

1 Introduction
Phosphorus ligands play important roles in both Organometallic and Coordination Chemistry. In recent years the synthesis of functionalized phosphanes, some of which have unusual stereoelectronic properties, has received a great deal of attention [1], since subtle changes in the structure of this type of ligand often lead to the appearance of interesting properties in the complexes [2].
Complexes that incorporate phosphorus ligands in which the phosphorus is adjacent to a functional group have rarely been reported [3] and those that contain a carbonyl group adjacent to the phosphorus atom, namely phosphamide ligands, are extremely rare [4]. This fact could be related to the stability of the P–C bonds, which have been shown to undergo degradation reactions readily in the presence of traces of water or oxygen [5]. However, DFT studies have established that the interaction with the metal should increase the stability of the phosphamide ligands [4]. Furthermore, the presence of a carbonyl group bound to the phosphorus atom may lead to an inductive electron-withdrawing effect and a tendency for the phosphorus atom to delocalize its lone electron pair towards the carbonyl group, as shown in Scheme 1.
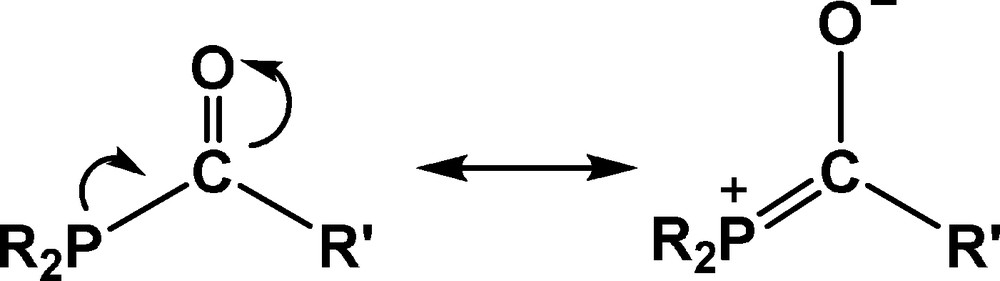
Several studies on the synthesis and reactivity of early transition metal phosphido complexes have been published [6] and most of these concern group 4 complexes. Our group recently developed the synthesis of new phosphido complexes [Nb(η5-C5H4SiMe3)2(L)(PPh2)], LCO (1) [7] or CNXylyl (2) [8], which are suitable precursors for the isolation of new niobocene complexes with phosphorus-containing functionalized ligands.
The niobium-phosphorus bond is susceptible to insertion reactions with unsaturated molecules such as carbon disulfide [7,8] or polarized alkynes [9]. Furthermore, the phosphorus atom of the phosphido terminal ligand has a lone electron pair, so it can be viewed as a Lewis base, and reacts with organic electrophiles to give electrophilic P-alkylation, as reported previously in the preparation of cationic d2 18-electron complexes [Nb(η5-C5H4SiMe3)2(PRPh2)(L)]X by reaction with several RX reagents [7,8]. The present paper concerns the reaction of the phosphido-niobocene complexes 1 and 2 with both alkyl and acyl chlorides to afford new P–C bonds. In this process, the former phosphido terminal ligand is transformed into a new functionalized phosphane ligand.
2 Results and discussion
2.1 Reaction with 2-chloroacetophenone
Our group recently reported the synthesis of new d2 cationic phosphane-containing niobocene complexes by nucleophilic substitution of the phosphorus atom in the terminal phosphido ligand of complexes [Nb(η5-C5H4SiMe3)2(L)(PPh2)], LCO (1) [7] or CNXylyl (2) [8], with alkyl halides. This reactivity seems to be a good synthetic pathway for the isolation of complexes that contain functionalized phosphane ligands by the presence of functional groups in one of the substituents on the phosphorus atom. As a result, we carried out the reaction of complexes 1 and 2 with 2-chloroacetophenone, which incorporates a ketone group in the alkyl chain bonded to phosphorus.
2.1.1 Synthesis of [Nb(η5-C5H4SiMe3)2(P(CH2CO(C6H5))Ph2)(L)]Cl, LCO (3), CNXylyl (4)
The reaction was performed by the addition of an equimolar amount of 2-chloroacetophenone to a solution of complexes 1 or 2 in hexane at room temperature. After the appropriate work up (see Experimental section), the new d2 cationic niobocene complexes 3 and 4 were isolated in good yield (85%), as shown in Scheme 2.
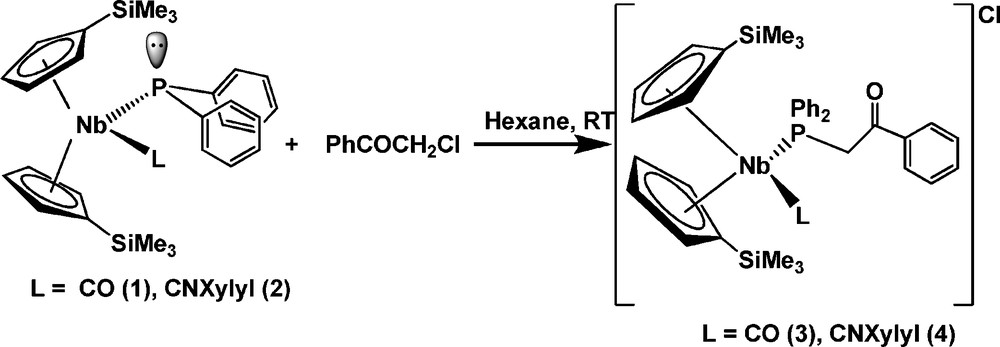
Complexes 3 and 4 were isolated as yellowish-orange and orange air-sensitive solids, respectively.
Complexes 3 and 4 were fully characterized by elemental analysis, IR spectroscopy and, in solution, by NMR spectroscopy. The ionic nature of these compounds was confirmed by measurement of the molar conductivity (ΛM: 71.9 and 77.4 Ω−1 cm2 mol−1 for 3 and 4, respectively) and the values are consistent with 1:1 electrolytes [10].
The most significant bands in the IR spectra appear at 1946 and 2046 cm−1, corresponding to the triple bond of the carbonyl-and isocyanide ancillary ligands for 3 and 4, respectively. Both spectra also contain a strong band at ca. 1677 cm−1 corresponding to the CO double bond of the ketone group.
The presence in the 1H-NMR spectra of doublet signals, which can be assigned to the CH2 protons of the alkyl moiety, at δ = 4.85 ppm (2JHP = 7.7 Hz) for 3 and δ = 5.09 ppm (2JHP = 7.3 Hz) for 4 confirms that alkylation has occurred at the phosphorus. The two spectra each contain a single signal for the methyl groups of the SiMe3 and four multiplets corresponding to the cyclopentadienyl ligands, a finding consistent with an ABCD spin system.
In agreement with the IR spectra, the 13C{1H}-NMR spectrum of 4 shows lowfield resonances for the ketone group, δ = 195.4 ppm, and the isocyanide group, δ = 199.0 ppm, as broad signals due to the quadrupolar moment of the niobium atom. The carbon atom of the alkyl moiety, CH2, is observed as a doublet due to coupling with the phosphorus atom, at δ = 42.6 ppm (1JCP = 23.8 Hz) for 3 and δ = 43.1 ppm (1JCP = 19.9 Hz) for 4.
In CDCl3 solution, the 31P{1H}-NMR broad resonances for 3 (δ = 41.0 ppm) and 4 (δ = 41.0 ppm) are shifted downfield with respect to the corresponding signals for the phosphido ligand in complexes 1 and 2. The signals lie in a range typical of d2 cationic niobocene complexes, as reported previously by our group [7,8]. This fact shows the effect of alkylation of the phosphorus by comparison with the 31P NMR chemical shift of the phosphido terminal ligand in complexes 1 and 2 [7,8].
The spectroscopic data are consistent with a pseudo-tetrahedral arrangement in these niobocene complexes where the phosphorus atom of the terminal phosphane ligand has a functionalized substituent, as shown in Scheme 2.
2.2 Reactions with acyl halides
Functionalization of the phosphanes can also be performed by the reaction of 1 and 2 with acyl halides, RCOX. In this case, direct attachment of the phosphorus atom and the carbonyl group by nucleophilic acyl substitution occurs to give the new cationic complexes 5–8.
2.2.1 Synthesis and characterization of [Nb(η5-C5H4SiMe3)2(P{CO(C6H5)}Ph2)(L)] Cl, LCO (5), CNXylyl (6)
An equimolar amount of benzoyl chloride was added to a solution of 1 or 2 in hexane at room temperature. After the appropriate work up (see Experimental section), the new d2 cationic niobocene complexes 5 and 6 were isolated in good yield (ca. 85%), as shown in Scheme 3.
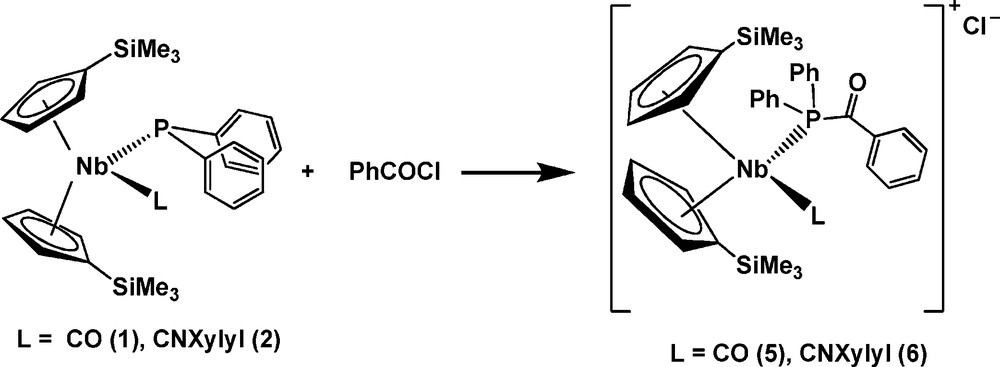
Complexes 5 and 6 were isolated as red air-sensitive solids.
The ionic nature of complexes 5 and 6 was confirmed by measuring the molar conductivity, ΛM = 78.6 and 76.4 Ω−1 cm2 mol−1 for 5 and 6, respectively. These values are consistent with 1:1 electrolytes [10].
Compounds 5 and 6 were also characterized by commonly used spectroscopic techniques. The most significant bands in the IR spectra appear at 1947 cm−1 for 5 and 2054 cm−1 for 6, and these correspond to the carbonyl and isocyanide ligands, respectively. The stretching vibration of the CO double bond of the ketone group also appears in the two spectra as a strong band at ca. 1650 cm−1.
In agreement with the IR spectra, the 13C{1H}-NMR spectra of 5 and 6 show low-field resonances for the carbon atom of the ketone group (ca. δ = 207 ppm), the quaternary carbon of the ancillary ligand carbonyl (δ 248.1 ppm) and isocyanide (δ = 194.7 ppm). These resonances are observed as broad signals, probably due to the quadrupolar moment of the niobium atom.
The 31P{1H}-NMR spectra each contain a broad resonance and these appear at ca. δ = 64.0 ppm (see Experimental section). These values are shifted to lower field in comparison with the 31P{1H}-NMR signals for complexes 3 and 4, and also the cationic phosphane niobocene complexes previously described by our group [7,8]. This change in shift is indicative of the decreasing electronic density at the phosphorus atom of complexes 5 and 6, probably due to the presence of the ketone group bonded directly to the phosphorus atom in the phosphamide moiety resulting from the P–C coupling process. Moreover, these data are consistent with the chemical shift values observed for other phosphamido complexes described previously [11].
The spectroscopic data are consistent with a pseudo-tetrahedral arrangement of these niobocenes containing an acylphosphane ligand, as shown in Scheme 3.
2.2.2 Synthesis of [Nb(η5-C5H4SiMe3)2(P(COCH(C6H5)2)Ph2)(L)]Cl, LCO (7), CNXylyl (8)
Complexes 7 and 8 were prepared in a similar way to complexes 5 and 6. An equimolar amount of diphenylacetyl chloride was added at room temperature to a solution of 1 or 2 in hexane. After the appropriate work up (see Experimental section), the new d2 cationic niobocene complexes 7 and 8 were isolated in good yield (ca. 85%), as shown in Scheme 4.
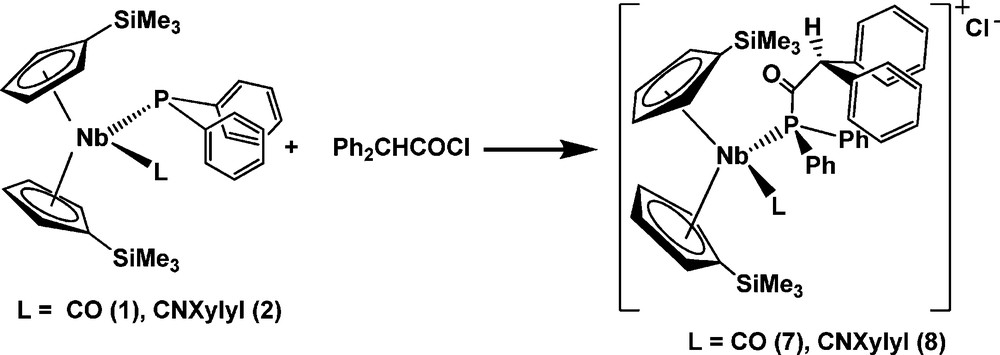
Complexes 7 and 8 were isolated as orange air-sensitive solids.
Once again the ionic nature of complexes 7 and 8 was established by measuring the molar conductivity, ΛM = 87.2 and 83.3 Ω−1 cm2 mol−1 for 7 and 8, respectively. As commented above, these values are consistent with 1:1 electrolytes [10].
The IR spectra of both complexes show the expected bands at 1950 cm−1 for the carbonyl ligand of 7 and at 2038 cm−1 for the isocyanide ligand of 8 (see Experimental section). Additionally, strong bands appear at 1682 and 1672 cm−1 for 7 and 8, respectively, and these correspond to the stretching vibration of the CO bond of the ketone group.
The 1H NMR spectra of the two complexes each contain a broad signal and these correspond to the methylenic proton of the diphenylacetylphosphane moiety, PC(O)CHPh2, at δ = 5.67 ppm for 7 and δ = 5.26 ppm for 8. The chemical shifts of these proton signals indicate their potentially acidic character.
In agreement with the IR spectrum, the 13C{1H}-NMR spectrum of complex 8 shows a broad low-field resonance for the carbon atom of the ketone group, at δ = 193.1 ppm. The methylenic carbon atom of the diphenylacetylphosphane moiety of each complex gives rise to a doublet due to coupling with the phosphorus atom, at δ = 63.0 ppm (2JCP = 31.4 Hz) for 7 and δ = 63.2 ppm (2JCP = 29.6 Hz) for 8.
The 31P{1H}-NMR spectra each contain a broad resonance at δ = 74.9 ppm for 7 and δ = 70.9 ppm for 8 (see Experimental section). These values are within the range found for complexes 5 and 6 (as mentioned above) and were shifted to low field in comparison with the 31P{1H}-NMR chemical shifts previously described for cationic phosphane niobocene complexes and for complexes 3 and 4 [7,8]. These chemical shift values are indicative of the direct connection between the ketone group and the phosphorus atom in the phosphamide unit resulting from the P–C coupling process.
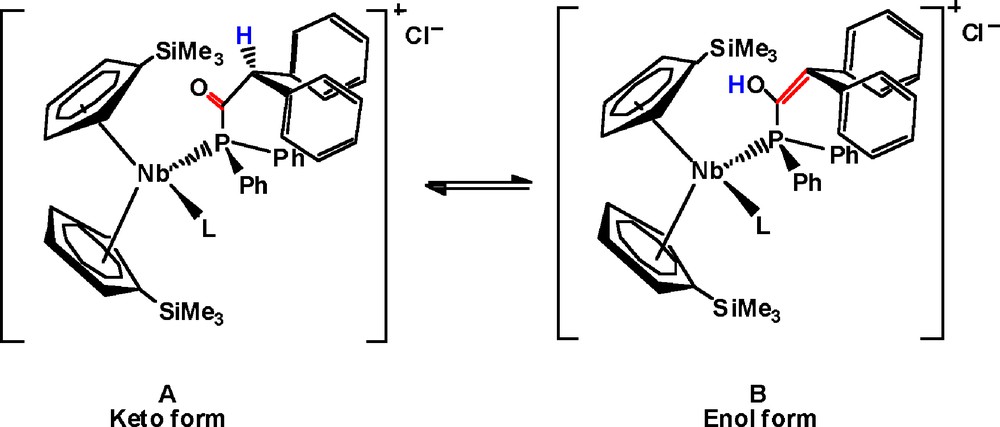
The spectroscopic data are consistent with the isolation of new niobocenes containing an acylphosphane ligand. These compounds are formed by SN2 reaction of the phosphido-niobocene complexes 1 and 2 with diphenylacetyl chloride. According to the behavior of some phosphamido complexes reported in the literature [12], which exhibit keto-enol tautomerism, the presence of the acidic methylenic proton of the acylphosphane ligand may give rise to such a tautomeric equilibrium in which enol and keto niobocene derivatives could be present (See Scheme 5). However, for complexes 7 and 8, this equilibrium was not observed according to the IR and 13C{1H}-NMR data discussed above. Thus, a pseudo-tetrahedral arrangement for these complexes containing a ketone-phosphane moiety is proposed.
3 Conclusions
The synthesis of new functionalized phosphane-containing niobocenes is described and this process involves a P–C bond formation reaction of phosphido-niobocene complexes 1 and 2 with both alkyl and acyl chloride reagents. The new complexes 3–8 can therefore be viewed as novel metallophosphanes, where the presence of the carbonyl group bound to the phosphorus atom may increase the coordination modes of the phosphane and therefore may enrich the applications of these complexes as building blocks.
4 Experimental section
4.1 General experimental conditions
All reactions were carried out using Schlenk techniques. Oxygen and water were excluded through the use of vacuum lines supplied with purified N2. Toluene was distilled from sodium. Hexane was distilled from sodium/potassium alloy. Diethyl ether and THF were distilled from sodium benzophenone. All solvents were deoxygenated prior to use.
4.2 Instrumentation
1H, 13C and 31P NMR spectra were recorded on a Varian Innova 500 MHz spectrometer at ambient temperature unless stated otherwise. 1H, 13C and 31P NMR chemical shifts (δ values) are given in ppm relative to the solvent signal (1H, 13C) or standard resonances (31P, external 85% H3PO4). IR spectra were recorded on a Perkin-Elmer 883 spectrophotometer as Nujol mulls on CsI windows. Electrical conductivity was measured on a Metrohm Herisau E587 Conductimeter, with a cell constant of 0.79 cm−1 and 5.10−4 M concentration of the samples in acetone.
4.3 Reagents
[Nb(η5-C5H4SiMe3)2(PHPh2)(CO)]Cl (1) [13] and [Nb(η5-C5H4SiMe3)2(PHPh2)(CNXylyl)]Cl (2) [13] were prepared as described in the literature. Deuterated solvents were dried over 4 Å molecular sieves and degassed prior to use. PhCOCH2Cl, PhCOCl, Ph2CHCOCl were used as supplied by Aldrich.
4.4 Synthesis of [Nb(η5-C5H4SiMe3)2(P(CH2CO(C6H5))Ph2)(L)]Cl, LCO (3), CNXylyl (4)
To a solution of 1 (0.35 g, 0.57 mmol) or 2 (0.40 g, 0.56 mmol) in anhydrous hexane (30 mL) at room temperature was added an equimolar quantity of PhCOCH2Cl (0.09 g, 0.57 mmol for 1, 0.09 g; 0.56 mmol for 2). A yellowish-orange solid for 3 and an orange solid for 4 precipitated immediately. The mixture was stirred for 1 h and the solids were filtered off and washed with anhydrous hexane (2 × 10 mL). Complexes 3 and 4 are soluble in polar solvents such as acetone, chloroform and dichloromethane, slightly soluble in THF and toluene and insoluble in apolar solvents like diethyl ether, pentane and hexane.
(3): Yield: 0.35 g (85% rel. 1). Electrical conductivity: ΛM (Ω−1cm2mol−1); 71.9. IR (Nujol/Polyethylene): ν (cm−1); 1946 (C≡O), 1679 (CO). 1H-NMR (CDCl3): δ (ppm); 0.24 (s, 18H, SiMe3), 4.85 (d, 2H, 2JHP = 7.7 Hz, CH2), 4.72, 5.44, 5.76, 5.79 (m, 2H, C5H4), 7.42–8.13 (m, 15H, Ph). 13C{1H}-NMR (CDCl3): δ (ppm); 0.1 (SiMe3), 42.6 (d, 1JCP = 23.8 Hz, CH2), 95.9, 96.7, 100.7, 101.7 (C5H4), 128.6–133.5 (Ph). 31P{1H}-NMR (CDCl3): δ (ppm); 41.0 (s). 31P-NMR (CDCl3): δ (ppm); 41.0 (m). Anal. Found: C, 60.27; H, 5.76. Calcd. (C37H43ClNbO2PSi2): C, 60.44; H, 5.89; M, 735.24.
(4): Yield: 0.40 g (85% rel. 2). Electrical conductivity: ΛM (Ω−1cm2mol−1); 77.4. IR (Nujol/Polyethylene): ν (cm−1); 2046 (C≡N); 1675 (CO). 1H-NMR (CDCl3): δ (ppm); 0.17 (s, 18H, SiMe3), 2.40 (s, 6H, CH3 Xylyl), 5.09 (d, 2H, 2JHP = 7.32 Hz, CH2), 4.50, 5.21, 5.82, 5.89 (m, 2H, C5H4), 7.11–8.78 (m, 14H, Ph), 8.19 (d, 4H, 2JHP = 7.3 Hz, Ph). 13C{1H}-NMR (CDCl3): δ (ppm); 0.3 (SiMe3), 19.3 (CH3 CNXylyl), 43.1 (d, 1JCP = 19.9 Hz, CH2), 91.4, 101.3, 102.5 (C5H4), 125.7–134.3 (Ph), 135.9 (d, 1JCP = 35.7 Hz, Ph), 137.1 (Cipso Ph’) 195.8 (CO), 199.0 (CN). 31P{1H}-NMR (CDCl3): δ (ppm); 38.3 (s). 31P-NMR (CDCl3): δ (ppm); 38.3 (m). Anal. Found: C, 63.36; H, 6.03; N, 1.67. Calcd. (C45H52ClNNbO2PSi2): C, 63.26; H, 6.13; N, 1.64; M, 854.40.
4.5 Synthesis of [Nb(η5-C5H4SiMe3)2(P(CO(C6H5))Ph2)(L)]Cl, LCO (5), CNXylyl (6)
Complexes 5 and 6 were prepared using a similar procedure to 3 and 4 by the reaction of complexes 1 and 2 (0.29 g, 0.47 mmol for 1, 0.57 g, 0.79 mmol for 2) in anhydrous hexane (30 mL) with a stoichiometric amount of PhCOCl (ρ = 1.211 g mL−1; 0.07 g, 0.47 mmol for 1, 0.11 g, 0.79 mmol for 2) for 1 h at room temperature. Complexes 5 and 6 were isolated as red solids after drying and they were washed twice anhydrous hexane (2 × 10 mL) at 0 °C. Complexes 5 and 6 are soluble in acetone, chloroform and dichloromethane but are insoluble in diethyl ether, pentane and hexane.
(5): Yield: 0.28 g (83% rel. 1). Electrical conductivity: ΛM (Ω−1cm2mol−1); 78.6. IR (Nujol/Polyethylene): ν (cm−1); 1947 (C≡O), 1649 (CO). 1H-NMR (CDCl3): δ (ppm); −0.06 (s, 18H, SiMe3), 3.96, 5.12, 5.26, 6.14 (m, 2H, C5H4), 6.80–7.39 (m, 15H, Ph). 13C{1H}-NMR (CDCl3): δ (ppm); 0.1 (SiMe3), 95.7, 98.4, 100.3, 101.0, 101.3 (C1) (C5H4), 128.5–137.7 (Ph), 208.6 (m, CO), 248.1 (C≡O). 31P{1H}-NMR (CDCl3): δ (ppm); 64.8 (s). 31P-NMR (CDCl3): δ (ppm); 64.8 (m). Anal. Found: C, 60.09; H, 5.76. Calcd. (C36H41ClNbO2PSi2): C, 59.95; H, 5.73; M, 721.21.
(6): Yield: 0.55 g (85% rel. 2). Electrical conductivity: ΛM (Ω−1cm2mol−1); 76.4. IR (Nujol/Polyethylene): ν (cm−1); 2054 (C≡N), 1649 (CO). 1H-NMR (CDCl3): δ (ppm); 0.21 (s, 18H, SiMe3), 2.40 (s, 6H, CH3 Xylyl), 4.39, 5.33, 5.36, 5.57 (m, 2H, C5H4), 7.01–7.51 (m, 18H, Ph). 13C{1H}-NMR (CDCl3): δ (ppm); 0.2 (SiMe3), 19.1 (CH3 CNXylyl), 90.4, 100.1, 100.6, 101.9 (C5H4), 125.6–133.9 (Ph), 194.7 (C≡N), 207.0 (m, CO). 31P{1H}-NMR (CDCl3): δ (ppm); 64.7 (s). 31P-NMR (CDCl3): δ (ppm); 64.7 (m). Anal. Found: C, 64.24; H, 6.21, N, 1.62. Calcd (C44H50ClNNbOPSi2): C, 64.11; H, 6.11; N, 1.70; M, 824.38.
4.6 Synthesis of [Nb(η5-C5H4SiMe3)2(P(COCH(C6H5)2)Ph2)(L)]Cl, LCO (7), CNXylyl (8)
Complexes 7 and 8 were prepared as described previously for complexes 3–6. To a solution of 1 or 2 (0.30 g, 0.49 mmol for 1 and 0.49 g, 0.68 mmol for 2) in anhydrous hexane (30 mL) was added the stoichiometric amount of Ph2CHCOCl (0.11 g, 0.49 mmol for 1, 0.16 g, 0.68 mmol for 2) at room temperature and the mixture was stirred for 1 hour. Orange solids were isolated after drying these were washed with anhydrous hexane (2 × 10 mL) at 0 °C. The compounds are slightly soluble in hexane, pentane, toluene and diethyl ether and very soluble in polar solvents such as acetone and dichloromethane.
(7): Yield: 0.34 g (86% rel. 1). Electrical conductivity: ΛM (Ω−1cm2mol−1); 87.2. IR (Nujol/Polyethylene): ν (cm−1); 1950 (C≡O), 1682 (CO). 1H-NMR (CDCl3): δ (ppm); 0.18 (s, 18H, SiMe3), 5.67 (s, 1H, CH), 4.15, 5.18, 5.55, 6.18 (m, 2H, C5H4), 7.05 (s, 5H, Ph), 7.24 (m, 10H, Ph), 7.49 (m, 5H, Ph). 13C{1H}-NMR (CDCl3): δ (ppm); 0.1 (SiMe3), 63.0 (d, 2JCP = 31.4 Hz, CH), 95.0, 98.5, 101.1, 103.7 (C5H4), 127.9, 129.4, 128.7, 131.6 (Ph), 129.4 (d, 2JCP = 9.1 Hz, Ph), 134.3 (d, 1JCP = 59.3 Hz, Cipso Ph). 31P{1H}-NMR (CDCl3): δ (ppm); 74.9 (s). 31P-NMR (CDCl3): δ (ppm); 74.9 (m). Anal. Found: C, 63.57; H, 5.83. Calcd. (C43H47ClNbO2PSi2): C, 63.66; H, 5.84; M, 811.34.
(8): Yield: 0.53 (84% rel. 2). Electrical conductivity: ΛM (Ω−1cm2mol−1); 83.3. IR (Nujol/Polyethylene): ν (cm−1); 2038 (C≡N); 1672 (CO). 1H-NMR (CDCl3): δ (ppm); 0.11 (s, 18H, SiMe3), 2.32 (s, 6H, CH3 Xylyl), 5.26 (s, 1H, CH), 4.64, 5.02, 5.26, 5.48 (m, 2H, C5H4), 6.82–7.78 (m, 23H, Ph). 13C{1H}-NMR (CDCl3): δ (ppm); –0.1 (SiMe3), 18.8 (CH3 CNXylyl), 63.2 (d, 2JCP = 29.6 Hz, CH), 91.7, 99.7, 100.0, 100.7, 101.4 (C5H4), 127.9–135.9 (Ph), 129.2 (d, JCP = 9.5 Hz, Ph), 133.0 (d, JCP = 9.9 Hz, Ph), 193.1 (CO), 213.9 (CN). 31P{1H}-NMR (CDCl3): δ (ppm); 70.9 (s). 31P-NMR (CDCl3): δ (ppm); 70.9 (m). Anal. Found: C, 66.81; H, 6.05; N, 1.49. Calcd. (C51H56ClNNbOPSi2): C, 66.98; H, 6.17; N, 1.53; M, 914.50.
Acknowledgements
The authors gratefully acknowledge financial support from the Ministerio de Ciencia e Innovación (MICINN), Spain (Grants CTQ2006-11845/BQU and CTQ2008-00318/BQU, Consolider Ingenio 2010 ORFEO CSD-2007-00006), UE Action COST CM0802 and the Junta de Comunidades de Castilla-La Mancha (Grant no PCI08-0010, PCIO8-0032).