

1 Introduction
The activation and functionalisation of small molecules is of great current importance but the formation of complex organic molecules directly from relatively inert, simple and abundant chemical feedstocks is chemically very challenging. Complexes of the low-valent f-elements provide a combination of high reduction potential with sterically demanding, robust ligands and have been shown to possess reactivities not observed elsewhere in the Periodic Table [1,2]. However, to effect these transformations cleanly, well-defined molecular complexes are required. The balance between the stabilisation of the metal centre in a low oxidation state and its accessibility towards the substrate can be a difficult one to strike.
It has been demonstrated that uranium(III) complexes of the type [U(η-C8H6{SiiPr3-1,4}2) (η-CpR)(THF)] (CpR = Cp* or C5Me4H) display high reactivity towards small molecules. The reaction of these complexes with CO to form C3O32− and C4O42− respectively, was the first example of selective, spontaneous, low-temperature, reductive homologation of CO [3,4]. Furthermore, the stoichiometric reaction of the desolvated complex [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)] with CO resulted in the isolation of [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)]2(μ-η1:η1-C2O2) [5]. The related mixed-sandwich complex [U(η-C8H4{SiiPr3-1,4}2)(η-Cp*)], which utilises the relatively less studied silylated dianionic pentalene ligand [C8H4{SiiPr3-1,4}2]2− [6], has been shown to reversibly bind N2 [7]; the resultant U(IV) complex contains a bridging, sideways-bound N22− unit.
Recent years have seen a move away from the domination by the cyclopentadienyl derivatives as other ligand systems have emerged as viable alternatives [8]. One such class is the scorpionate ligand, pioneered by Trofimenko [9]. A number of half-sandwich complexes of the general formula [Ln(η-C8H8)(Tp)] (Tp = hydrotris(pyrazolyl)borate, Ln = Ce, Pr, Nd, Sm) were synthesised by Edelmann et al. who found that the use of the bulkier TpMe2, with substituents on the 3- and 5- positions of the pyrazolyl ring, improved handling and solubility. These complexes have been used to study the bonding between the metal centre and the cyclooctatetrenyl ring [10]. Poly(pyrazolyl)borate complexes of uranium have been studied since the early 1980s, with a range of first and second generation scorpionates [11]. The size and the electronic properties of substituents on the pyrazolyl ring have a profound influence upon molecular structure, solution behaviour and reactivity [12].
Takats et al. reported the synthesis and full characterisation of [U(κ3-TpMe2)I2(THF)2] and [U(κ3-TpMe2)(κ2-η2-TpMe2)I], the latter displaying an unusual structure in the solid state with an interaction between the uranium-and the NN double bond of one of the pyrazolyl rings on one of the TpMe2 ligands [13]. These complexes provided a clean, high-yielding entry into both amido and hydrocarbyl derivative chemistry of low-valent uranium. In particular, the complex [U(κ3-TpMe2)(CH{SiMe3}2)2(THF)] was shown to display reactivity towards H2 and CO under mild conditions, though the reaction products were not characterised [14a]. More recently the derivatisation of [U(κ3-TpMe2)(κ2-h2-TpMe2)I] was also demonstrated including the structures of [U(κ3-TpMe2)2(N{C6H5}2)] and [U(κ3-TpMe2)2(N{SiMe3}2)] [15].
In the work presented herein, we examine the effect of substituting the CpR ligand for the TpMe2 ligand on the stability and reactivity of uranium(III) half-sandwich complexes containing the derivatised cyclotoctatetrenyl [C8H6{SiiPr3-1,4}2]2− and pentalenyl [C8H4{SiiPr3-1,4}2]2− ligands.
2 Results and discussion
The reaction of UI3 in THF with KTpMe2, separation of KI by toluene workup and the subsequent slow addition of 0.75 equivalents of [K2(C8H6{SiiPr3-1,4}2)] at 0 °C in THF, followed by extraction with pentane yielded [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1 as a dark red microcrystalline solid in a moderate yield (30% w.r.t. UI3). Using the synthetic strategy described for 1 and 0.8 equivalents of [K2(C8H4{SiiPr3-1,4}2)], it was possible to synthesise [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2 as a purple crystalline solid in a poor isolated yield (11% w.r.t UI3). This isolated yield reflects the difficulty of separating 2 from attendant side-products of similar solubility. The elemental analysis and spectroscopic data for both complexes are in agreement with the molecular formation. 1 and 2 are stable at room temperature under inert atmosphere over a period of months, both in the solid state and in solution (Scheme 1).
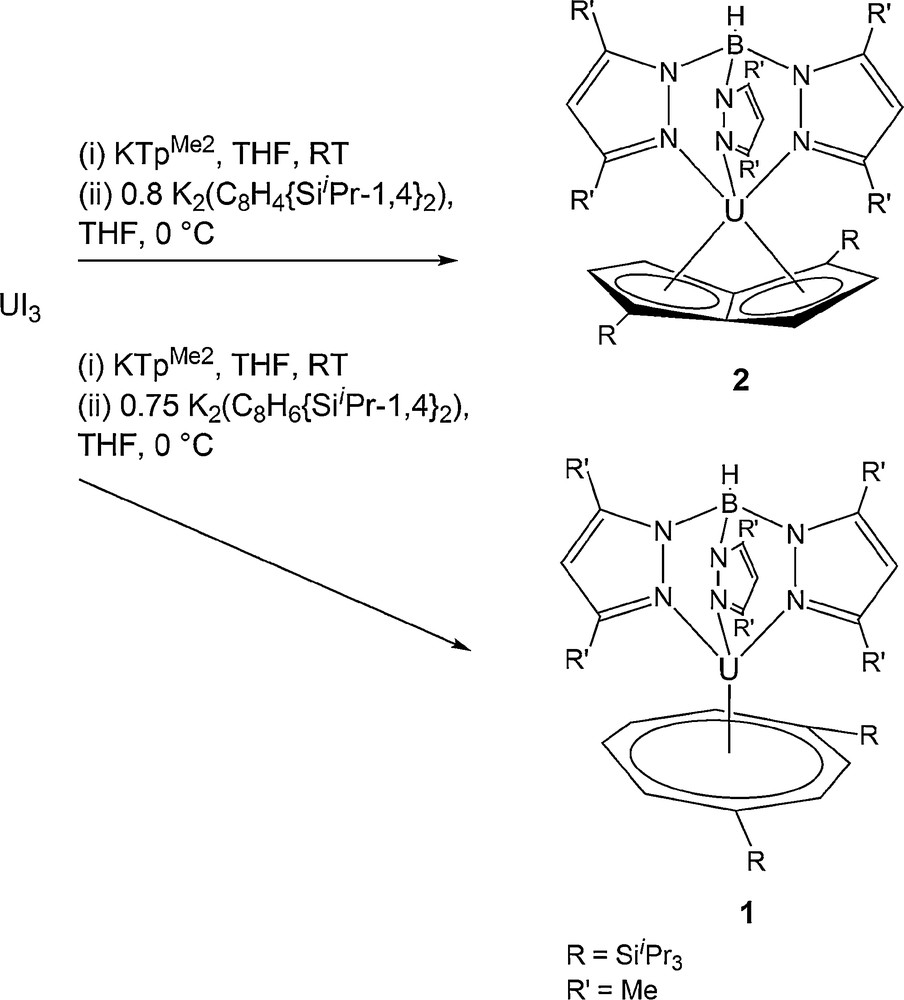
Synthesis of [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1 and [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2.
The 1H NMR spectrum of 1 in polar and non-polar solvents at room temperature is not in accordance with a 9:9:3 ratio for the 3-Me, 5-Me and 4-H proton environments of a fully equilibrated TpMe2 ligand [9b] but rather was consistent with an idealised pseudo-staggered conformation, where only two of the pyrazolyl rings are chemically equivalent giving rise to a 3:3:1:6:6:2 ratio. This static conformation remains well-defined up to 80 °C, above which the 3-Me, 5-Me and 4-H resonances were seen to broaden, though the high-temperature limiting spectra were not able to be obtained. In contrast, the 1H NMR spectrum of 2 at room temperature does exhibit a 9:9:3 ratio for the bound TpMe2. The range over which the resonances associated with both complexes are observed is broad, spanning 74 ppm for 1 and 41 ppm for 2, as expected for the paramagnetic U(III) centre. The 1H and 29Si NMR resonances associated with the [C8H6{SiiPr3-1,4}2]2− and [C8H4{SiiPr3-1,4}2]2− ligands are consistent with a single SiiPr3 environment rendered by mirror plane symmetry on an NMR time scale. The B-H resonance is also clearly identifiable in the room temperature 1H NMR spectra as a very broad multiplet, at 18.9 ppm for 1 and 18.0 ppm for 2. Though the integration of this resonance is complicated by its broadness, the assignment was confirmed by the loss of multiplicity of this resonance in the 1H{11B} spectrum. 11B NMR spectroscopy is a useful tool for looking at such complexes and can provide important information about the coordination environment of the boron [16]. The 11B{1H} NMR spectrum at room temperature displays a broad resonance at 32.8 ppm for 1 and 38.2 ppm for 2. A combination of paramagnetic and quadrupolar broadening produces a larger line-width and the coupling of this signal is not resolved in the 1H coupled spectrum.
Due to the limited solubility of 1 in common solvents, it was difficult to obtain crystals suitable for X-ray diffraction. However, eventually suitable crystals were grown from a saturated solution of diethylether at 5 °C over ten days. Crystals of 2 were grown from a saturated pentane solution at −20 °C over 48 h (Tables 1 and 2). The molecular structures of 1 and 2 are displayed in Figs. 1 and 2, respectively.
Crystal structure and refinement data for [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1, [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2 and [U(C8H6{SiiPr3-1,4}2)(κ2-dmpz)2(η1-CNMe)] 3.
| 1 | 2 | 3 | |
| Formula | C41H70BN6Si2U | C41H68BN6Si2U | C38H65N5Si2U |
| Formula weight | 952.05 | 950.03 | 886.16 |
| Temperature/K | 173(2) | 173(2) | 173(2) |
| Wavelength/Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal size/mm | 0.18 × 0.16 × 0.05 | 0.30 × 0.28 × 0.02 | 0.28 × 0.14 × 0.08 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c (No. 14) | P21/c (No. 14) | |
| a/Å | 14.7539(3) | 11.1026(2) | 11.1426(1) |
| b/Å | 16.6976(3) | 26.7178(6) | 15.3770(2) |
| c/Å | 18.9057(3) | 16.7781(3) | 25.9618(3) |
| α/° | 78.877(1) | 90 | 90 |
| β/° | 80.450(1) | 117.665(1) | 109.926(1) |
| γ/° | 78.412(1) | 90 | 90 |
| V/Å3 | 4437.53(14) | 4408.02(15) | 4181.98(8) |
| Z | 4 | 4 | 4 |
| Dc/Mg m−3 | 1.43 | 1.43 | 1.41 |
| Absorption coefficient/mm−1 | 3.75 | 3.77 | 3.97 |
| θ range for data collection/° | 3.43 to 27.12 | 3.43 to 27.48 | 3.50 to 27.86 |
| Reflections collected | 79,818 | 53,016 | 68,954 |
| Independent reflections | 19,539 [Rint = 0.092] | 9985 [Rint = 0.095] | 9892 [Rint = 0.058] |
| Reflections with I > 2σ(I) | 11,968 | 8271 | 8796 |
| Data/restraints/parameters | 19,539/7/959 | 9985/7/488 | 9892/0/420 |
| Goodness-of-fit on F2 | 1.019 | 1.239 | 1.097 |
| Final R indices [I > 2σ(I)] | R1 = 0.045 | R1 = 0.064 | R1 = 0.025 |
| R indices (all data) | R1 = 0.097 | R1 = 0.081 | R1 = 0.032 |
| Largest peak/hole/e Å−3 | 1.10 and −2.24a | 4.14 and −2.78a | 0.97 and −1.18 |
a Close to uranium.
Selected bond distances (Å) and angles (°) for [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1 (M1 is the centroid of the [(C8H6{SiiPr3-1,4}2)]2− ring; M2 is the centroid defined by N1, N3 and N5) and [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2 (M1 is the mid-point of the C4–C5 bond; M2 is the centroid defined by N1, N3 and N5).
| 1 | 2 | |
| U–N1 | 2.573(5) | 2.604(7) |
| U–N3 | 2.647(4) | 2.611(7) |
| U–N5 | 2.652(5) | 2.572(7) |
| U–Nave | – | 2.596(12) |
| U–M1 | 2.000(5) | 2.417(7) |
| U–M2 | 1.841(5) | 1.804(7) |
| U–C1 | – | 2.775(7) |
| U–C3 | – | 2.736(7) |
| U–C6 | – | 2.791(7) |
| U–C8 | – | 2.755(7) |
| M1–U–M2 | 176.18(1) | 166.60(2) |
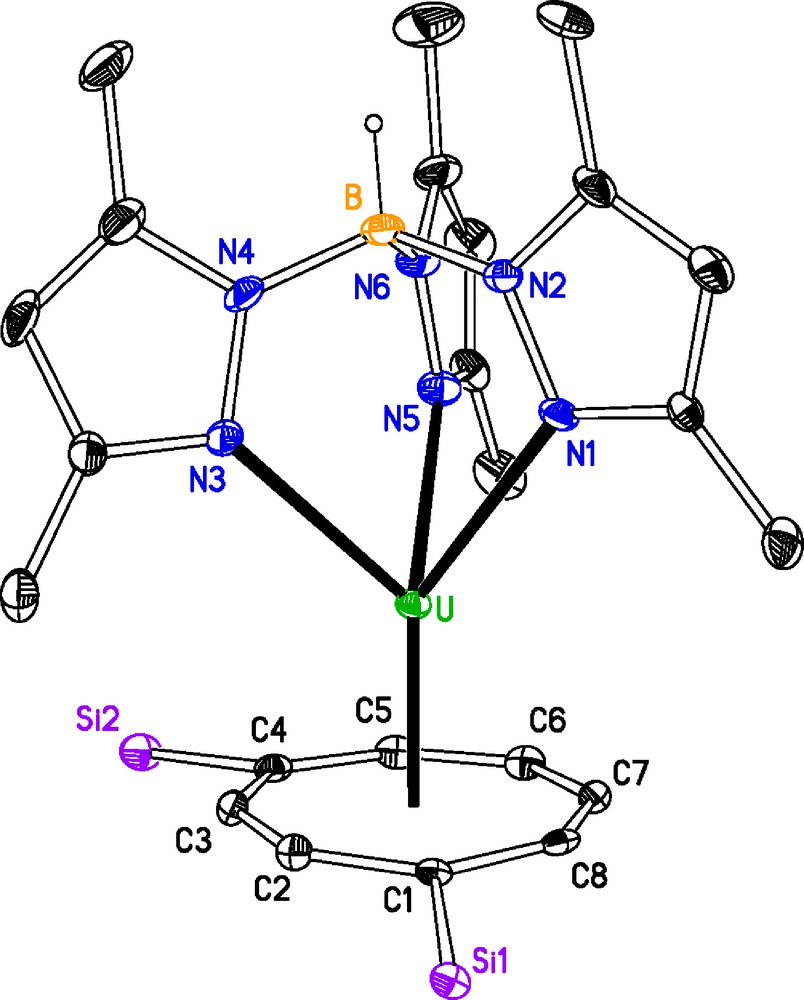
Molecular structure of [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1, ellipsoids at 30% probability, iPr groups and H atoms, except B-H, omitted for clarity. M1 is the centroid of the [(C8H6{SiiPr3-1,4}2)]2− ring.
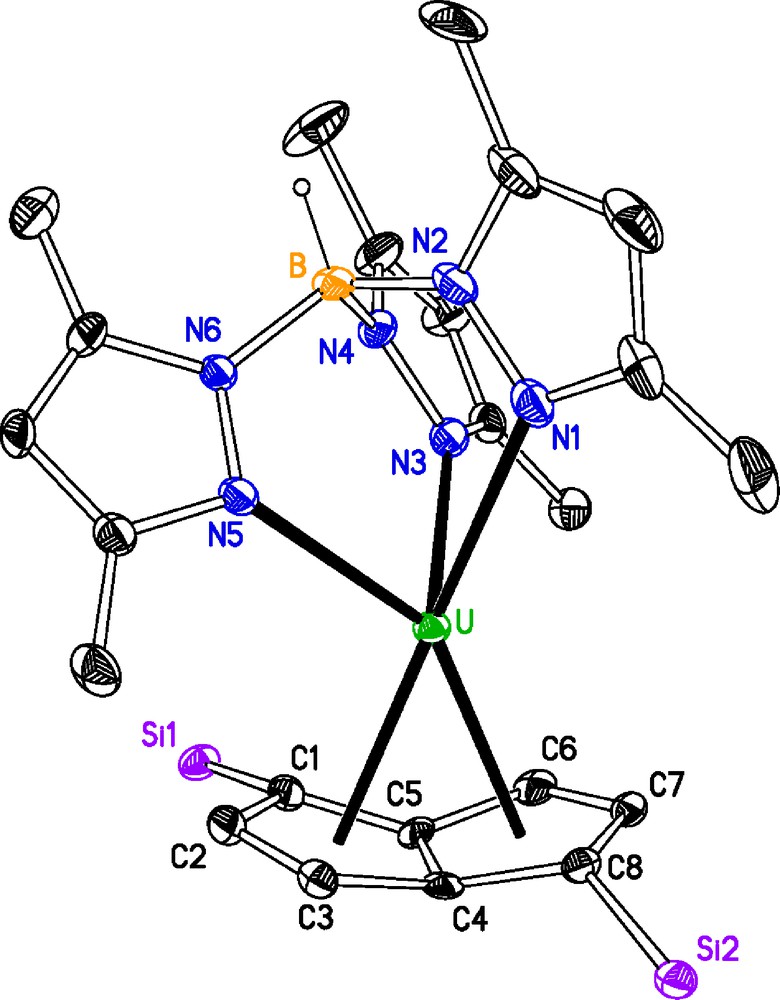
Molecular structure of [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2, ellipsoids at 30% probability, iPr groups and H atoms, except B-H, omitted for clarity.
In 1 although U–N1 is shorter than U–N3 and U–N5, it does not appear structurally significant. The U–Nave in 2 is shorter than those found in 1, this is presumably the result of the fold around the bridgehead in the pentalene ligand vide infra, thus easing the steric crowding. The U–M1 distance in 1, however, is essentially identical to that observed in [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*(THF))] U–M1 (1.975(6) Å). The U–N distances in both 1 and 2 are longer than the U–Nave (2.53(3) Å) found in the [U(κ3-TpMe2)I2(THF)2] precursor [14]. These U–N distances are comparable to those found other U(III) complexes and can cover a large range, for example, U–N (2.480(6) Å – 2.802(6) Å) in [U(κ3-TpMe2)2(N{SiMe3}2)] [15]. The fold angle between least squares planes defined by C5-rings of the pentalene ligand is 24.2(6)°, this is closer to the value of 24° observed in the sterically crowded [Th(η-C8H4{SiiPr3-1,4}2)2] [17], than the 26° of [U(η-C8H4{SiiPr3-1,4}2)(η-Cp*)] [7]. The U–C1,C3,C6,C8 distances that represent the major bonding interactions [18] are likewise longer than those found in [U(η-C8H4{SiiPr3-1,4}2)(η-Cp*)] U–C1 (2.733(7) Å), U–C3 (2.721(7) Å), U–C6 (2.683(7) Å) and U–C8 (2.722(7) Å). Both the [C8H6{SiiPr3-1,4}2]2− and [C8H4{SiiPr3-1,4}2]2− ligands are substituted in the 1 and 4 positions, in [C8H6{SiiPr3-1,4}2]2− case the bulky substituents are on the same side of the ring, whereas they are on opposite sides of the bridgehead in the [C8H4{SiiPr3-1,4}2]2− case. This allows the staggered conformation found in 1 but in 2 the positioning of the N3–N4 and N5–N6 rings either side of Si1 brings the N1–N2 ring and Si2 into proximity and their mutual repulsion results in a M1–U–M2 angle which is significantly more acute than that in 1.
The complexes are both base-free, unlike their Cp* analogues, when their preparation was undertaken in THF. The substitution of TpMe2 for Cp* is reflected by a less acute fold angle and by a lengthening of the U–C interactions in 2 but not by a lengthening of the U–M1 in 1. The solution NMR spectra of unsolvated Tp lanthanide complexes have been shown to approximate closely to their solid-state structure [19,16c]. In complex 1 the positioning of the TpMe2 ligand prevents the free rotation of the [C8H6{SiiPr3-1,4}2]2−, even in solution. The longer bonding interactions between the uranium centre and the [C8H4{SiiPr3-1,4}2]2− and the positioning of the SiiPr3 groups in 2 may lead to it being fluxional in solution. Thus, their solution behaviour would seem to be a reflection of their solid state molecular structures.
In marked contrast to [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)], 1 displays no reactivity towards CO, CO2 or MeNC under mild conditions by 1H, 13C, 11B and 29Si NMR and MS. The reaction of 1 with an overpressure of CO, although accompanied by a colour change, did not yield any material suitable for X-ray diffraction studies and showed only decomposition products by mass spectrometry. The lack of reactivity of 1 is similar to that of complexes [Ln(TpMe2)2] (Ln = Sm, Yb) which, in spite of the reducing nature of divalent lanthanides, display no reactivity towards CO, isocyanides or alkynes [20]. This lack of reactivity was attributed to the lack of an available oxidative reaction pathway. There is no relative electronic trend that describes the electron-donating properties of the TpMe2 and Cp* ligands across the periodic table [21]. The donation is dependent rather on the overall composition of the specific complex. The bonding modes adopted by the two ligands are quite different, the Tp is a σ donor, whereas the Cp ligand is capable of σ, π and δ donation [9b]. The unsubstituted Tp and Cp* are of similar steric bulk [22], though the larger TpMe2 is often used as an alternative to Cp*, its Tolman cone angle of 236° [23] is significantly larger than that of Cp* (182°) [24]. A further difference is that the Tp ligand adopts octahedral geometry preferentially, whereas the Cp ligand is capable of several hapticities [9b].
Binding of CO to [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)] involves a high degree of reduction of the CO ligand, with two unpaired electrons partially occupying the CO π* orbitals [3]. One possible reason for the lack of reactivity of 1 with CO might be that the uranium centre is less reducing than [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)]. In order to test this hypothesis, we have used DFT to estimate the relative reducing power of the two compounds. In order to make the two systems computationally accessible, the SiiPr3 groups attached to the cyclooctatetraenyl ligands were replaced by SiH3 groups, but full methylation of both Cp* and TpMe2 was maintained. The calculated structures [U(η-C8H6{SiH3-1,4}2)(η-Cp*)] I and [U(η-C8H6{SiH3-1,4}2)(κ3-TpMe2)] II were optimised with S = 3/2 and are shown in Fig. 3. Selected structural parameters are listed in Table 3. The three unpaired electrons occupied 5f orbitals in both cases with the spin density on the uranium being 3.0. The energies of the half occupied 5f orbitals (Fig. 3) were higher for the TpMe2 complex than the Cp* complex indicating a higher reducing power for the former. The calculated structures of the CO adducts, [U(η-C8H6{SiH3-1,4}2)(η-Cp*)(η1-CO)] III and [U(η-C8H6{SiH3-1,4}2)(κ3-TpMe2)(η1-CO)] IV, were optimised; selected structural parameters and spin densities are given in Table 3. The CO SCF binding energies were calculated as 1.02 eV for III and 0.84 eV for IV. Thus, in spite of the higher f orbital energies for the TpMe2 complex, the CO is less tightly bound. This is confirmed by the U–C distance which is longer in the TpMe2 complex and the spin densities, where less unpaired spin density is transferred to the CO group in the TpMe2 complex (Table 3). The distances between the uranium and the supporting ligands increase on CO binding for both complexes. For the bulkier TpMe2 group, the energetic cost appears to be too great to support the entropic barrier to CO binding (Fig. 4).
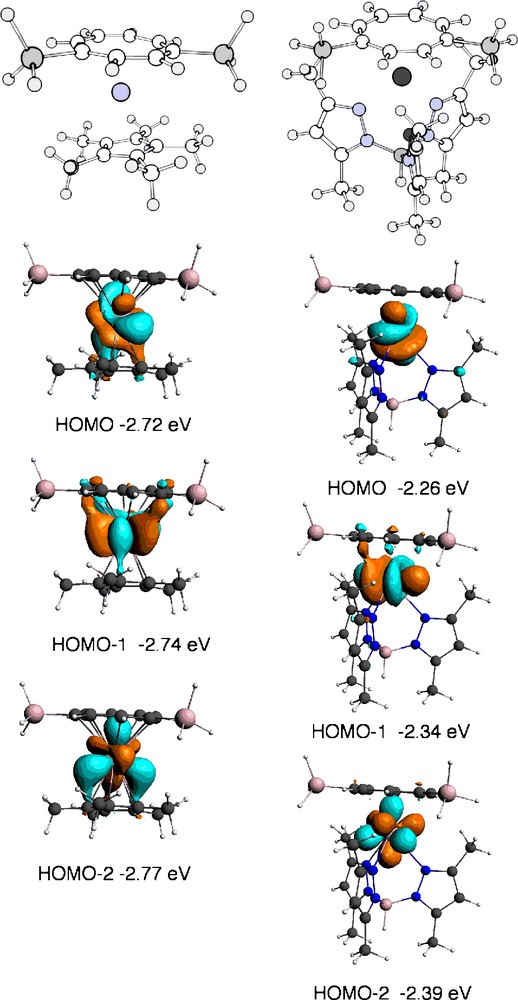
Calculated structures and half occupied 5f orbitals of [U(η-C8H6{SiH3-1,4}2)(η-Cp*)] I and [U(η-C8H6{SiH3-1,4}2)(κ3-TpMe2)] II.
Selected structural parameters (Å) and spin densities calculated for structures [U(η-C8H6{SiH3-1,4}2)(η-Cp*)] I, [U(η-C8H6{SiH3-1,4}2)(κ3-TpMe2)] II, [U(η-C8H6{SiH3-1,4}2)(η-Cp*)(η1-CO)] III and [U(η-C8H6{SiH3-1,4}2)(κ3-TpMe2)(η1-CO)] IV.
| I | II | III | IV | |
| U-C (C8H6{SiH3-1,4}2) | 2.65–2.70 | 2.71–2.75 | 2.66–2.82 | 2.72–2.82 |
| U-C (Cp*) | 2.62–2.76 | 2.70–2.77 | ||
| U-N (TpMe2) | 2.57–2.59 | 2.55–2.72 | ||
| U-C (CO) | 2.35 | 2.40 | ||
| C-O (CO) | 1.17 | 1.17 | ||
| U spin density | 3.0 | 3.0 | 2.72 | 2.79 |
| C spin density | 0.26 | 0.22 | ||
| O spin density | 0.14 | 0.19 |
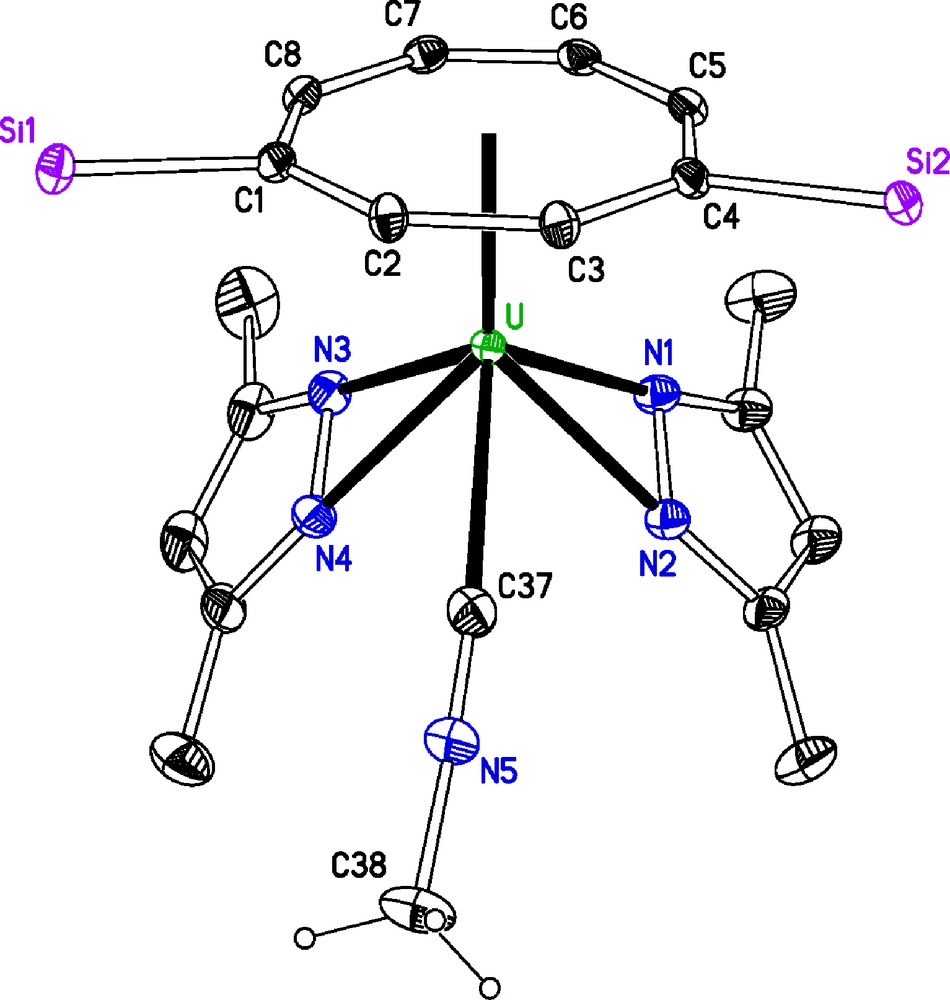
Molecular structure of [U(η-C8H6{SiiPr3-1,4}2)(η2-dmpz)2(η1-CNMe)] 3, ellipsoids at 30% probability, iPr groups and H atoms, except MeNC, omitted for clarity. Selected bond distances (Å) and angles (°): U–N1 2.353(2), U–N2 2.397(2), U–N3 2.360(2), U–N4 2.387(2), U–M1 1.987(7) Å, U–C37 2.675(3), C37–N5 1.140(3), N5–C37–U 170.0(2) (M1 is the centroid of the [C8H6{SiiPr3-1,4}2]2− ring).
However, 1 reacts in the presence of a tenfold excess of MeNC when heated at 80 °C on an NMR scale in d8-toluene to yield [U(η-C8H6{SiiPr3-1,4}2)(η2-dmpz)2(η1-CNMe)] 3. (see Scheme 2) The elemental analysis and mass spectrum of 3 are in agreement with the molecular formation shown, given the facile loss of the coordinated isocyanide.
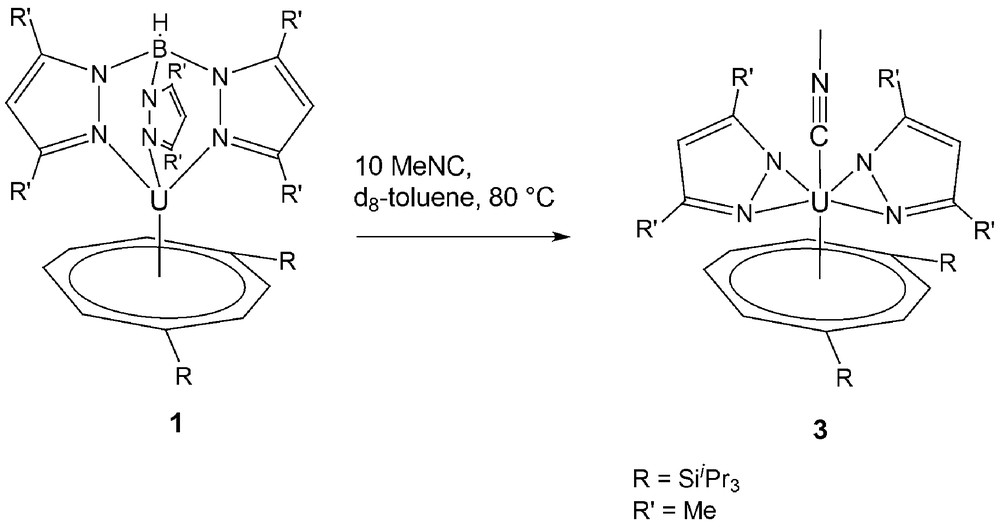
Reaction of [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)]1 with excess MeNC at 80 °C.
The U–M1 distance in 3 and in 1 are essentially identical within esds. That the change in formal oxidation state from U(III)/U(IV) is not necessarily accompanied by a change in structural parameters has been observed for other mixed-sandwich complexes and attributed to steric congestion [3,7]. The two pyrazolide rings are bound in an endo-bidentate fashion to the metal centre, the distances in 3 are closer to those found in [U(Cp*)2(η2-pz)2] and [U(Cp)3(η2-pz)] U–N (2.4 Å) and (2.36 Å) [25], than the longer averaged distances in [U(κ3-TpMe2)(N{SiMe3}2)(η2-dmpz)] U–Nave (2.440(8) Å) and [U(κ3-TpMe2)2(η2-dmpz)] U–Nave (2.444(11) Å) [14]. This endo-bidentate binding mode was first observed in [U(Cp)3(η2-pz)] and considered a consequence of the more ionic bonding in the actinides as opposed to the bridging mode observed in the d-block elements, which allows for directional covalent bonding.
The relevant bonding interactions between the metal centre and the (η1-CNMe) in 3 are essentially identical within esds to those found in the [U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)(η1-CNMe)] [26], though the U-C-N angle (178.4°), is less acute than that observed in 3. This U–C distance in 3 is significantly longer than those found in the literature; much longer than the U–C (2.464(4) Å) observed for [U(CpMe4H)3(CNC6H4-p-OMe)] in which π-back-bonding is thought to occur [27]. These observations are in keeping with an unactivated, loosely coordinated isocyanide in 3. The 1H NMR spectrum of complex 3 at room temperature is unexpectedly complex, showing two SiiPr3 environments and significant broadening and merging of the dmpz resonances. It has not been possible to determine whether this reflects different conformations in solution, or fast association and dissociation of the isocyanide, or a mixture of 3 and [U(η-C8H6{SiiPr3-1,4}2)(η2-dmpz)2] resulting from partial loss of the isocyanide on drying 3 in vacuo. The resonance for the methyl group of the isocyanide could not be located, mostly likely as a result of rapid exchange between free and bound MeNC in solution, as has been suggested for the CO ligands in U(III) carbonyl complexes which are also NMR silent in 13C NMR spectra [27a].
The fragility of the B–N bond is well documented and fragmentation of the TpMe2 ligand especially when coordinated to an electropositive metal has been a recurrent problem in lanthanide chemistry [28], though in some cases this fragmentation is the result of adventitious water or oxygen [29]; in most instances the mechanistic details of the fragmentation pathway have been neither investigated or reported. Fragmentation, however, is not usually accompanied by redox behaviour. When [Sm(κ3-TpMe2)(κ2-TpMe2)(η-Cp)] was heated to 165 °C under vacuum overnight, it yielded [Sm(κ3-TpMe2)2(dmpz)] and [Sm(HB(dmpz)2(C5H4))(κ3-TpMe2)] [30]. For low-valent uranium, the availability of the +4 oxidation state has led to a few examples of redox behaviour: the themolysis of [U(κ3-TpMe2)(κ2- TpMe2)(Cp)] at 160 °C, under vacuum for 3 days resulted in the oxidation of the metal centre to yield [U(Cp)3(dmpz)], [U(TpMe2)(dmpz)3] and [(HB(dmpz)2)2] [31]. Redox behaviour was also observed during the thermal decomposition of [U(κ3-TpMe2)(N{SiMe3}2)2] [14a]. In both of these examples, there is more than one U(IV) species produced and the mechanism is unclear, but B–N cleavage is proposed to dominate. The observed reactivity for 1, mediated by a combination of heating and the excess isocyanide, results in the single U(IV) species 3, suggesting that it is the oxidation of the uranium centre, that takes precedence over B–N cleavage.
3 Conclusion
The substitution of the CpR ligand for the TpMe2 ligand has led to the synthesis of two novel uranium(III) half-sandwich complexes: [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1 and [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2. The X-ray structures of these complexes reveal that the increase in steric congestion resulting from the use of the TpMe2 ligand, in place of Cp*, is not reflected in an increase in the U-COT in 1 but in 2 results in a lengthening of the U–C distances to the pentalene ligand. This is corroborated by the solution NMR behaviour of the complexes: 1 retains a static structure up to 80 °C, whereas 2 is rapidly equilibrated at room temperature. The ability of the pentalene ligand to fold around the bridgehead allows greater access to the metal centre in 2. 1 displays no reactivity towards CO, CO2, MeNC under mild conditions, though initial reactivity studies on 2 show it is reactive towards MeNC under the same conditions. DFT calculations on the model systems [U(η-C8H6{SiH3-1,4}2)(η-Cp*)] I and [U(η-C8H6{SiH3-1,4}2)(κ3-TpMe2)] II show that II is more reducing than its Cp* analogue, but on CO binding the energetic cost for the bulkier system is too great for a reaction to occur. When heated at 80 °C in the presence of a tenfold excess of MeNC, 1 reacts to yield [U(η-C8H6{SiiPr3-1,4}2)(η2-dmpz)2(η1-CNMe)] 3.
4 Experimental
4.1 General
The manipulation of air-sensitive compounds were undertaken using standard Schlenk-line techniques, under an atmosphere of catalytically dried and deoxygenated argon, or under catalytically dried and deoxygenated nitrogen in an MBraun glove box (< 1 ppm H2O and < 1 ppm O2). NMR spectra were obtained using Varian Direct Drive 400 MHz or 600 MHz spectrometers.
Elemental Analyses were carried at the Elemental Analysis Service, London Metropolitan University. Mass spectra were recorded using a VG Autospec Fisons instrument (electron ionisation at 70 eV). Solvents were purified by standard procedures and degassed prior to use. d8-toluene was dried by refluxing over potassium. Isotopically enriched 13CO (99%) was supplied by Cambridge Isotopes, and added via Toepler pump. Depleted uranium turnings were supplied by CERAC, and also kindly donated by BNFL. KTpMe2 was generously donated by Dr. I.R. Crossley, University of Sussex. The following compounds were prepared according to published procedures: UI3 [32], MeNC [33], [K2(C8H6{SiiPr3-1,4}2)] [34] and [K2(C8H4{SiiPr3-1,4}2)] [35].
4.2 X-ray diffraction
The data for crystals 1–3 were collected at 173 K on a Enraf-Nonius CAD4 diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Data collection was handled using KappaCCD software, final cell parameter calculations performed using program package WinGX. The data were corrected for absorption using the MULTISCAN program. Refinement was performed using SHELXL-97, and the thermal ellipsoid plots drawn using Shelxtl-XP. SADI restraints, isotropic C atoms, iPr groups and H atoms omitted, except where specified. One of the iPr groups on one of the [(C8H6{SiiPr3-1,4}2)]2− rings is disordered in 1 and it contains two molecules in the unit cell and. In 2 one of the iPr groups is disordered over two positions. CCDC 758454-758456 contain the supplementary crystallographic data for this paper. These can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request.cif.
4.3 Computational methods
Quantum chemical calculations were performed using density functional methods of the Amsterdam Density Functional (Version ADF2008.01) package [36]. TZP basis sets were used with triple-ξ accuracy sets of Slater-type orbitals, with polarisation functions added to all atoms. Relativistic corrections were made using the zero-order relativistic approximation (ZORA) formalism [37] and the core electrons were frozen up to 1 s for B, C and N, 2p for Si, and 5d for U. For U the 6p electrons were included in the valence set. The energies of the structures were calculated using the local density approximation (LDA) [38] due to Vosko et al. with the non-local exchange terms of Becke [39], and the non-local correlation correction of Perdew et al. [40] being applied to the calculated LDA densities (BP86).
4.4 Synthesis of [U(κ3-TpMe2)(C8H6{SiiPr3-1,4}2)] 1
An ampoule was charged with UI3 (0.386 g, 0.624 mmol) and THF (ca. 50 ml) added, the dark purple solution was stirred overnight at ambient temperature. To this a colourless solution of KTpMe2 (0.210 g, 0.624 mmol) in THF (ca. 20 ml) was added dropwise over 45 min and the reaction mixture stirred for a further 2 h 30 min after which time a white precipitate was observed. Volatiles were removed at reduced pressure, solids extracted with toluene and filtered on a frit through dry Celite®. The dark purple solution was stripped to dryness, taken up in THF (ca. 60 ml) and cooled to 0 °C. To this, a yellow solution of K2(C8H6{SiiPr3-1,4}2) (0.75 equivalents, 0.232 g, 0.469 mmol) in THF (25 ml) was added dropwise over the course of an hour. The solution was warmed to ambient temperature overnight under a partial vacuum after which the colour of the solution was observed to be a deep red and a white precipitate was observed. Volatiles were removed at reduced pressure and solids extracted with pentane and filtered on a frit through dry Celite®. Yield: 175 mg, 29.4% 1H NMR (399.5 MHz, d8-toluene, 303 K): δH 18.9 (br, m, 1H, B-H), 10.1 (s, 2H, TpMe2-CH), 4.1 (s, 1H, TpMe2-CH), 3.5 (s, 6H, TpMe2-CH3), −0.2 (s, 3H, TpMe2-CH3), −1.8 (d, JHH = 5.2 Hz, 18H, iPr-CH3), −2.3 (d, JHH = 5.3 Hz, 18H, iPr-CH3), −2.8 (br, s, 6H, iPr-CH), −15.4 (s, 3H, TpMe2-CH3), −17.5 (s, 6H, TpMe2-CH3), −22.0 (br, s, 2H, COT ring CH), −50.3 (br, s, 2H, COT ring CH), −54.8 (br, s, 2H, COT ring CH). 11B{1H} NMR (599.7 MHz, d8-toluene, 328 K): δB 32.8; 29Si NMR (599.7 MHz, d8-toluene, 328 K): δSi −115.6 MS (EI): m/z = 951 (M+).
4.5 Synthesis of [U(κ3-TpMe2)(C8H4{SiiPr3-1,4}2)] 2
In an analogous manner to that described in Section 4.4, 2 was synthesised using UI3 (0.770 g, 1.24 mmol), KTpMe2 (0.418 g, 1.24 mmol) and K2(C8H4{SiiPr3-1,4}2) (0.80 equivalents, 0.490 g, 0.992 mmol). Yield: 129 mg, 11%. Analysis calculated (found) for C41H68BN6Si2U: % C 51.58 (51.91), % H 7.17 (7.12), % N 8.85 (8.81). 1H NMR (399.5 MHz, d8-toluene, 303 K): δH18.0 (br, m, 1H, B-H), 7.7 (s, 3H, Tp-CH), 7.3 (br, s, 2H, pentalene ring CH), 2.5 (s, 9H, Tp-CH3), −5.3 (s, 18H, iPr-CH3), −9.9 (s, 18H, iPr-CH3), −11.6 (br, s, 6H, iPr-CH), −17.1 (s, 9H, Tp-CH3), −23.2 (br, s, 2H, pentalene ring CH). 11B{1H} NMR (399.5 MHz, d8-toluene, 303 K): δB 38.2; 29Si (399.5 MHz, d8-toluene, 303 K): δSi −159.3 MS (EI): m/z = 949 (M+).
4.6 Synthesis of [U(C8H6{SiiPr3-1,4}2)(κ2-dmpz)2(η1-CNMe)] 3
1 (61 mgs, 6.41 mmol) was dissolved in d8-toluene and placed in an NMR tube. MeNC (34 μL, 64.1 mmol) was added by syringe and the tube inverted to aid mixing. The dark red solution was heated at 80 °C in a heating block and monitored by 1H and 11B NMR. After 24 hrs, the colour of the solution was observed to lighten to a dark orange colour and bright red crystals were visible. The solution was heated for a further 24 hrs after which time the reaction was observed to be complete. Crystalline material for characterisation was washed into a schlenk, cooled, washed with cold toluene and dried in vacuo.
Analysis calculated (found) for C38H65N4Si2U: % C 51.18 (51.38), % H 7.35 (7.29), % N 6.64 (6.59). 1H NMR (399.5 MHz, d8-toluene, 303 K): δH 1.1 (m, 6H, COT ring CH, dmpz-H), −1.9 (v br, m, 12H, dmpz-CH3), −4.8 (br, s, 9H, iPr-CH3), −5.0 (br, s, 3H, iPr-CH), −5.46 (br, s, 9H, iPr-CH3), −9.0 (br, s, 9H, iPr-CH3), −10.0 (br, s, 3H, iPr-CH), −31.4 (s, 2H, COT ring CH). One iPr-CH3 resonance is obscured by solvent. MS (EI): m/z = 844 (M+ - MeNC).
Acknowledgements
We thank the EPSRC for financial support, Dr. P.B. Hitchcock for assistance with the refinement of crystallographic data and Drs I.R. Crossley and I.J. Day for useful discussions.