

1 Introduction
The f-element metallocenes M(η8–C8H8)2 (M = Ln, An) have received considerable attention, both experimental and theoretical, since the original synthesis of uranocene, UCOT2 (COT = C8H8), in 1968 by Streitwieser and Müller-Westerhof [1]. Subsequently, Th, (substituted) Pa, Np and Pu analogues of uranocene have been synthesized [2–4], along with the lanthanide analogue, cerocene, CeCOT2 [5,6]. Compounds incorporating the other example of an aromatic eight-membered ring system, pentalene (Pn = C8H6), have, however, received comparatively little attention, “in part due to synthetic difficulties” [7]. Compounds with substituted pentalene ligands have been reported for Th and U [7,8] and Ce [9,10], but computational investigations have not been performed to the same theoretical level as for the COT-containing systems. This is of particular importance for the cerium compounds, for whilst there is little doubt that the actinocenes synthesized to date contain a tetravalent actinide ion, opinion with regard to cerocene is divided. We have discussed this is in detail in recent publications [11,12], and so only briefly summarise here.
Streitwieser et al. [6] and Raymond et al. [13,14] have argued strongly for a +4 oxidation state in cerocene. This interpretation, however, implies the presence of two highly reducing COT2− rings in close proximity to the strongly oxidizing Ce4+ cation. An alternative to this (somewhat unlikely) formulation was proposed by Neumann and Fulde [15] and supported by sophisticated ab initio calculations on cerocene and the actinide analogue [16–19] by Dolg et al. They concluded that the ground states of both systems is 1A1g, and that thorocene is clearly a Th(IV) compound. Cerocene, however, exhibits strong multiconfigurational character, with the dominant (c. 80%) configuration involving the occupation of a single metal-localised valence 4fδ level, with a corresponding hole in the highest occupied ring-based π orbitals of e2u symmetry. Thus CeCOT2 is best described as a Ce(III) compound containing two COT rings each carrying a formal 1.5– charge. This view has received (strong) experimental support via XANES measurements [20,21], and optical spectroscopic properties of substituted cerocenes have been shown to be more consistent with a bonding model based on Ce(III) than Ce(IV) [22].
Recent attention has turned to the pentalene (Pn, η8–C8H6) analogue of CeCOT2. Balazs et al., after detailed spectroscopic, magnetic and density functional theory (DFT) [23,24] studies, conclude that for CePn2, “a classification of a formal oxidation state IV is recommended” [9]. Interestingly, some of the same authors have also studied the permethylated analogue, CePn*2 (Pn* = η8–C8Me6) using similar methodology, and concluded that “the [XANES spectroscopy] gives strong evidence for a formal valency close to Ce(III) in this molecule” [10]. The continuing division in opinion regarding the formal oxidation state of these cerium compounds has led us to perform a CASPT2 study of CePn2. We have recently carried out similar studies of cerocene, thorocene and protactinocene [11], and find that in the case of cerocene strong hybridization between the metal fδ and ligand π(e2u) levels can lead to an ambiguous interpretation of the degree of ‘f1’ character in the ground state wavefunction in terms of configurational admixture. We instead use the natural orbital occupations [25] (NOOs) along with the corresponding natural orbitals (NOs) in order to interpret the multiconfigurational nature of the cerocene ground state and obtain a value for the total f occupation, nf. We find active space NOOs of 1.751 and 0.216 for the ‘ligand’ and ‘metal’ NOs of e2u symmetry, respectively. Such deviation from integer occupation is an indicator of strong multiconfigurational character [26], yet under the assumption of localized orbitals, in which there is no hybridization between the ligand and metal e2u levels, yields an nf value of only = 0.43. The natural orbitals, however, show approximately 20% metal character in the ‘ligand’ level and vice versa. Taking this hybridization into account allows us to calculate a revised value of nf = 0.90 ± 0.04, in excellent agreement with that found experimentally [21], yet without near-integer occupation of a localised fδ level.
In this contribution, we apply the same high-level methodology we have successfully employed on cerocene to establish if a similar description of the ground state is appropriate for CePn2.
2 Computational details
The Gaussian 03 code [27] was used to investigate CePn2 at the DFT level, using the TPSS exchange-correlation functional [28]. All results from DFT calculations were obtained using the small core SDD pseudopotential [29] and associated basis set for cerium, and the cc-pVDZ set [30] for carbon and hydrogen.
Complete active space self-consistent field (CASSCF) [31] calculations were performed using the MOLCAS 6.4 code [32], and dynamic correlation was accounted for using complete active space second order perturbation theory (CASPT2) [33]. Details of the active spaces employed for both the D2h and D2d isomers are given in the main text (and Tables 2 and 4). For the D2h isomer, the number of orbitals kept frozen in each irrep during the CASPT2 procedure were: ag: 11, b1g: three, b2g: four, b3g: four, au: one, b1u: six, b2u: five, b3u: five. For the D2d isomer which, as discussed in the main text, was calculated in the C2v point group, the number of orbitals kept frozen in each irrep during the CASPT2 procedure were: a1: 17, a2: four, b1: nine, b2: nine.
The (12,16) active space used in the CASPT2 calculations on the D2d conformer of CePn2, defined in terms of the occupied and virtual IVO-RHF input orbitals within the C2v point group.
| A1 | A2 | B1 | B2 | |
| Inactive (occupied) | 31 | 10 | 18 | 18 |
| Active (occupied) | 2 | 2 | 1 | 1 |
| Active (virtual) | 2 | 2 | 3 | 3 |
The (12,16) active space used in the CASPT2 calculations on the D2h conformer of CePn2, defined in terms of the occupied and virtual IVO-RHF input orbitals within the D2h point group.
| Ag | B1g | B2g | B3g | Au | B1u | B2u | B3u | |
| Inactive (occupied) | 18 | 6 | 7 | 9 | 4 | 13 | 11 | 9 |
| Active (occupied) | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 |
| Active (virtual) | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 |
Scalar relativistic effects were included using the second order Douglas-Kroll Hamiltonian [34], whereas spin-orbit coupling, which we have previously shown to be very weak in CeCOT2 [11], was neglected. All electron ANO basis sets were used for these calculations. For cerium, the ANO set was constructed by Roos [35], contracted as (25s22p15d11f)/[9s8p5d4f], and is of approximate cc-VQZ quality. The basis sets were of cc-pVDZ quality for C and H. This basis set combination has been shown [11] to produce results of comparable accuracy to those obtained using sets of cc-pVQZ quality for Ce and of cc-pVTZ quality for C and H. To aid comparison with CASPT2 results, MOLCAS 6.4 was also used to perform DFT calculations using the B3LYP exchange-correlation functional [36] and the same ANO basis in order to consider the differences in f-orbital occupation when using the CASPT2 and DFT methods.
3 Results and discussion
3.1 DFT study
Crystalline
Structural parameters for calculated CePn2 and synthesized substituted compounds.
| Parameter | Calculated | Experimental | |||
| Ref [9] | Ref [10] | Present Work | |||
| Ce-C (br) | 2.51 | - | 2.50 | 2.49–2.52 | - |
| Ce-C (α) | 2.74 | - | 2.72 | 2.73–2.76 | - |
| Ce-C (β) | 2.88 | - | 2.86 | 2.89–2.93 | - |
| 2.72 | 2.67 | 2.70 | 2.73 | 2.68 | |
| Ligand fold ∠ | 24.4 | 24.0 | 25.6 | 23.0 | 24.7 |
| Twist ∠ | 87.72 | 90a | 90a (90.04b) | 85.0 | 79.8 |
a Optimization performed under constraints of D2d symmetry.
b Optimised twist angle, differing from 90° due to convergence criteria.
We find the DFT calculated electronic structure of the D2d conformer to be as reported previously [9,10]. The ground state is 1A1, with the a2 symmetry HOMO and b1 HOMO-1 displaying significant fδ and dδ character respectively. The LUMO is of e-symmetry and is an almost completely localized f-level. The ground state of the higher energy D2h conformer is analogous: the state symmetry is 1Ag, with the au symmetry HOMO and b3g HOMO-1 displaying significant fδ and dδ character respectively. The lowest lying unoccupied levels are localized f-levels. We defer consideration of the degree of f-character in the ground state until Section 3.2.3, where comparison is made between DFT and CASPT2 calculations using identical basis sets.
3.2 CASPT2 study
CASPT2 calculations were performed on both the D2h and D2d conformers at the TPSS-optimised geometries. In the case of the D2d conformer, computational restrictions enforced in MOLCAS required calculations to be performed in the C2v point group (an Abelian subgroup of D2d). Such a reduction in symmetry can, in principle, allow mixing of orbitals that would be forbidden in the full point group, and such mixing was monitored and eliminated during the calculations. Throughout the remainder of this study we will refer to orbitals and states in terms of their C2v symmetry labels and, where appropriate, will give the corresponding D2d labels in parentheses. In the case of the D2h conformer, the full point-group could be employed in the calculations, and so no complication arises.
Although CePn2 can be considered to be analogous to CeCOT2, we have used a larger active space in the present calculations than was used in our CeCOT2 study. In the previous study, an active space explicitly correlating eight electrons in 14 orbitals, denoted CASPT2(8,14), was used. This active space included the eight electrons occupying the highest energy π2 ligand orbitals spanning the doubly degenerate e2u and e2g irreps of the D8h point group, and was sufficient since the energy gap between these orbitals and those below them (π1 orbitals of e1g and e1u symmetry) was large. The lower point groups employed here strongly affect the relative energies of these orbitals, resulting in a significant reduction in the π2-π1 energy gap when the CePn2 complexes are considered. This effect is shown graphically for the tetra-anionic ligand systems in Fig. 1, where the π2-π1 energy gap reduces from 2.97 eV (CeCOT2) to 1.06 eV (CePn2, D2h) and 1.17 eV(CePn2, D2d), and led us to expand the active space in the present calculations to include the higher lying of the π1 levels. We therefore present CASPT2(12,16) calculations, using our adopted notation. It should be noted that this active space allows for occupation of any of the twelve 4f and 5d levels.

Energy levels and molecular orbitals of COT24− and the D2h and D2d conformers of Pn24−. The blue and red levels indicate the lowest lying π2 and highest lying π1 level respectively.
3.2.1 The D2d conformer
The two energetically lowest singlet and triplet states of each irrep of C2v (the working point-group) were obtained at the CASPT2 level, using the active space defined in Table 2. Input orbitals were generated from a restricted Hartree-Fock (RHF) [38,39] calculation, with the improved virtual orbital (IVO) method [40] used to produce more compact virtual orbitals.
Our CASPT2 calculations find a 1A1 ground state, in agreement with our DFT results. This state lies 0.87 eV below the doubly degenerate first excited state, of B1/B2 (E) symmetry and singlet multiplicity. We quantify the multiconfigurational character in terms of the natural occupation numbers; NOOs which deviate from integer values by more than 0.1 are indicative of a strongly multiconfigurational system [26]. Table 3 shows the energies and the largest deviation from integer orbital occupancy within the active space for each of the sixteen states we consider here. The ground state is strongly multiconfigurational, with significant (0.383) occupation of the anti-bonding a2 level of dominant fδ character. This occupation is a direct result of the deoccupation of the corresponding bonding a2 level (with a deoccupation of 0.391). This state bears a strong resemblance to that which we have previously reported as the cerocene ground state (as can be seen from the natural orbital diagrams of Fig. 2), with the difference here that the lower symmetry splits the degeneracy of the two fδ orbitals, resulting in the multiconfigurational character being almost exclusively attributable to charge transfer between the bonding and antibonding a2 orbitals with fδ character, as opposed to contributions from both components of the doubly degenerate e2u cerocene orbitals. The largest deviation from integer occupancy in the other active space orbitals is just 0.052, confirming the origin of the multiconfigurational character. By contrast to the CASPT2 ground state, the fifteen other states under consideration here have much lower multiconfigurational character. Again, this is strongly reminiscent of our cerocene results.
Relative energies and maximum deviation from integer natural orbital occupancy (ΔNOO) of the 16 CASPT2(12,16) calculated states of the D2d isomer of CePn2.
| Singlet | Triplet | |||
| Energy (eV) | ΔNOO | Energy (eV) | ΔNOO | |
| A1 | 0.00 | 0.391 | 1.16 | 0.054 |
| 2.34 | 0.086 | 2.24 | 0.106 | |
| A2 | 2.72 | 0.068 | 2.72 | 0.068 |
| 3.19 | 0.072 | 3.21 | 0.073 | |
| B1 | 0.87 | 0.054 | 0.88 | 0.054 |
| 2.01 | 0.086 | 1.94 | 0.086 | |
| B2 | 0.87 | 0.054 | 0.88 | 0.054 |
| 2.01 | 0.086 | 1.94 | 0.086 |

CePn2 ground state natural orbitals obtained from CASSCF(12,16) calculations, and CeCOT2 ground state natural orbitals obtained from a CASSCF(8,14) calculation [11]. Only NOs with signifcantly non-integer occupation are shown.
Experimentally, an “intense and broad absorption” is observed at 2.34 eV in the UV/Vis spectrum [10]. Time-dependent DFT calculations find a transition with oscillator strength f = 0.071 at an energy of 2.85 eV, due to excitation from a largely ligand based orbital with some Ce dδ character into an Ce fδ orbital, with the intensity of the transition arising from the d to f character of the transition [10]. Our CASPT2 calculations also identify this transition, but give better agreement with experiment. We find an oscillator strength of f = 0.345 for the transition, which we calculate to occur at 2.35 eV. Whilst this extremely good agreement is no doubt somewhat fortuitous, it again highlights the fact that multiconfigurational methods are required in order to model systems such as CePn2 to a high level of accuracy.
3.2.2 The D2h conformer
The two energetically lowest singlet and triplet states of each irrep of D2h were obtained at the CASPT2 level, using the active space defined in Table 4. Input orbitals were generated from a restricted Hartree-Fock (RHF) [38,39] calculation, with the improved virtual orbital (IVO) method [40] used to produce more compact virtual orbitals.
As with the G03 DFT calculations we find an 1Ag ground state for the D2h conformer. At the CASPT2 level this state lies 0.24 eV (23.2 kJ/mol) above the D2d ground state. The D2h ground state bears strong similarities to both that of the D2d conformer and cerocene, i.e. a strongly multiconfigurational wavefunction resulting from the strong (0.378) occupation of the antibonding au orbital of dominant fδ character. Again, this occupation results almost exclusively from deoccupation of the corresponding bonding au level (deoccupation of 0.388). The ground state is stable by 0.93 eV relative to the first excited state of 1B2g symmetry. The low lying excited states typically have only small deviations from integer occupancy, as is the case for the D2d conformer (compare Tables 3 and 5). There are, however, several excited states (of B1g, Au and B1u symmetry and both spin multiplicities) which are strongly multiconfigurational. These multiconfigurational excited states exhibit the strong occupation of the antibonding au orbital of dominant fδ character, in common with the ground state, but also correspond to ligand to metal charge transfer excitations. This is in contrast to cerocene, where all excited states were found to be well described by a single-configuration wavefunction.
Relative energies and maximum deviation from integer natural orbital occupancy (ΔNOO) of the 32 CASPT2(12,16) calculated states of the D2h isomer of CePn2.
| Singlet | Triplet | |||
| Energy (eV) | ΔNOO | Energy (eV) | ΔNOO | |
| Ag | 0 | 0.388 | 1.21 | 0.060 |
| 2.36 | 0.194 | 3.96 | 0.710 | |
| B1g | 2.21 | 0.054 | 2.22 | 0.055 |
| 2.78 | 0.578 | 2.55 | 0.561 | |
| B2g | 0.93 | 0.052 | 0.97 | 0.050 |
| 2.08 | 0.066 | 2.11 | 0.061 | |
| B3g | 0.96 | 0.054 | 0.98 | 0.052 |
| 2.09 | 0.066 | 2.10 | 0.054 | |
| Au | 1.60 | 0.241 | 1.60 | 0.231 |
| 3.38 | 0.060 | 3.41 | 0.083 | |
| B1u | 2.45 | 0.087 | 2.30 | 0.109 |
| 2.63 | 0.434 | 2.63 | 0.424 | |
| B2u | 2.18 | 0.076 | 2.06 | 0.085 |
| 4.59 | 0.148 | 3.26 | 0.097 | |
| B3u | 2.13 | 0.092 | 2.01 | 0.093 |
| 3.70 | 0.017 | 3.66 | 0.125 |
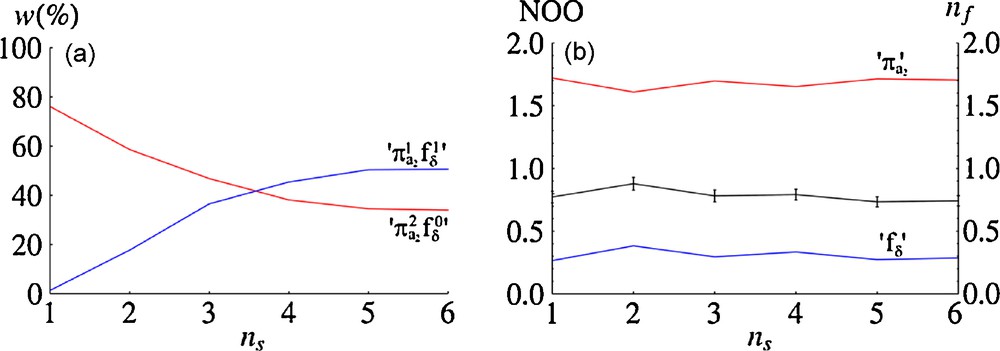
3.2.3 Evaluation of the f-density
Fig. 3a shows the variation in the configurational weights of the
In order to compare this result with that obtained from a single-configuration DFT calculation, MOLCAS was used to generate a set of Kohn-Sham orbitals for CePn2 using the B3LYP functional and the same basis set and molecular geometries as was used for the CASPT2 calculations. The D2d conformer was again found to be the lowest energy structure, and was stable by 0.21 eV (20 kJ/mol) with respect to the D2h conformer. Inspection of the Kohn-Sham orbitals for the D2d conformer reveals the a2 HOMO and LUMO to be almost indistinguishable from the partially-occupied natural orbitals shown in Fig. 2, and nf is calculated to be 0.62 ± 0.01. This value, whilst significant, is substantially lower than that obtained from the natural orbitals, and is a result of the inability of the single-configurational method to represent the partial occupation of the a2 LUMO of dominant fδ character.
4 Summary and conclusions
CePn2 has been shown experimentally [9,10] to bear a striking resemblance to cerocene, for which the debate as to the oxidation state of the cerium ion continues. We have shown computationally that the electronic structure of CePn2 is extremely similar to that of cerocene, and that whilst multiconfigurational methods are required to accurately describe the ground state wavefunction, it is not dominated by a ‘π3fδ1’ configuration. Instead, our analysis via natural orbital occupancies suggests that CePn2, like CeCOT2 [11], is best described as a Ce(IV) system in which the transfer of electron density from ligand to metal through partial occupation of a metal dominated orbital results in a significant metal 4f density. This conclusion is not in conflict with the XANES data of [9,10]: in fact, our calculated value for the Ce ion f-density is in good agreement with that found experimentally. However, our analysis suggests that experimental measures of the effective (as opposed to formal) oxidation state cannot necessarily be used to unambiguously define the detailed electronic structure of complexes such as CePn2.
Acknowledgements
We are grateful to the EPSRC for financial support (via grants EP/C533054 and GR/S06233) and for the use of its National Service for Computational Chemistry Software (http://www.nsccs.ac.uk). We also thank UCL for computing resources via the Research Computing “Legion” cluster and associated services.
1 The used of inverted commas is to remind the reader that these orbitals are not localized, and the labels refer to dominant character.
Vous devez vous connecter pour continuer.
S'authentifier