

1 Introduction
Deficiencies of α- and β-hexosaminidases are responsible for a number of lysosomal storage diseases; lack of enzymes to cleave a terminal N-acetyl-β-D-glucosamine unit gives rise to Sanfilippo [1], Tay-Sachs and Sandhoff [2] diseases whereas lack of an enzyme to hydrolyze a terminal N-acetyl-α-D-galactosamine component is associated with Schindler-Kanzaki disease [3]. Inhibition of hexosaminidases may also provide strategies for the treatment of osteoarthritis [4], cancer [5], and type II diabetes [6]; thus the design of specific and potent hexosaminidase inhibitors provides an attractive synthetic target.
Iminosugars – in which the ring oxygen of the carbohydrate is replaced by nitrogen – constitute a family of both natural and synthetic inhibitors of glycosidases [7]. DNJNAc 1 [8], the corresponding analogue of N-acetyl-α-D-glucosamine, and its N-alkyl analogues [9] – as well as other synthetic piperidine pyranose mimics [10] – are potent inhibitors of β-hexosaminidases (Fig. 1). The natural product nagstatin 2 [11], with a galacto-configuration in the piperidine ring, is also a potent inhibitor of β-hexosaminidases [12]. The analogue of N-acetyl-α-D-galactosamine 3 is the only powerful inhibitor of N-acetyl-α-D-galactosaminidases (GalNAcases) yet reported [13]. Hydroxyazepane [14] mimics of N-acetyl-hexosamines, such as the azepanes 4 and 5, are specific sub-micromolar inhibitors of β-hexosaminidases [15]. Pochonicine 6 [or its enantiomer], a nanomolar pyrrolizidine β-hexosaminidase inhibitor, has been isolated from Pochonia suchlasporia var. suchlasporia TAMA 87 [16].
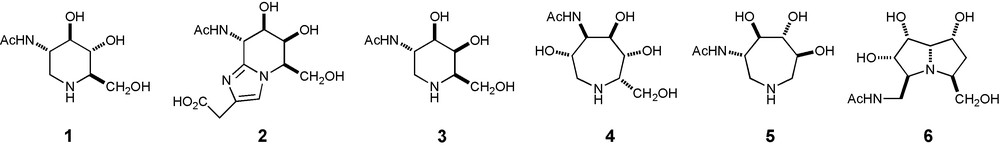
Potent inhibitors of hexosaminidases.
Pyrrolidine analogues of pentoses provide examples of powerful glycosidase inhibitors. The natural product DAB 7D, isolated from Arachniodes standishii and Angylocalyx boutiqueanus [17], shows strong inhibition of α-glucosidases and weaker inhibition of several other glycosidases; the unnatural enantiomer LAB 7L is also a potent – but highly specific – inhibitor of α-glucosidases [18] (Scheme 1). A significant number of L-enantiomers of imino-D-sugar mimics show inhibition of the same glycosidases [19]. Both DABNAc 8D and LABNAc 8L, the NAc analogues of DAB 7D and LAB 7L were synthesised from the enantiomers of lyxonolactone [20]. LABNAc 8L and its N-alkyl analogues [such as the N-benzyl derivative] were micromolar – whereas DABNAc 8D analogues were weak – specific inhibitors of β-hexosaminidases; neither showed any inhibition of α-GalNAcases [21].
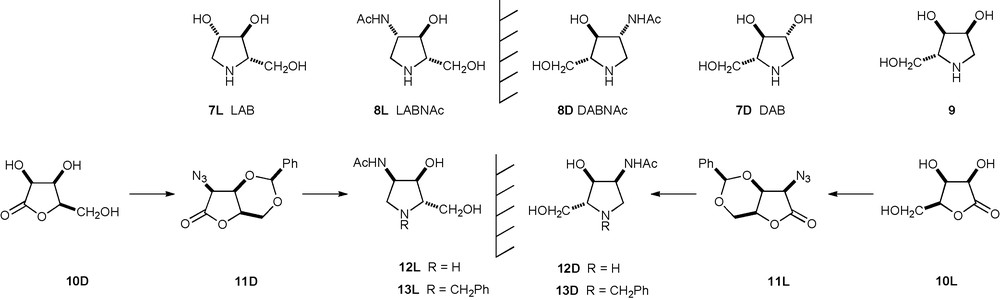
Relationships between pyrrolidine glycosidase inhibitors.
The imino ribitol 9, isolated from the mulberry tree Morus alba [22] and synthesized from carbohydrate [23] and non-carbohydrate [24] starting materials, was a weak inhibitor of several glycosidases (IC50 = 230–610 μM) [13], but was shown to mimic a galactose motif by its moderate inhibition of a β-D-galactosidase (rat intestinal lactase, IC50 17 μM) [25]. LRBNAc 12L and DRBNAc 12D are related to 9 in the same way that LABNAc 8L and DABNAc 8D are related to LAB 7L and DAB 7D. Accordingly, in the search for GalNAcase inhibitors, the syntheses of LRBNAc 12L from D-lyxonolactone 10D and of DRBNAc 12D from L-lyxonolactone 10L (Scheme 1) – which depend on the efficient introduction of an azide at C2 with retention of configuration in suitably protected intermediates 11L and 11D – are herein described. However, this paper reports that neither DRBNAc 12D nor LRBNAc 12L showed significant inhibition of any glycosidase.
The key steps in the synthesis of LABNAc 8L from D-lyxonolactone 10D were introduction of nitrogen by nucleophilic substitution with inversion at C2, and formation of the pyrrolidine ring by nucleophilic substitutions by benzylamine between C5 and C2 [with inversion of configuration] of the D-lyxo sugar to give the required L-ribo configuration. Reaction of D-lyxonolactone 10D with benzaldehyde and hydrochloric acid gave the highly crystalline benzylidene derivative 14D (90% yield) in which only the C2-OH group of the lactone is unprotected (Scheme 2).
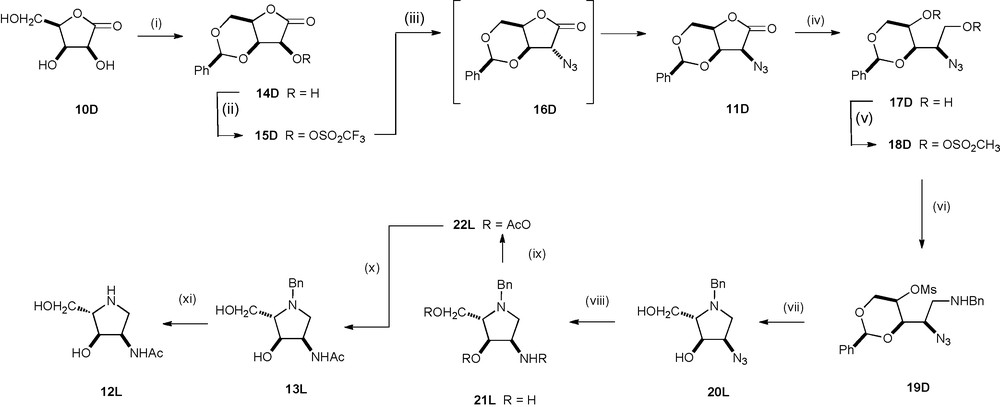
Reagents and conditions: (i) PhCHO, HCl, 90% (ii) Tf2O, pyridine, −50 °C, 2.5 h, 100%; (iii) NaN3, PPTS, DMF, RT, 20 h, 86% 11D, 4% 16D; (iv) L-SelectrideØ, THF, −78 °C, 2 h, 57%; (v) MsCl, Et3N, DCM, 0 °C, 2 h, 92%; (vi) BnNH2, 95 °C, 20 h, 80%; (vii) AmberlystØ 15 beads, 1,4-dioxane, H2O, 95 °C, 14 h; (viii) PPh3, H2O, THF, RT, 14 h, 89%; (ix) Ac2O, pyridine, RT, 14 h, 85%, (x) NaOMe, MeOH, RT, 2 h, 91%; (xi) H2/Pd black, 1,4-dioxane, H2O, r.t., 18 h, 88%.
There are two strategies for the introduction of azide at C2 of 10D with retention of configuration. One route involves two successive SN2 reactions; initial inversion by trifluoroacetate displacement of a triflate to afford the epimeric alcohol [26], with a subsequent SN2 displacement of the epimeric triflate by azide to give overall retention of configuration [27]. An alternative approach depends on the observation that, in general, azides which are cis to all the other substituents on a lactone ring are thermodynamically more stable than epimers in which the azide is the sole trans substituent [28]; there are even examples where base catalysed epimerization of the less stable kinetic triflate is so rapid that it is not possible to isolate the kinetic azide but only the thermodynamic azide with retention of configuration [29]. Esterification of 14D with triflic (trifluoromethanesulfonic) anhydride in pyridine gave the corresponding triflate 15D in quantitative yield [extra care must be taken in the handling of this triflate, as a co-worker experienced a strong allergic response to this compound or its decomposition products]. Treatment of the lyxono-triflate 15D with sodium azide in DMF produced 83% yield of the xylono-azide 16D under kinetic control using a short reaction time (50–60 min) and just under stoichiometric amounts of sodium azide (0.97 equivalents) during the reaction [21,28]. However, when the triflate 15D was left for 20 h at room temperature in the presence of excess sodium azide and pyridinium p-toluenesulfonate (PPTS) the thermodynamic lyxono-azide 11D was formed in 86% yield with a trace of the easily separable xylono-azide 16D (4%).
The sensitivity of α-azidolactones to base frequently results in low yields of direct conversion of the lactone to the corresponding diol by hydrides [27]. Attempted reductions of the azidolactone 11D with lithium borohydride, sodium borohydride or Super-Hydride® gave low yields of the lyxo-diol 17D. However, L-Selectride® in THF at −30 °C gave acceptable yields for the formation of lyxo-diol 17D (57%); after the reduction was complete, it was necessary to employ an oxidative workup procedure using sodium hydroxide and aqueous hydrogen peroxide to remove the borane adducts in order to obtain a reasonable yield of pure material after flash chromatography. Esterification of the lyxo-diol 17D with mesyl (methanesulfonyl) chloride in dichloromethane in the presence of triethylamine at 0 °C afforded the dimesylate 18D in 90% yield. Reaction of the dimesylate 18D with benzylamine at 95 °C for 20 h gave the aminobenzylmesylate 19D in 80% yield; there was no subsequent intramolecular cyclization since the resulting pyrrolidine would have a benzylidene acetal trans to the pyrrolidine ring. Deprotection of the benzylidene acetal of 19D which would allow subsequent cyclization to the pyrrolidine 20L, was achieved by heating 19D, in aqueous dioxane in the presence of acid ion-exchange resin at 95 °C for 14 h. After filtering off the resin, washing with 2 M ammonium hydroxide solution, solvent removal and flash chromatography, the L-ribo-pyrrolidine 20L was isolated in 89% yield. Staudinger reduction [30] of the azide 20L by triphenylphosphine and water in THF gave, after purification by ion-exchange chromatography, the L-ribo-amine 21L in 89% yield. Reaction of the aminodiol 21L with acetic anhydride in pyridine afforded the triacetyl derivative 22L (85% yield). The O-acetyl groups in 22L were selectively removed by treatment with catalytic sodium methoxide in methanol to give N-benzyl-LRBNAc 13L, in 91% yield; the structure of 13L was unequivocally established by X-ray crystallographic analysis [31]. Removal of the N-benzyl group in 13L by hydrogenolysis in the presence of palladium black in aqueous 1,4-dioxane gave LRBNAc 12L in 88% yield. The preparation of LRBNAc 12 from D-lyxonolactone was achieved in 18% overall yield over the 12 steps.
DRBNAc 12D was prepared from 3,5-benzylidene-L-lyxonolactone 10L by an identical procedure; the benzylidene lactone 10L was formed in 95% yield [21] from 2,3-isopropylidene-L-lyxonolactone, readily available on a large scale from D-ribose [32].
2 Inhibition studies
Neither N-benzyl-DRBNAc 13D nor N-benzyl-LRBNAc 13L showed significant inhibition of the hydrolysis of the corresponding p-nitrophenyl glycosides by any of the following range of enzymes: α-glucosidase (yeast), β-glucosidase (almond), α-galactosidase (coffee bean), β-galactosidase (jack bean), α-mannosidase (jack bean), β-mannosidase (snail), α-fucosidase (bovine), β-xylosidase (Aspergillus niger), β-N-acetylhexosaminidase (jack bean), and α-N-acetylgalactosaminidase (Charonia lampas). In particular, there was no inhibition of any α- or β-hexosaminidases.
3 Summary
The synthesis of LRBNAc 12L from D-lyxonolactone 10D over 12 steps proceeded in a good overall yield of 18%. The enantiomer DRBNAc 12D was also prepared from L-lyxonolactone. Assays of N-benzyl-LRBNAc 13L and N-benzyl-DRBNAc 13L showed there was no significant inhibition of either β-N-acetylhexosaminidase (jack bean) or α-N-acetylgalactosaminidase (Charonia lampas).
4 Experimental
THF was distilled from sodium benzophenone ketyl or purchased dry from Aldrich in Sure/Seal™ bottles and recapped with Oxford Sure/Seal™ valve-caps. N,N-Dimethylformamide (DMF) and pyridine were purchased dry from Aldrich in Sure/Seal™ bottles and were recapped with Oxford Sure/Seal™ valve caps. Water was distilled. All other solvents were used as supplied (analytical or HPLC grade) without prior purification. Reactions performed under an atmosphere of nitrogen, argon, or hydrogen gas were maintained by an inflated balloon. Imidazole was recrystallised from dichloromethane. All other reagents were used as supplied without prior purification. Thin-layer chromatography (TLC) was performed on aluminium-backed sheets coated with 60 F254 silica. Sheets were developed by dipping in a solution of cerium(IV) sulfate (0.2% w/v) and ammonium molybdate (5%) in 2 M sulfuric acid and heating with a heat gun. Flash column chromatography was performed using Sorbosil® C60 40/60 silica. Ion-exchange chromatography was performed using either Amberlite® CG-150 or Dowex® 50WX8–200, as stated. NMR spectra were recorded on a Bruker DQX 400 (1H: 400 MHz, 13C: 100 MHz) spectrometer at room temperature in the deuterated solvent stated. All chemical shifts (δ) are quoted in ppm. Coupling constants (3J) are quoted in Hz. Residual signals from the solvents were used as an internal reference. The 13C resonances were assigned using either DEPT or APT sequences or from 1H–13C HMQC and HMBC spectra. Melting points were recorded on a Kofler hot block and are uncorrected. IR spectra were recorded at room temperature on a PerkinElmer 1750 IR Fourier transform spectrophotometer or a Bruker Tensor 27 FTIR spectrometer, using thin films on NaCl or Ge plates, as stated, and were recorded over 16 or 32 scans at a resolution of 4 cm−1. Only the characteristic peaks are quoted. Mass spectrometry: high-resolution mass spectra (HRMS) were recorded on a micromass Autospec 500 OAT spectrometer using the technique of chemical ionisation (CI), or on a Waters 2790-Micromass LCT mass spectrometer using the technique of electrospray ionisation (ESI). Optical rotations were recorded on a PerkinElmer 241 polarimeter with a path length of 1 dm. Concentrations are quoted in g(100 mL)−1. The wavelength at which the rotations were measured corresponds to the sodium D line (ca. 589 nm). Elemental analyses were performed by the microanalysis service of the Inorganic Chemistry Laboratory, University of Oxford.
5 Enzyme inhibition assays
All enzymes and corresponding PNP substrates were obtained from Sigma, apart from coffee bean α-D-galactosidase, jack bean β-D-galactosidase, α-D-mannosidase and β-N-acetyl-D-hexosaminidase, and Charonia lampas α-N-acetyl-D-galactosaminidase, which were purified from the natural sources in the Oxford Glycobiology Institute. Enzyme solutions: For the purposes of this investigation, all enzyme solutions were made up using 50 mM citrate–phosphate buffer (anhydrous disodium hydrogen phosphate dissolved in citric acid) at pH 5 containing 1 mg mL−1 bovine serum albumin (BSA) and 0.02% NaN3, except α-D-glucosidase, for which Dulbecco's phosphate buffered saline (PBS) buffer at pH 7.4 was used. These solutions were stored on ice whilst in use and at −20 °C when not in use. Inhibitor solutions: NBn-LRBNAc 13L and NBn-DRBNAc 13D were dissolved in distilled water at a concentration of 10 mg mL−1. These solutions were stored on ice whilst in use and at −20 °C when not in use. Enzyme substrate solutions: Substrate solutions were made from a stock solution (3 mM) in 10 mL batches, as required, by dissolving the appropriate mass of substrate in the appropriate buffer solution for the enzyme. These were kept at 4 °C when not in use. Method: Assays were carried out in triplicate, using H2O as a blank in place of the inhibitor. The concentrations of the enzyme were adjusted so that the reading for the final absorbance was in the range of 0.5–1.5 units over a reaction time of 20 min. For example, β-N-acetyl-D-hexosaminidase was diluted from 6 U mL−1 to 0.4 U mL−1 by experiment. Linearity over the time course of the reaction was checked using a series of incubation times. The following were combined in the well of a flat-bottomed 96-well (300 mL) microtitre plate: 5 μL enzyme solution, 5 μL inhibitor solution, and 40 μL substrate solution. The reaction mix was incubated at 37 °C for 20 min (Denley Wellwarm 1) and was quenched by the addition of 200 μL of 0.5 M Na2CO3. Absorbance at 405 nm was measured immediately using a microtitre plate reader (Molecular Devices UVmax kinetic microplate reader and SOFTmax 2.35 software).
6 2-Azido-3,5-O-benzylidene-2-deoxy-D-lyxono-1,4-lactone 11D
Triflic anhydride (2.44 mL, 14.5 mmol, 1.6 equivalents) was added dropwise to a stirred solution of the benzylidene lactone 14D21 (2.14 g, 9.06 mmol) in pyridine (20 mL) at −50 °C under an atmosphere of nitrogen. The temperature was allowed to rise to room temperature during the course of the reaction. After 2.5 h, analysis by TLC (ethyl acetate) showed no residual starting material (Rf 0.07) and a major spot due to the product (Rf 0.50). The solvents were removed in vacuo, by coevaporation with toluene (high vacuum). The residue was partitioned between ethyl acetate (80 mL) and 0.1 M HCl (50 mL). The resulting biphasic solution was separated and the organic layer washed with brine (50 mL). The aqueous phase was extracted with ethyl acetate (3 × 15 mL) and the combined organic phase was dried (magnesium sulfate) and the solvents removed in vacuo. Sodium azide (1.46 g, 22.6 mmol, 2.5 equivalents) was added to a stirred solution of crude triflate 15D (ca. 3.34 g, 9.06 mmol) in DMF (35 mL) at room temperature under an atmosphere of nitrogen. After 1 h, PPTS (2.73 g, 10.9 mmol) was added. After a further 18 h, analysis by TLC (1:1, ethyl acetate/cyclohexane) showed a major product spot (Rf 0.17) and a minor spot (Rf 0.41). The solvents were removed in vacuo, by coevaporation with toluene (high vacuum); the residue was purified by flash chromatography (3:1 → 1:1, cyclohexane/ethyl acetate) to give the azido-lyxono-lactone 11D (2.04 g, 7.81 mmol, 86%) as a white solid, m.p. 122-123 °C, 128-130 °C (recrystallised from ethyl acetate/cyclohexane) [Lit. [28] 121-122 °C (acetone)];
The enantiomeric azidolactone 11L, m.p. 128-130 °C;
7 2-Azido-3,5-O-benzylidene-2-deoxy-D-lyxitol 17D
A 1 M solution of L-Selectride® in THF (2.87 mL, 2.5 equivalents) was added dropwise to a stirred solution of the lyxono-lactone 11D (300 mg, 1.15 mmol) in THF (3.0 mL) at −78 °C under an atmosphere of argon in the presence of molecular sieve (4 Å). After 2 h, analysis by TLC (ethyl acetate) showed a major spot due to the product (Rf 0.50) and no residual starting material (Rf 0.59). Methanol (0.5 mL) was added slowly to destroy the excess hydride and after 15 min a further portion (2.5 mL) was added. After a further 2 h, the mixture was filtered over Celite®; diethyl ether (10 mL) and a 50% v/v solution of 4 M NaOH/35% aqueous H2O2 (5 mL) were added to the filtrate and the reaction mixture allowed to stir for 1 h. The resulting biphasic solution was separated and the aqueous phase was extracted using ethyl acetate (3 × 2 mL). The combined organic phase was dried (magnesium sulfate) and the solvents were removed in vacuo. The residue was passed through a silica plug (9:1 ethyl acetate/methanol) to yield the diol 17D (175 mg, 57%) as a white amorphous solid, m.p. 102–103 °C;
The enantiomeric diol 17L, m.p. 101-102 °C;
8 2-Azido-3,5-O-benzylidene-2-deoxy-1,4-di-O-methanesulfonyl-D-lyxitol 18D
Triethylamine (0.60 mL, 4.32 mmol, 4 equiv.) was added to a stirred solution of the diol 17D (287 mg, 1.08 mmol) in dichloromethane (6.0 mL) at 0 °C under an atmosphere of nitrogen. After 20 min, methanesulfonyl chloride (0.34 ml, 4.32 mmol, 4 equiv.) was added dropwise. After 2 h, analysis by TLC (ethyl acetate) showed no residual starting material (Rf 0.56) and a major spot due to the product (Rf 0.67) [Note: change in the colour of the spot made it easier to see the progress of the reaction]. The solvents were removed in vacuo and the residue was partitioned between ethyl acetate (20 mL) and brine (30 mL). The layers were separated and the aqueous phase was extracted using ethyl acetate (3 × 10 mL). The combined organic phase was dried (magnesium sulfate), the solvents were removed in vacuo, and the remaining residue was purified by flash chromatography (1:1, cyclohexane/ethyl acetate) to afford the dimesylate 18D (421 mg, 92%) as a white crystalline solid, m.p. 139-140 °C;
The enantiomeric dimesylate 18L, m.p. 138-140 °C;
9 1-Aminobenzyl-2-azido-3,5-O-benzylidene-1,2-dideoxy-4-O-methanesulfonyl-D-lyxitol 19D
The dimesylate 18D (669 mg, 1.59 mmol) was dissolved in benzylamine (14.0 mL) and the resulting solution heated at 95 °C. After 20 h, analysis by TLC (1:1, cyclohexane/ethyl acetate) showed one major spot (Rf 0.32) and a UV active spot (Rf 0.59) due to residual benzylamine. The solvents were removed in vacuo by co-evaporation with toluene. The resulting orange residue was partitioned between ethyl acetate (30 mL) and brine (40 mL). The layers were separated and the aqueous phase was extracted using ethyl acetate (3 × 15 mL). The combined organic phase was dried (magnesium sulfate), the solvents were removed in vacuo and the resulting residue was purified by flash chromatography (9:1 → 1:1, cyclohexane/ethyl acetate) to yield the aminomesylate 19D (551 mg, 1.27 mmol, 80%) as a white amorphous solid, m.p. 89-91 °C;
The enantiomeric aminomesylate 19L, m.p. 91-92 °C;
10 N-Benzyl-2-azido-1,4-imino-1,2,4-trideoxy-L-ribitol 20L
Amberlyst® 15 (wet) beads were added to a stirred solution of the aminomesylate 19D (540 mg, 1.25 mmol) in 1,4-dioxane (20 mL) and water (20 mL) and the resulting mixture was heated at 95 °C under an atmosphere of nitrogen. After 14 h, analysis by TLC (ethyl acetate) showed one spot (Rf 0.23) due to the product. After cooling to room temperature, 2 M NH4OH (aq) solution (10 mL) was added and the mixture was stirred for 2 h. The beads were filtered off, washing thoroughly with 2 M NH4OH (3 × 5 mL) and 1,4-dioxane (10 mL). The solvents were removed in vacuo and the remaining residue was purified by flash chromatography (ethyl acetate) to give the imino-L-ribitol 20L (275 mg, 89%) as a pale brown oil;
The enantiomeric benzylamine 20D, oil;
11 2-Amino-N-benzyl-1,4-imino-1,2,4-trideoxy-L-ribitol 21L
Triphenyl phosphine (169 mg, 0.644 mmol) was added to a stirred solution of the azide 20L (80.0 mg, 0.322 mmol) and water (12 μL, 0.322 mmol) in THF (1.0 mL) at room temperature under an atmosphere of nitrogen. After 14 h, analysis by TLC (ethyl acetate) showed a major product spot (baseline) and no residual starting material (Rf 0.23). The solvents were removed in vacuo and the remaining residue was partitioned between chloroform (3 mL) and water (2 mL). The aqueous layer was extracted with chloroform (3 × 1 mL) to remove excess triphenylphosphine oxide. The organic layer was extracted with water (3 × 1 mL); the combined aqueous phase was purified by ion exchange chromatography (Dowex® 50WX8-200) to afford the amine 21L (64.0 mg, 0.288 mmol, 89%) as a colourless oil;
The enantiomeric diamine 21D, oil;
12 2-Acetamido-N-benzyl-3,5-di-O-acetyl-1,4-imino-1,2,4-trideoxy-L-ribitol 22L
Acetic anhydride (0.15 mL, 1.620 mmol) was added dropwise to a stirred solution of the amine 21L (60.0 mg, 0.270 mmol) in pyridine (2.0 mL) at room temperature under an atmosphere of argon. After 14 h, analysis by TLC (ethyl acetate) showed no residual starting material (baseline) and a spot due to the product (Rf 0.21). The solvents were removed in vacuo using toluene to aid by coevaporation; the remaining residue was purified by flash chromatography (98:2, ethyl acetate/triethylamine) to form the triacetate 22L as a colourless oil (79.8 mg, 0.229 mmol, 85%);
The enantiomeric triacetate 22D, oil;
13 2-Acetamido-N-benzyl-1,4-imino-1,2,4-trideoxy-L-ribitol (N-benzyl-LRBNAc) 13L
Sodium methoxide (3.2 mg, 0.059 mmol) was added to a stirred solution of the triacetate 22L (103 mg, 0.296 mmol) in methanol (1.0 mL) at room temperature under an atmosphere of nitrogen. After 2 h, analysis by TLC (acetone) showed no residual starting material (Rf 0.55) and a spot due to the product (Rf 0.29). The solvents were removed in vacuo and the resulting residue was purified by flash chromatography (ethyl acetate, then acetone) to give the N-benzyl-acetamide 13L (71.1 mg, 91%) as a pale yellow oil that was recrystallised from acetonitrile, m.p. (acetonitrile) 81–82 °C;
The enantiomeric acetamide 13D, oil;
14 2-Acetamido-1,4-imino-1,2,4-trideoxy-L-ribitol (LRBNAc) 12L
Palladium black (10.0 mg) was added to a stirred solution of N-benzyl derivative 13L (40.0 mg, 0.151 mmol) in 1,4-dioxane (0.5 mL) and water (0.5 mL) at room temperature under an atmosphere of H2. After 18 h, analysis by TLC (acetone) showed no residual starting material (Rf 0.15) and a spot due to the product on the baseline. The palladium black was filtered off over Celite®, washing with methanol. The solvents were removed in vacuo and the remaining residue was purified by ion-exchange chromatography (Dowex® 50WX8-200) to afford LRBNAc 12L (23.3 mg, 88%) as an orange oil;
The enantiomeric DRBNAc 12D, oil;