L′)(N″)(R)], which are intractable. Treatment of 4 with the thermodynamically more basic, but less nucleophilic phosphazene superbase ButNP{NP(NMe2)3}3 results in indiscriminate deprotonations at iso-propyl methyl and methine sites and the terphenyl aryl para position, which we ascribe to the greater kinetic accessibility of these sites compared to the amide–proton linkage.
4 Experimental
4.1 General
All manipulations were carried out using standard Schlenk techniques, or an MBraun UniLab glovebox, under an atmosphere of dry nitrogen. Hexane, diethyl ether and THF were dried by passage through activated alumina towers and degassed before use. Benzene and tert-butyl methyl ether were distilled from potassium under an atmosphere of dry nitrogen. All solvents were stored over potassium mirrors (with the exception of THF which was stored over activated 4 Å molecular sieves). Deuterated benzene was distilled from potassium, degassed by three freeze–pump–thaw cycles and stored under nitrogen. The compounds [MeK] [18], [Me3SiCH2K] [11b], [Ar′N(H)K] [12], Ar∗NH2 [14], and 1 [7a] were prepared by published literature procedures [Ar′ = 2,6-Pri2C6H3; Ar∗ = 2,6-(2,4,6-Pri3C6H2)2C6H3]. ButNP{NP(NMe2)3}3 (1.0 M in hexane), [MeLi] (1.6 M in diethyl ether) and [Me3SiCH2Li] (1.0 M in pentane) were purchased from Aldrich and used as supplied.
1H, 13C, and 31P NMR spectra were recorded on a Bruker 300 spectrometer operating at 300.1, 75.5, and 121.5 MHz, respectively; chemical shifts are quoted in ppm and are relative to TMS (1H and 13C) and external 85% H3PO4 (31P). Elemental microanalyses were carried out by Mr. Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK.
4.2 Preparation of [Ar∗N(H)K] (2)
THF (60 ml) was added to a mixture of Ar∗NH2 (6.92 g, 13.90 mmol) and KH (1.50 g, 37.00 mmol) and the resulting suspension was refluxed for 5 days. The mixture was filtered to give a green-brown solution and volatiles were removed in vacuo to give a sticky yellow solid. The solid was washed with hexane (50 ml) and dried in vacuo to afford 2 as a free flowing yellow powder. The hexane washing was concentrated until precipitation occurred and then stored at −30 °C overnight. The resultant yellow powder was collected by filtration and dried. Combined yield: 5.77 g, 78%. Yellow crystals of [2(OEt2)2] were obtained from a saturated solution in diethyl ether stored at −30 °C overnight; the diethyl ether is weakly bound, as evidenced by the collapse of single crystals to a yellow powder on exposure to vacuum and the CHN microanalysis. Anal. Calcd for C36H50KN: C, 80.69; H, 9.40; N, 2.61. Found: C, 80.58; H, 9.56; N, 2.63. 1H NMR (benzene-d6, 295 K): δ 1.28 (d, 3JHH = 6.91 Hz, 12H, para-CH(CH3)2), 1.35 (d, 3JHH = 6.86 Hz, 24H, ortho-CH(CH3)2), 2.25 (s, br, 1H, NH), 2.94 (sept, 3JHH = 6.91 Hz, 2H, para-CH(CH3)2), 3.41 (sept, 3JHH = 6.86 Hz, 4H, ortho-CH(CH3)2), 6.62 (t, 3JHH = 7.20 Hz, 1H, para-Ar-CH), 7.08 (d, 3JHH = 7.20 Hz, 2H, meta-Ar-CH) and 7.22 (s, 4H, meta-Tripp-CH). 13C{1H} NMR (benzene-d6, 295 K): δ 24.29 (ortho-CH(CH3)2), 24.93 (para-CH(CH3)2), 30.36 (ortho-CH(CH3)2), 34.34 (para-CH(CH3)2), 107.10 (para-Ar-CH), 120.43 (meta-Tripp-CH), 123.71 (meta-Ar-CH), 129.59 (para-Tripp-C), 141.15 (ortho-Ar-C), 146.98 (ortho-Tripp-C), 148.72 (ipso-Tripp-C) and 161.59 (ipso-Ar-C).
4.3 Preparation of [Nd(L′)(N″)(NHAr′)] (3)
A solution of [Ar′N(H)K] (0.43 g, 2.01 mmol) in THF (10 ml) was added dropwise to a solution of 1 (1.46 g, 2.01 mmol) in THF (5 ml). The mixture was stirred overnight prior to volatiles being removed in vacuo. The mixture was extracted with diethyl ether (30 ml), filtered, and volatiles removed in vacuo to give a ‘foamy’ pale blue oil. The oil was dissolved in tert-butyl methyl ether (1.5 ml) and stored at −30 °C for 5 days to afford a crop of blue crystals of 3. Yield: 0.60 g, 38%. Anal. Calcd for C34H68N5NdSi3: C, 52.66; H, 8.84; N, 9.03. Found: C, 52.47; H, 8.78; N, 9.00. 1H NMR (benzene-d6, 295 K): δ −14.45 (s, 9H, C(CH3)3), −7.71 (s, 9H, C(CH3)3), −2.28 (s, 18H, Si(CH3)3), −0.03 (s, 9H, Si(CH3)3), 0.43 (s, 1H, CH(CH3)2), 1.00 (s, 1H, CH(CH3)2), 3.21 (s, 6H, CH(CH3)2), 4.59 (s, 6H, CH(CH3)2), 7.97 (t, 3JHH = 7.29 Hz, para-Ar′-CH), 12.20 (d, 3JHH = 7.29 Hz, 2H, meta-Ar′-CH), 19.00 (s, 1H, CH2), 24.03 (s, 1H, CH2), 24.48 (s, 1H, CCH), 46.83 (s, br, 1H, NH), 63.95 (s, 1H, CH2) and 65.09 (s, 1H, CH2).
4.4 Preparation of [Nd(L′)(N″)(NHAr∗)] (4)
THF (80 ml) was added to a cold (−78 °C) mixture of 1 (5.48 g, 7.55 mmol) and 2 (4.05 g, 7.55 mmol) and the resulting mixture was allowed to warm slowly to room temperature with stirring overnight. The solution was filtered, and volatiles removed in vacuo from the orange-brown solution to give a ‘foamy’ pale blue oil. The oil was dissolved in diethyl ether (6 ml) and stored at 5 °C overnight to afford blue crystals of 4·1⅓OEt2; the diethyl ether is weakly held in the crystal lattice as evidenced by the collapse of crystals of 4 to a ‘foamy’ blue oil on prolonged exposure to vacuum and the CHN microanalysis. Yield: 7.48 g, 83%. Anal. Calcd for C58H100N5NdSi3: C, 63.56; H, 9.20; N, 6.39. Found: C, 63.48; H, 9.27; N, 6.22. 1H NMR (benzene-d6, 295 K): δ −19.81 (s, br, 9H, C(CH3)3), −7.71 (s, br, 9H, C(CH3)3), −1.14 (s, 18H, Si(CH3)3), 0.22 (s, 4H, ortho-CH(CH3)2), 0.97 (s, br, 12H, para-CH(CH3)2), 0.99 (s, 9H, Si(CH3)3), 2.35 (s, 2H, para-CH(CH3)2), 3.40 (s, br, 24H, ortho-CH(CH3)2), 7.12 (s, 4H, meta-Tripp-CH), 8.87 (s, 1H, para-Ar-CH), 11.89 (s, 2H, meta-Ar-CH), 16.56 (s, br, 2H, CH2), 25.63 (s, br, 1H, CCH), 47.10 (s, br, 1H, NH) and 53.57 (s, br, 2H, CH2).
4.5 General procedure for reaction of 3 or 4 with group 1 alkyls
A solution of diethyl ether (20 ml) was added to a cold (−78 °C) mixture of 3 or 4 (1.00 mmol) and solid [Me3SiCH2M] or [MeM] (M = Li or K) (1.00 mmol). The mixture was allowed to slowly warm to room temperature with stirring overnight. Only [Ar∗N(H)M] (M = Li or K) could be isolated from reaction mixtures, as confirmed by comparison of their NMR spectra with authentic samples and by a unit cell determination of the latter crystallised as 2(OEt2)2.
4.6 Reaction of 4 with ButNP{NP(NMe2)3}3
A solution of ButNP{NP(NMe2)3}3 (1.32 g, 2.08 mmol) in benzene (10 ml) was added dropwise to a solution of 4 (2.27 g, 2.08 mmol) in benzene (10 ml) to give a green-red solution. The mixture was stirred overnight, then volatiles were removed in vacuo. Crude 31P{1H} NMR (benzene-d6, 295 K): δ 5.90 (d, 2JPP = 17.98 Hz) and −24.28 (q, 2JPP = 17.98 Hz). Following exhaustive attempts at recrystallisation, a portion of the sample was hydrolysed with D2O and a 2H NMR spectrum (benzene, 295 K) was recorded, which exhibited broad resonances at δ 1.82 (ortho-CH(CH2D)(CH3)), 3.14 (para-CH(CH2D)(CH3)), 4.16 (ortho- and para-CD(CH3)2) and 6.91 (para-Ar-CD).
4.7 X-ray crystallography
Crystal data for compounds 2(OEt2)2, 3, and 4·1⅓OEt2 are given in Table 2. Crystals were examined on a Bruker AXS 1000 or Apex CCD area detector diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Intensities were integrated from a sphere of data recorded on narrow (0.3°) frames by ω rotation. Cell parameters were refined from the observed positions of all strong reflections in each data set. Semi-empirical absorption corrections were applied, based on symmetry-equivalent and repeat reflections. The structures were solved by direct methods and were refined by least-squares methods on all unique F2 values, with anisotropic displacement parameters, and with constrained riding hydrogen geometries; U(H) was set at 1.2 (1.5 for methyl groups) times Ueq of the parent atom. The largest features in final difference syntheses were close to heavy atoms. A highly disordered diethyl ether solvent molecule of crystallisation in 3 could not be modelled and was treated with the Platon SQUEEZE procedure [19]. Programs were Bruker AXS SMART (control) and SAINT (integration) [20], and SHELXTL for structure solution, refinement, and molecular graphics [21]. The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC# 658923–658925) and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number).
Crystal data for 2(OEt2)2, 3, and 4·1⅓OEt2
| 2(OEt2)2 | 3 | 4·1⅓OEt2 | |
| Formula | C44H70KNO2 | C34H68N5NdSi3 | C63.33H113.33N5NdO1.33Si3 |
| Formula weight | 684.11 | 775.44 | 1194.77 |
| Crystal size, mm | 0.22 × 0.27 × 0.32 | 0.31 × 0.33 × 0.38 | 0.37 × 0.44 × 0.48 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P– | P– | P– |
| a, Å | 9.741(2) | 12.4161(8) | 16.7142(9) |
| b, Å | 12.294(3) | 12.7123(8) | 26.6355(14) |
| c, Å | 18.274(4) | 15.7210(10) | 27.1609(15) |
| α, ° | 99.954(3) | 109.104(2) | 65.480(2) |
| β, ° | 93.269(3) | 99.689(2) | 74.110(2) |
| γ, ° | 90.137(3) | 108.166(2) | 89.535(2) |
| V, Å3 | 2151.7(8) | 2124.4(2) | 10,503.7(10) |
| Z | 2 | 2 | 6 |
| ρcalcd, g cm−3 | 1.056 | 1.212 | 1.133 |
| μ, mm−1 | 0.157 | 1.333 | 0.832 |
| No. of reflections measured | 14,669 | 14,864 | 97,847 |
| No. of unique reflections, Rint | 7528, 0.0572 | 7429, 0.0238 | 36,779, 0.0695 |
| No. of reflections F2 > 2σ(F2) | 3862 | 6620 | 4207 |
| Transmission coefficient range | 0.703–0.970 | 0.631–0.683 | 0.891–0.919 |
| R, Rwa (F2 > 2σ) | 0.0585, 0.1309 | 0.0417, 0.1042 | 0.0659, 0.1377 |
| R, Rwa (all data) | 0.1412, 0.1655 | 0.0481, 0.1086 | 0.1234, 0.1544 |
| Sa | 0.982 | 1.076 | 0.925 |
| Parameters | 449 | 410 | 1945 |
| Max., min. difference map, e Å−3 | 0.304, −0.372 | 4.111, −1.202 | 2.463, −2.389 |
a Conventional R = Σ‖Fo| − |Fc‖/Σ|Fo|; Rw = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2; S = [Σw(Fo2 − Fc2)2/(No. data − No. parameters)]1/2 for all data.
Acknowledgements
We thank the UK EPSRC, The Leverhulme Foundation, and the Royal Society for funding.