

1 Introduction
Chelating Werner-type ligands have played a very important role in the coordination chemistry of transition metals [1,2]. Employing chelating macrocyclic ligands to Ln and An metal ions has the advantage of stabilizing these large and reactive metal centers through the chelate effect. We have shown that stabilization of U(III) ions can be achieved with the hexadentate tris-aryloxide functionalized triazacyclononane ligand system (RArOH)3tacn (R = t-Bu L1, Ad = L2) and that the U(III) precursors show unusual reactivity towards small molecules. [3,4] The influence of steric pressure from the (RArOH)3tacn ligand system on the reactivity of the trivalent uranium complexes has been well demonstrated. [5,6] Varying the tert-butyl substituents at the ortho position of the aryl rings in the first-generation ligand (t-BuArOH)3tacn (Fig. 1, L1) to adamantyl substituents in the second-generation ligand (AdArOH)3tacn (Fig. 1, L2) significantly alters the steric environment. As previously reported, the U(III) complex [(L1)U] (Fig. 2, left) possesses a more open and shallow axial reactive cavity compared to [(L2)U] (Fig. 2, right). As a result, the coordinated axial ligand is insufficiently protected by L1 in [(L1)U] leading to the splitting of CO2 to form [(L1)U(μ-O)] [7] whereas L2 can promote stabilization of a U(IV) species with a radical anionic CO2− ligand [(L2)U(CO2−)] (Scheme 1) [8].

Chelating ligands for uranium coordination chemistry.
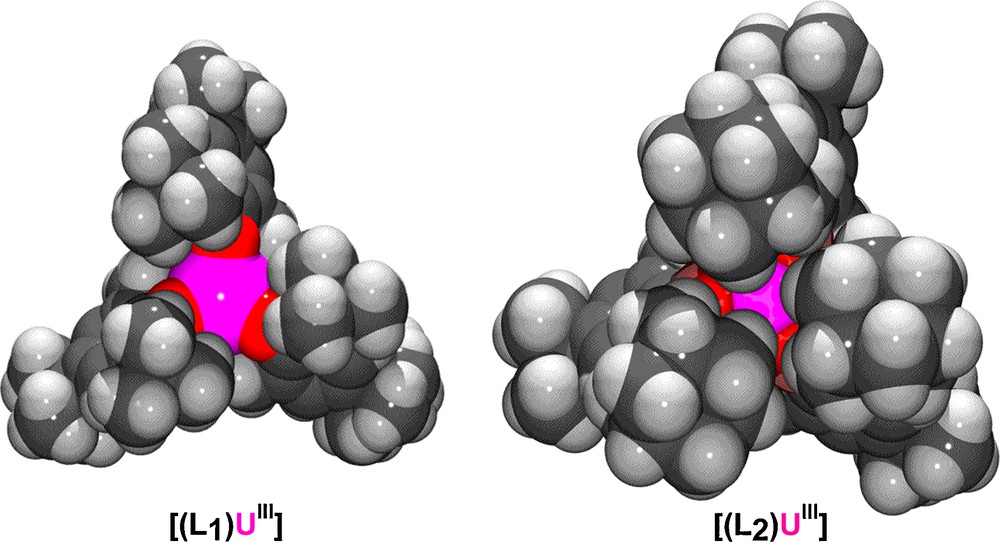
Space-filling representations of U(III) precursors of previously reported ligand systems L1 and L2.
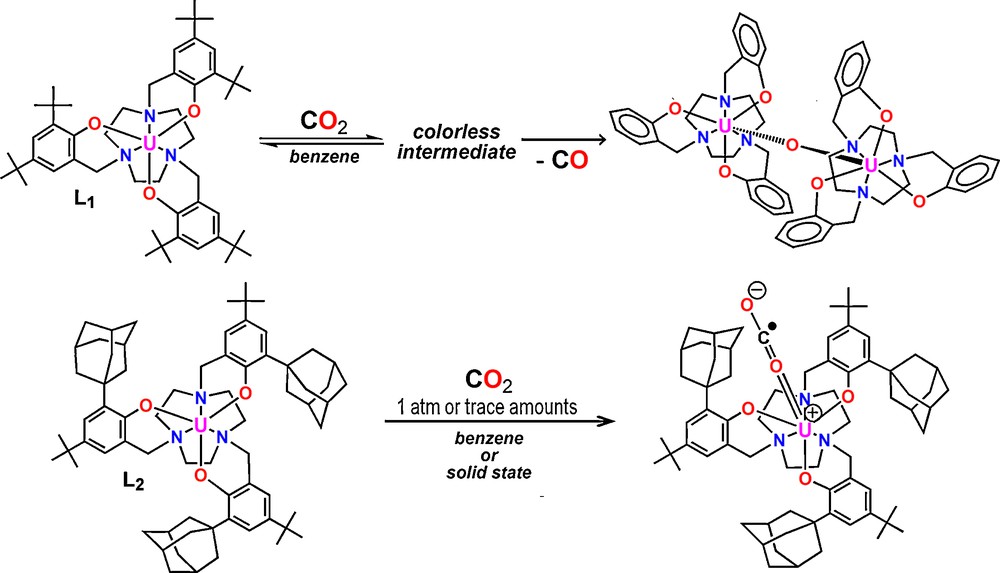
Reactivity of (L1)U and (L2)U with CO2.
Herein, we report the synthesis of a third-generation diamantyl functionalized ligand system (DiaArOH)3tacn (Fig. 1, L3), designed to deliver a sterically more demanding environment along with an enhanced hydrophobic reactive pocket around the uranium center. A deep hydrophobic reactive site was envisioned as part of the initial investigations into activation and functionalization of hydrocarbons. Syntheses of the U(III) precursor [(L3)U] (1), U(IV) chlorocomplex [(L3)U(Cl)] (2), and U(V) imido complex [(L3)U(NTMS)] (3) along with XRD and spectroscopic measurements are also presented.
2 Results and discussion
2.1 Synthesis and spectroscopic measurements of 1
Similarly to the syntheses of L1 and L2, [9,10] the diamantyl functionalized ligand L3 is synthesized from a Mannich condensation of triazacyclononane, paraformaldehyde, and 2-diamantyl-4-tert-butylphenol. The U(III) precursor complex [((DiaArO)3tacn)U] (1) is obtained by treating [U(N(SiMe3)2)3] with L3 in 1,2-dimethoxyethane (DME) yielding 1 as a red-brown solid (Scheme 2). Although X-ray diffraction (XRD) quality single crystals could not be obtained of 1, its spectroscopic behavior and reactivity are in accordance with the six-coordinate precursor complex.

Synthesis of complex 1 bearing the new diamantane ligand L3.
For instance, the 1H-NMR spectrum shows 13 resonances, consistent with a molecule possessing C3 symmetry. Variable temperature (VT) magnetization data clearly identifies 1 as a trivalent species exhibiting magnetic moments of 1.26 B.M. and 3.04 B.M. at 5 K and 300 K, respectively (Fig. 3). The temperature dependence of the magnetic moment is similar to those seen in other U(III) precursors. [3] The electronic absorption spectrum exhibits a Laporte-allowed, metal-centered 5f 3 to 5f 26d1 transition in the visible region (460 nm, ɛ = 766 M−1cm−1) typically observed for complexes with U(III) ions (Fig. 3, bottom).
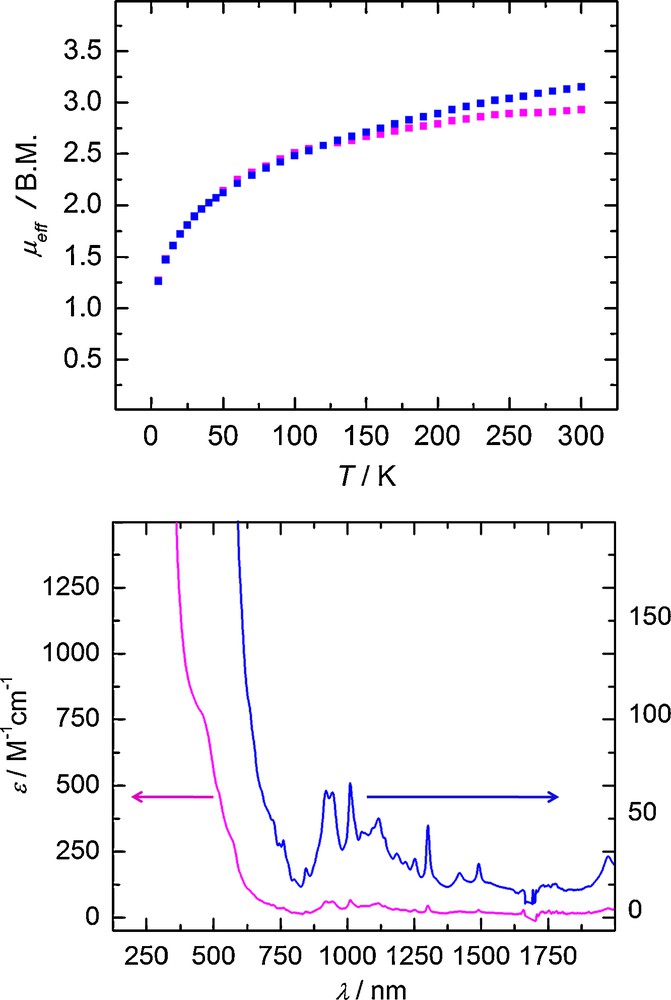
Temperature-dependent SQUID magnetization data of two independently synthesized samples of 1 (at 1 T) plotted as a function of magnetic moment (μeff) vs. temperature (T) (top). The electronic absorption spectra of 1 are recorded in toluene (bottom) at two different concentrations.
The CW X-band EPR spectrum of 1, recorded in frozen toluene solution at 6 K, exhibits an isotropic signal at g = 2.08 (Fig. 4), which is nearly identical to the one recorded for [((RArO)3tacn)UIII] (R = t-Bu, Ad) and similar to the one that was measured for the parent U(III) tris-amido system [U(N(SiMe3)2)3] [10]. A feature clearly visible on the low-field part of the signal could not be simulated with a set of anisotropic g-values.
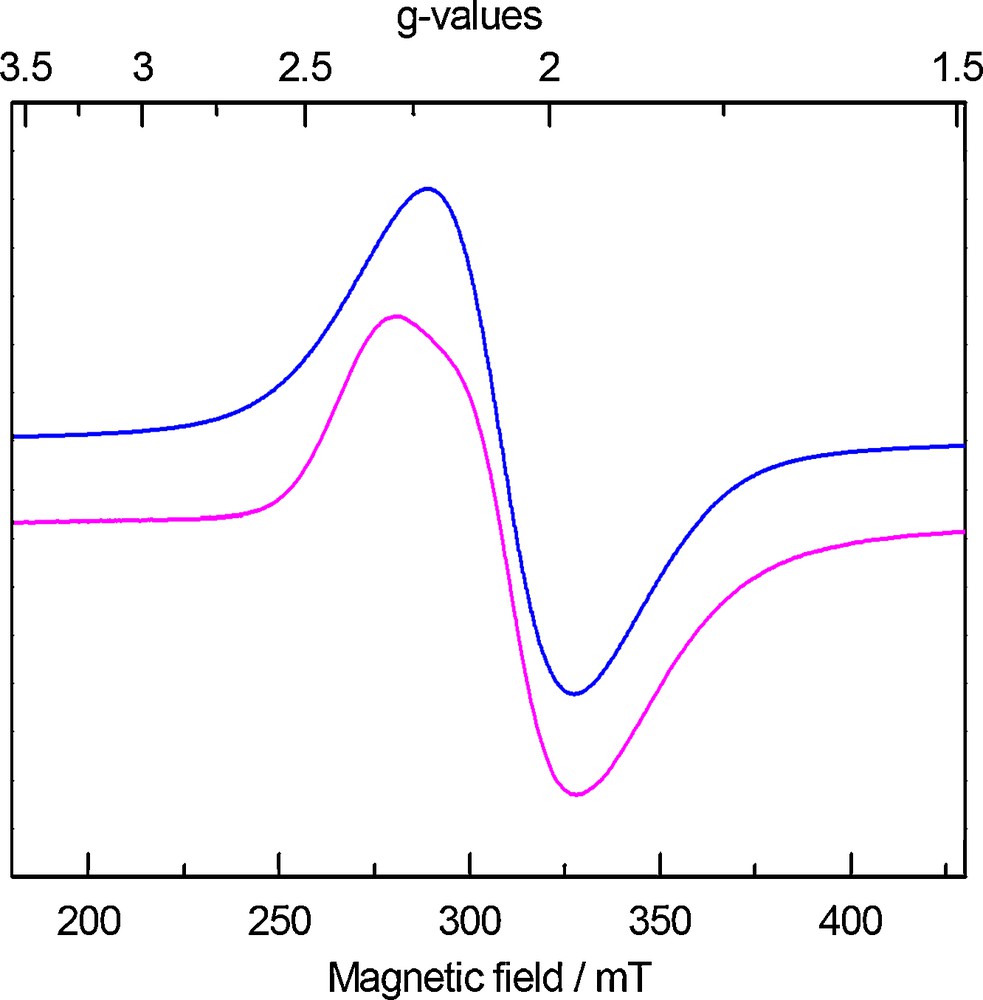
CW X-band EPR spectrum of 1 recorded in frozen toluene solution at T = 6 K. Experimental spectrum (magenta-lower): frequency, 8.9828 GHz; power, 0.20 mW; modulation amplitude, 5 G. Simulation (blue-upper): g = 2.08, WFWHM = 280 G.
Furthermore, the observed reactivity of complex 1 with small molecules is also in accordance with trivalent uranium such as the one-electron activation of methylene chloride to form 2 and two-electron reduction of azidotrimethylsilane to form 3. Synthesis and spectroscopic characterization of both complexes are discussed in detail in the following sections.
2.2 Synthesis and molecular structure of 2
Treatment of a red-brown solution of complex 1 with methylene chloride in DME immediately results in decolorization of the reaction solution to green. The solution was filtered and volatiles were removed giving green solids of [((DiaArO)3tacn)U(Cl)] (2) (Scheme 3). Green crystals of XRD quality were obtained from diffusion of acetonitrile into a solution of 2 in methylene chloride. The molecular structure of 2 reveals a seven-coordinate complex where the chloride ligand is coordinated in the axial position and the uranium center is located 0.26 Å below the plane formed by the three phenolate oxygens (Fig. 5). The average UOavg and UNavg bond distances in complex 2 of 2.174(6) Å and 2.652(8) Å are comparable to those of the corresponding U(IV) chloride complex derived from the adamantyl functionalized ligand [((AdArO)3tacn)U(Cl)] (2.160(6) Å, 2.654(7) Å). [11] The UCl bond distance of 2.691(3) Å also compares well with that of the [((AdArO)3tacn)U(Cl)] complex (2.708(3) Å). [11] A notable feature observed in the molecular structure of 2 is the hydrogen bonding interaction (2.706 Å) between a co-crystallized dichloromethane hydrogen with the coordinated axial chloride (Fig. 5). The observation of hydrogen bonding suggests that non-polar small molecules such as methane could also be coaxed into the reactive pocket of U(V) terminal oxo species, a necessary pathway towards CH activation.
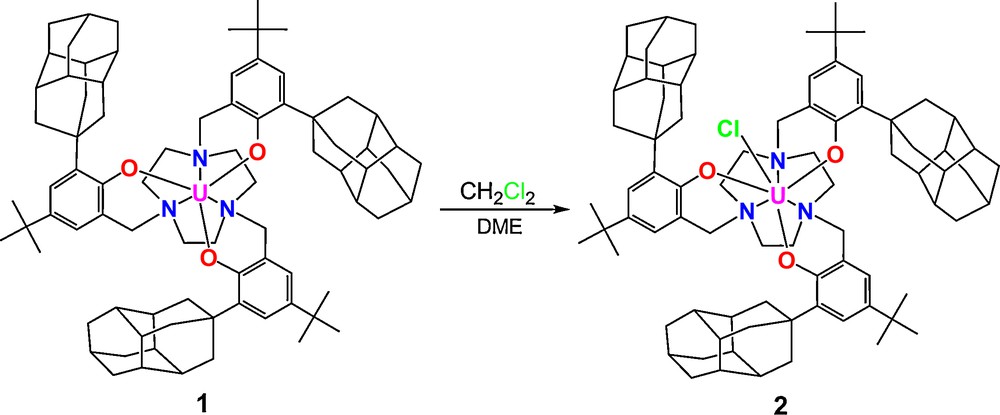
Synthesis of U(IV) chloride complex 2.
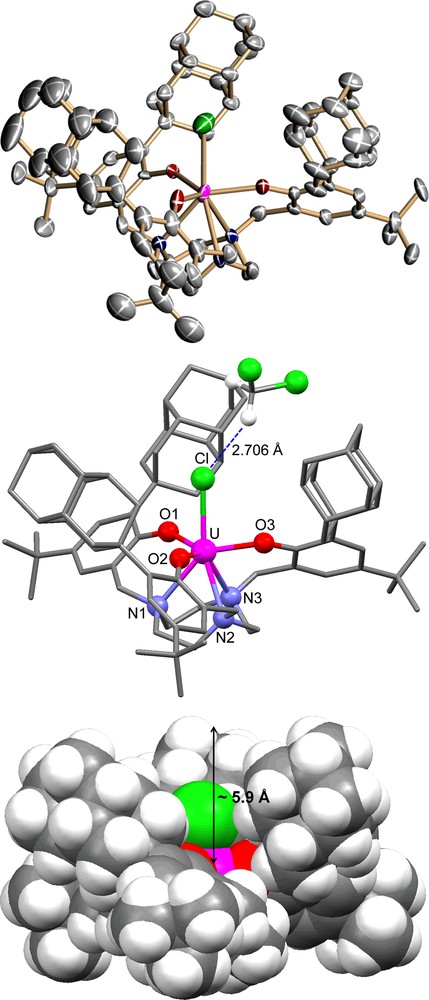
Molecular structure of U(IV) chloro complex 2; the space-filling representation is shown at bottom.
As expected, the three diamantyl substituents form a deep cavity in [((DiaArO)3tacn)U(Cl)] (2) of approximately 5.9 Å, significantly deeper than that of [((AdArO)3tacn)U(Cl)], where the adamantyl groups create a cavity depth of only 4.7 Å. The depth of the hydrophobic pocket can be expected to be even greater in the coordinatively unsaturated six-coordinate complex 1. Presumably, the strongly bound axial chloride ligand in complex 2 bends the diamantyl substituents away from the uranium center.
2.3 Magnetism and electronic absorption of 2
Variable temperature SQUID magnetization measurements are needed to identify U(III) f 3 and U(IV) f 2 complexes since their magnetic moment values at room temperature are nearly indistinguishable (calc.: U(III): 3.69 μB; U(IV): 3.58 μB). Due to a non-magnetic singlet ground state at low temperatures, U(IV) f 2 complexes exhibit temperature independent paramagnetism (TIP), resulting in low magnetic moments at low temperatures, typically ranging from 0.4–0.8 μB at 2 K. The VT SQUID plot of 2 clearly reveals a U(IV) species, where temperature dependent magnetic moments of 3.13 μB and 0.69 μB are observed at 300 K and 2 K, respectively (Fig. 6, top). By contrast, in U(III) f 3 complex 1, a steady decline from 3.04 to 1.26 μB is observed as the temperature decreases from 300 K to 5 K.
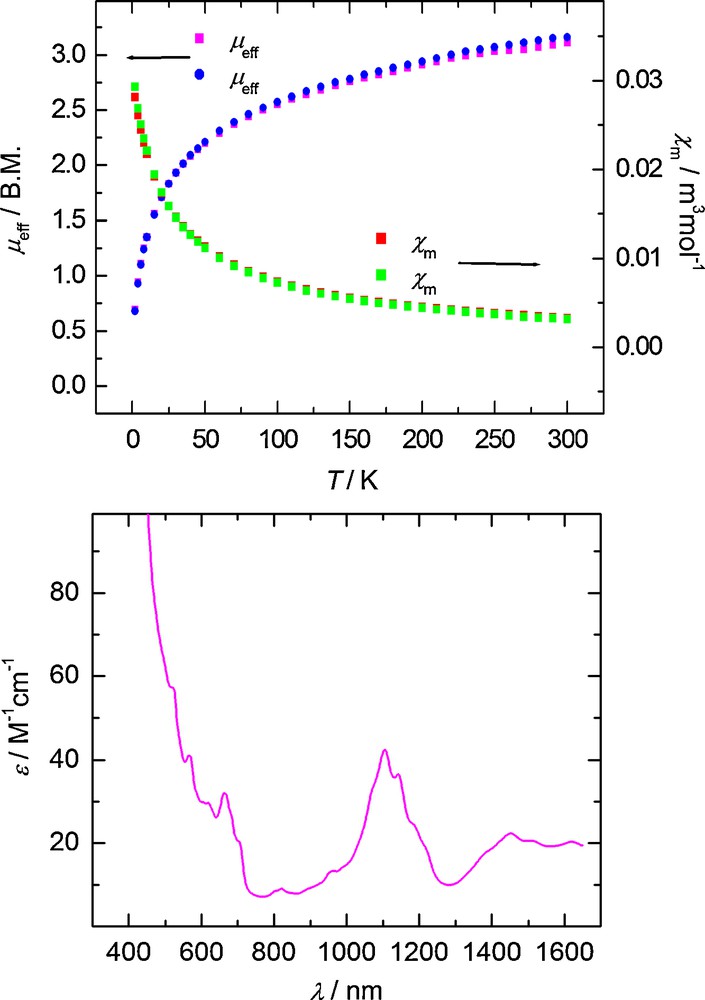
Temperature-dependent SQUID magnetization data (at 1 T) plotted as a function of magnetic moment (μeff) vs. temperature (T) (top, blue and magenta) and molar susceptibility (χm) vs. temperature (T) (top, green and red). The electronic absorption spectrum of 2 recorded in toluene is shown at bottom.
Two distinctive features observed in the electronic absorption spectrum further support that complex 2 is a U(IV) species. Upon oxidation of the uranium center, the color-giving f–d absorption band observed in the visible part of the spectrum found for the U(III) f 3 complex 1 shifts to the UV region and is no longer visible in the spectrum of 2, rendering the U(IV) species of this ligand system pale-colored. Only a series of weak absorption bands (ɛ ≈ 20–50 M−1cm−1) spanning the region of ca. 600–1600 nm are observed (Fig. 6, bottom). These bands arise from Laporte-forbidden f–f transitions and are characteristic of complexes containing tetravalent uranium centers [12].
2.4 Synthesis and molecular structure of 3
Precursor complex 1 undergoes two-electron redox chemistry when treated with azidotrimethylsilane to form a U(V) imido complex [((DiaArO)3tacn)U(NTMS)] (3) and dinitrogen (Scheme 4). Brown single crystals suitable for XRD analysis were obtained from a saturated solution of 3 in hexane. The molecular structure features a seven-coordinate species, where the trimethylsilylimide ligand is bound at the axial position and the uranium center is nearly coplanar with the three phenolate oxygens (0.066 Å below O3 plane) (Fig. 7).
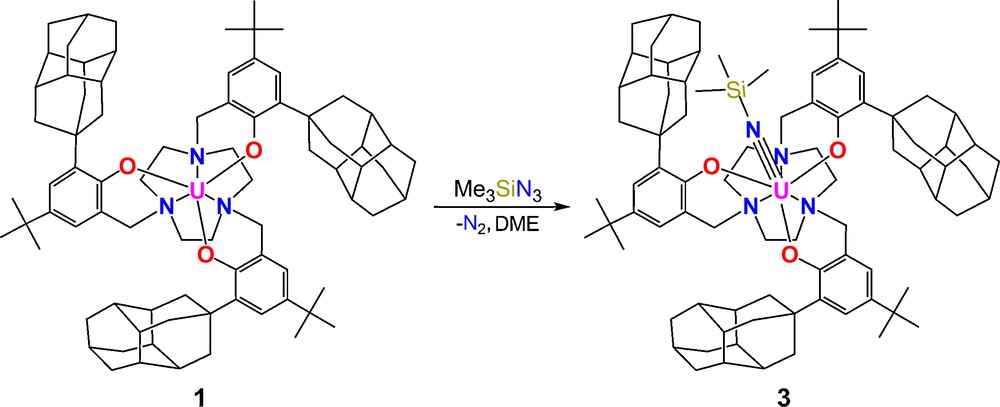
Synthesis of U(V) imide complex 3.
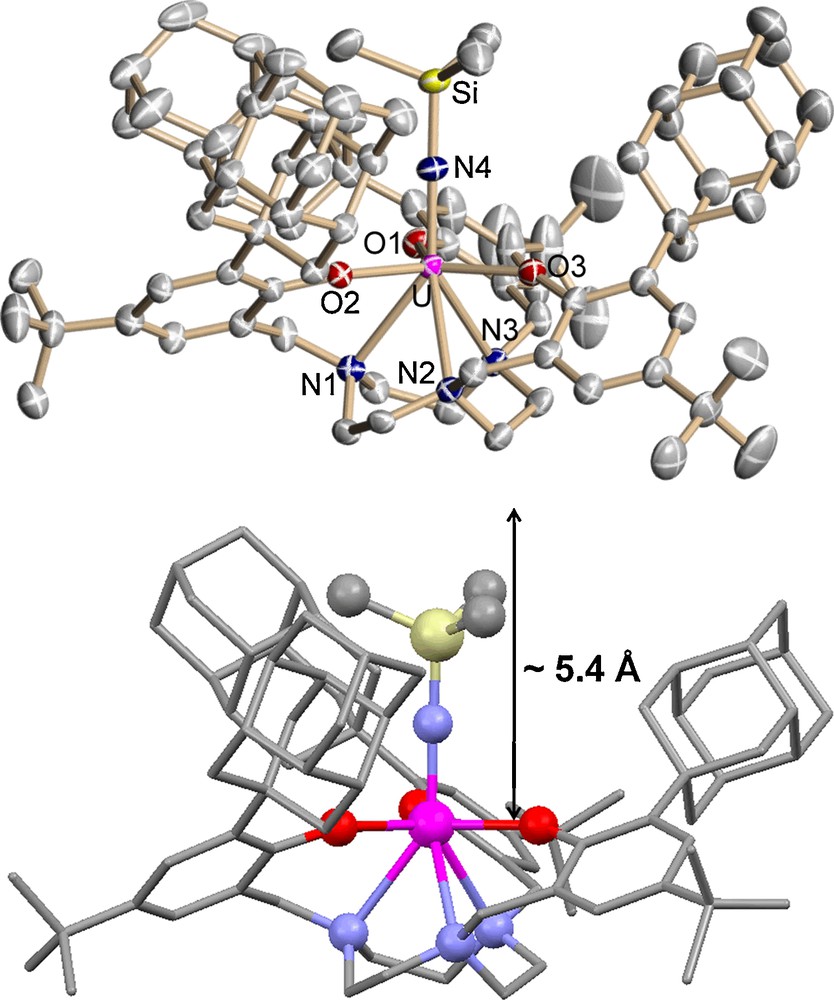
Molecular structure of U(V) imido complex 3. Thermal ellipsoids are at 50% probability.
The average UOavg and UNavg bond lengths of 2.191(3) Å and 2.700(3) Å in complex 3 compare well with those of the previously published U(V) imido complex [((AdArO)3tacn)U(NTMS)] (2.211(2) Å and 2.696(2) Å). [11] Remarkably, the UN4 bond distance of 1.935(3) Å is significantly shorter in complex 3 compared to the unprecedentedly long uranium imido bond of 2.122(2) Å in [((AdArO)3tacn)U(NTMS)] and even shorter than that of [((t-BuArO)3tacn)U(NTMS)] (1.985(5) Å).[11] The length of the UNTMS is an indication of the strength and covalency of the uranium imido bond and is reflected by the UNSi angle. A more linear UNSi angle allows for better orbital overlap of the uranium with the imide nitrogen resulting in a shorter bond length. The UNSi angle in 3 is nearly linear, measuring 177.5(2)° compared to that of [((AdArO)3tacn)U(NTMS)] (162.6(1)°), [11] accounting for a significantly shorter UNimido bond distance and a small displacement of the uranium center below the tris-aryloxide plane (0.066 Å). This latter U out-of-plane parameter shift is an exceedingly sensitive measure of the strength of the UX bond in these complexes. Concomitant with a short U≡N bond, the SiN4 bond length in complex 3 is 1.722(3) Å, considerably longer than that observed for the analogous [((AdArO)3tacn)U(NTMS)] (1.623(2) Å) [11]. In order to obtain an elusive and highly desirable uranium terminal nitride species, cleavage of the SiN is required and hence, a significantly weakened SiN bond is desirable. Strategies to accomplish such a synthesis are currently being undertaken. As observed for complex 2, the pocket in complex 3 of ∼ 5.4 Å is much deeper than that of [((AdArO)3tacn)U(NTMS)] (4.6 Å). The shallower cavity in complex 3 compared to complex 2 illustrates that the more the uranium center is bound to a stronger ligand (TMSN2− > Cl−), the more the diamantyl groups are driven away from the uranium center. The difference in pocket depth (Δdepth ≈ 0.5 Å) in the complexes of the diamantyl functionalized ligand is significantly larger than in the complexes supported by the adamantyl functionalized ligand (Δdepth ≈ 0.1 Å). This suggests that although the diamantyl substituents are more sterically cumbersome, the ligand system is more flexible and accommodating than the adamantyl derivatized ligand system.
2.5 Magnetism and electronic absorption of 3
Variable temperature SQUID magnetization data can be used to clearly differentiate U(V) f 1 complexes from the data of their f 2 and f 3 counterparts. Based on the LS coupling scheme, the calculated μeff value for a U(V) f 1 complex at room temperature is 2.54 μB. [13] The VT SQUID magnetization data for complex 3 shows a temperature dependent magnetic moment ranging from 2.55 μB to 1.26 μB in the temperature range of 300 K to 5 K, identifying 3 as a U(V) complex (Fig. 8, top).
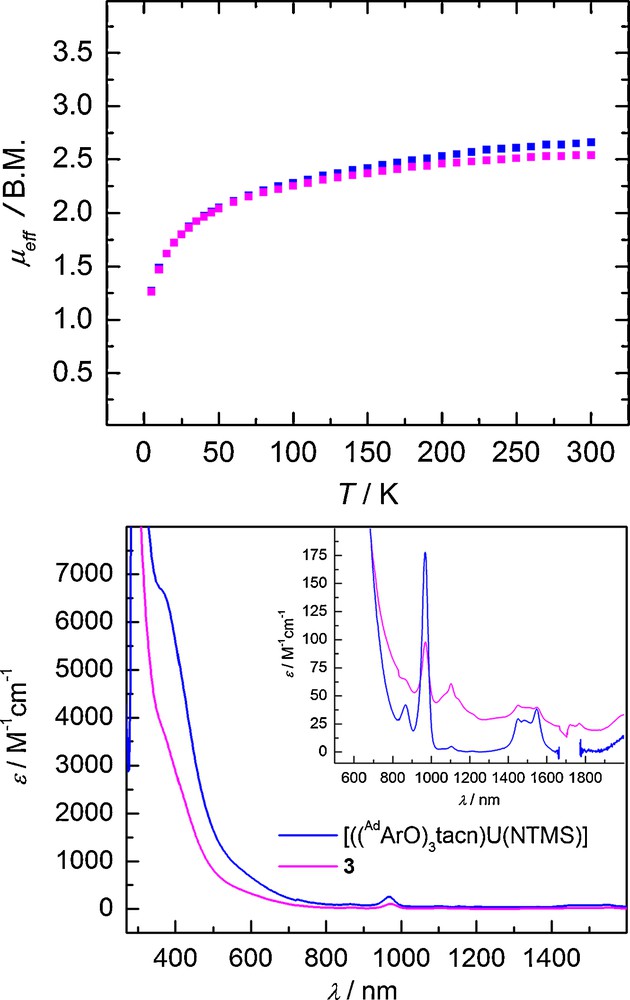
Temperature-dependent SQUID magnetization data (at 1 T) plotted as a function of magnetic moment (μeff) vs. temperature (T) (top) and electronic absorption spectrum of 3 (magenta) recorded in toluene (bottom) and comparison to [((AdArO)3tacn)U(NTMS)] (blue).
From the ligand field theory for U(V) complexes, the 2F manifold of a 5 f electron will be split by the 5 f spin-orbit interaction (approx. 2200 cm−1 for the free ion) into two J multiplets, J = 5/2 and 7/2. In C3v symmetry, the J = 5/2 ground state splits into three magnetic doublets, two EPR active μ = ± 1/2 and one EPR inactive μ = ± 3/2 state, where μ is the crystal ground state number. [14] Despite their f 1 electron configuration, the U(V) imido complex 3, [((DiaArO)3tacn)U(NTMS)], and all other reported U(V) imido complexes, are found to be EPR-silent. This observation suggests that the ground state crystal field of 3 must be μ = ± 3/2. Interestingly, the corresponding U(V) oxo complexes [((RArO)3tacn)U(O)] (R = t-Bu, Ad) are EPR active. These species show EPR spectra of axial symmetry; thus, confirming their μ = ± 1/2 ground state. Preliminary data of a U(V) oxo species, [((DiaArO)3tacn)U(O)], of the presented diamantane derivatized system are in agreement with this observation; the details of this species will be reported later.
Additionally, the electronic absorption spectrum of 3 displays absorption bands characteristic of U(V) imido complexes. The intense brown-orange color of 3 arises from intense LMCT transitions in the visible region ranging from ∼ 800 to 350 nm. Weaker Laporte-forbidden f–f transitions spanning from 850 nm to 2000 nm (ɛ ≈ 25–100 M−1cm−1) are also observed (Fig. 8, bottom). The absorption pattern of 3 is nearly identical to that of the corresponding U(V) imido complex bearing the adamantyl functionalized ligand [((AdArO)3tacn)U(NTMS)] (Fig. 8, bottom).
3 Conclusion
In our continued investigation of the effect of ligand environment on the reactivity of uranium complexes, we have synthesized a new ligand system, (DiaArO)3tacn3−, bearing sterically demanding diamantyl functionalized tris-phenolate pendent arms on a triazacylononane anchor. The initial reactivity studies are promising in that they show different results from the known systems [(L1)U] and [(L2)U]. In particular, the linear binding of the trimethylsilylimide ligand in [((DiaArO)3tacn)U≡N-SiMe3] results in the shortest UN imide bond observed. This raises hope for a greater potential for formation of a uranium terminal nitride species through cleavage of a significantly weakened SiN bond. In addition, the cavity depth is greatly increased as intended. The deeper hydrophobic pocket could potentially provide a site for alkane activation and functionalization such as monooxygenation of methane. These studies are currently underway.
4 Experimental section
4.1 General methods
All experiments were performed under dry nitrogen atmosphere using standard Schlenk techniques or an MBraun inert-gas glovebox. Solvents were purified using a two-column solid-state purification system (Glasscontour System, Irvine, CA) and transferred to the glovebox without exposure to air. NMR solvents were obtained from Cambridge Isotope Laboratories, degassed, and stored over activated molecular sieves prior to use.
4.2 Spectroscopic methods
Magnetization data of crystalline powdered samples (20 – 30 mg) were recorded with a SQUID magnetometer (Quantum Design) at 10 kOe (5 – 300 K for 1 and 3) and (2 – 300 K for 2). Values of the magnetic susceptibility were corrected for the underlying diamagnetic increment (χdia = -878.98 × 10−6 cm3 mol−1 (1), −888.08 × 10−6 cm3 mol−1 (2), −906.48 × 10−6 cm3 mol−1 (3)) by using tabulated Pascal constants and the effect of the blank sample holders (gelatin capsule/straw). Samples used for magnetization measurement were recrystallized multiple times and checked for chemical composition and purity by elemental analysis (C, H, and N) and 1H NMR spectroscopy. Data reproducibility was also carefully checked on independently synthesized samples.
The EPR measurement was performed in a quartz tube with a J. Young valve. Frozen solution EPR spectrum were recorded on a JEOL continuous wave spectrometer JES-FA200 equipped with an X-band Gunn oscillator bridge, a cylindrical mode cavity, and a helium cryostat as well as a Bruker ELEXSYS E500 spectrometer equipped with a helium flow cryostat (Oxford Instruments ESR 910) and Hewlett-Packard frequency counter HP5253B. Spectral simulation was performed using the program QCMP 136 by Prof. Dr. Frank Neese from the Quantum Chemistry Program Exchange [15].
1H NMR spectra were recorded on JEOL 270 and 400 MHz instruments operating at respective frequencies of 269.714 and 400.178 MHz with a probe temperature of 23 °C in C6D6 or CD2Cl2. Chemical shifts were referenced to protio solvent impurities (δ 7.15 (C6D6), δ 5.32 (CD2Cl2) and are reported in ppm.
Electronic absorption spectra were recorded from 200 to 2000 nm (Shimadzu (UV-3101PC)) in the indicated solvent.
Results from elemental analysis were obtained from the Analytical Laboratories at the Friedrich-Alexander-University Erlangen-Nürnberg (Erlangen, Germany) on Euro EA 3000.
4.3 Starting materials
Precursor complexes [(THF)4UI3] and [U(N(SiMe3)2)3] were prepared as described by Clark et al. [16,17] Uranium turnings were purchased from Oak Ridge National Laboratory (ORNL) and activated according to literature procedures. [16] The 4-hydroxydiamantane (99+%) was obtained from Chevron Technology Ventures as a generous gift and used as received. Thionyl chloride (> 99%) and azidotrimethylsilane (95%) were purchased from Aldrich and used as received. The 4-tert-butylphenol (97%) was purchased from Acros Organics and also used as received.
4.4 Synthesis of 4-chlorodiamantane
In a flask fitted with a reflux condenser, thionyl chloride (18.0 mL, 0.0248 mol) was added to 4-hydroxydiamantane (5.00 g, 0.0245 mol. The mixture was heated to reflux at 90 °C for 2 h. At room temperature, the remaining thionyl chloride is removed in vacuo. The residue is taken up in dichloromethane (50 mL) and washed three times (50 mL) with distilled water. The volatiles in the organic fraction were removed in vacuo to yield a white powder. Yield: 5.24 g (0.0235 mol, 96%). Elemental analysis (%) calcd: C, 75.49; H, 8.60. Measured: C, 75.67; H, 8.65.
4.5 Synthesis of 2-diamantyl-4-tert-butylphenol
In a flask equipped with a reflux condenser, 4-chlorodiamantane (5.00 g, 0.0224 mol) and 4-tert-butylphenol (8.41 g, 0.056 mol) were heated to reflux at 140 °C. The melt was stirred for 12 hours. At room temperature, the solid mass was placed on a Kugelrohr device at 100 °C for 20 hours or until a pure white product was obtained, checked by 1H-NMR. Yield: 5.88 g (0.0175 mol, 78%). 1H-NMR (400 MHz, dichloromethane-d2, 20 °C) δ = 7.27 (d, 1H), 7.07 (dd, 1H), 6.58 (d, 1H), 4.80 (s, 1H) 2.20 – 1.70 (m, 19H).
4.6 Synthesis of (DiaArOH)3tacn (L3)
In a flask fitted with a reflux condenser, triazacyclononane (0.43 g, 3.3 mmol) and paraformaldehyde (0.30 g, 9.9 mmol) were heated to 80 °C in 1-propanol. After 2 hours, the 2-diamantyl-4-tert-butylphenol (5.00 g, 14.9 mmol) was added and the reaction mixture was allowed to stir at 80 °C for an additional 14 hours. During the cooling process to room temperature, a white precipitate started to form. A few drops of water were added to the reaction mixture to facilitate complete precipitation of the product. The white precipitate was collected by filtration and washed three times (10 mL) with ethanol. Yield: 2.83 g (2.4 mmol, 72%). 1H-NMR (400 MHz, dichloromethane-d2, 20 °C) δ = 7.19 (d, 3H), 6.79 (d, 3H), 3.70 (s, 6H), 2.77 (s, 12H), 2.22 – 1.68 (m, 57H), 1.27 (s, 27H).
4.7 Synthesis of [((DiaArO)3tacn)U] (1)
A solution of [U(N(SiMe3)2)3] (0.643 g, 0.89 mmol) in 1,2-dimethoxyethane (∼8 mL) was added dropwise to a stirring solution of (DiaArOH)3tacn (L3) (1.00 g, 0.85 mmol) in DME (∼7 mL). Within 10 minutes, a red-brown solution is observed. The reaction was allowed to stir for 4 hours. The reaction mixture was filtered through Celite and the volatiles were removed in vacuo to obtain 1 as a red-brown powder. Yield: 0.823 g (0.70 mmol, 82%).
4.8 Synthesis of [((DiaArO)3tacn)U(Cl)] (2)
Dichloromethane (7 μL, 0.11 mmol) diluted in DME (∼2 mL) was added dropwise to a stirring solution of 1 (0.150 g, 0.11 mmol) in DME (∼6 mL). Immediately the red-brown reaction solution turned pale green. The reaction was allowed to proceed at room temperature for 5 hours. The reaction mixture was filtered and volatiles were removed yielding 2 as pale green solids. Yield: 0.123 g (0.085 mmol, 77%).
4.9 Synthesis of [((DiaArO)3tacn)U(NTMS)] (3)
Azidotrimethylsilane (14 μL, 0.11 mmol) diluted in hexane (∼2 mL) was added dropwise to a stirring solution of 1 (0.150 g, 0.11 mmol) in DME (∼6 mL). The reaction solution turns dark brown-orange and evolution of dinitrogen was observed. Stirring was continued for 4 hours. The reaction mixture was filtered and volatiles were removed to obtain 3 as brown-orange solid. Yield: 0.121 g (0.081 mmol, 76%).
4.10 Crystallographic details for 2
Green block crystals, grown from slow diffusion of acetonitrile into a dichloromethane solution of 2 at room temperature, were coated with isobutylene oil on a microscope slide. A crystal of approximate dimensions 0.25 × 0.21 × 0.14 mm3 was selected and mounted on a nylon loop. A total of 87361 reflections (–30 ≤ h ≤ 30, –15 ≤ k ≤ 16, –32 ≤ l ≤ 32) were collected at T = 150(2) K in the θ range from 3.29 to 25.68°, of which 16647 were unique (Rint = 0.0726) and 13714 were observed [I > 2σ(I)] on a Bruker-Nonius KappaCCD diffractometer using MoKα radiation (λ = 0.71073 Å). The structure was solved by direct methods (SHELXTL NT 6.12, Bruker AXS, Inc., 2002). All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated idealized positions. One of the t-Bu groups is disordered, with two refined alternative positions being occupied by 36(2) and 64(2)% for C54 – C56 and C54A – C56A, respectively. The compound crystallizes with a number of solvent molecules of which the positions of one acetonitrile and two dichloromethane molecules were located. Apart from this the crystal structure contains two solvent accessible voids that are occupied by heavily disordered solvents. Here, the Squeeze algorithm was applied [18]. The residual peak and hole electron density were 3.293 and –3.787e.Å−3. The absorption coefficient was 2.026 mm−1. The least-squares refinement converged normally with residuals of R1 = 0.1045, wR2 = 0.2070, and GOF = 1.241 (all data). C85H115N4O3Cl5U, monoclinic, space group P21/c, a = 24.996(5), b = 13.380(2), c = 27.038(3) Å, β = 101.300(9)°, V = 8867(2) Å3, Z = 4, ρcalcd = 1.240 Mg/m3, F(000) = 3416, R1(F) = 0.0910, wR2(F2) = 0.2014 [I > 2σ(I)]. CCDC reference number: 763284.
4.11 Crystallographic details of 3
Brown plate crystals, grown from a saturated solution of 3 in hexane at room temperature, were coated with Paratone N oil on a microscope slide. A crystal of approximate dimensions 0.13 × 0.10 × 0.04 mm3 was selected and mounted on a glass fiber. A total of 112504 reflections (–14 ≤ h ≤ 14, −23 ≤ k ≤ 23, –30 ≤ l≤ 30) were collected at T = 150(2) K in the θ range from 3.15 to 26.73°, of which 18581 were unique (Rint = 0.1249) and 14448 were observed [I > 2σ(I)] on a Bruker-Nonius KappaCCD diffractometer using MoKα radiation (λ = 0.71073 Å). The structure was solved by direct methods (SHELXTL NT 6.12, Bruker AXS, Inc., 2002). All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated idealized positions. One of the t-Bu groups is disordered, with two refined alternative positions being occupied by 38(3) and 62(3)% for C79 – C81 and C79A – C81A, respectively. The asymmetric unit contains two molecules of n-hexane both of which are disordered. The residual peak and hole electron density were 1.348 and –0.752 e.Å−3. The absorption coefficient was 1.917 mm−1. The least-squares refinement converged normally with residuals of R1 = 0.0748, wR2 = 0.0885, and GOF = 1.055 (all data). C96H145N4O3SiU, triclinic, space group P-1, a = 11.3040(6), b = 18.194 (2), c = 24.048(2) Å, α = 111.877(6)°, β = 91.959(7)°, γ = 105.210(7)°, V = 4380.6(6) Å3, Z = 2, ρcalcd = 1.266 Mg/m3, F(000) = 1758, R1(F) = 0.0458, wR2(F2) = 0.0797 [I > 2σ(I)]. CCDC reference number: 763285.
Acknowledgements
The authors wish to thank Dr. M. K. Robert Carlson (Molecular Diamond Technologies, Chevron Technology Ventures) for a generous gift of 4-hydroxydiamantane and Dr. Susanne Mossin and Michaela Scheffler for their contributions and insightful discussions. Research was supported by the DFG through the Sonderforschungsbereich SFB 583.