

1 Introduction
The simultaneous emission of volatile organic compounds (VOCs), nitrogen oxides (NOx) and carbonaceous particles concerns many industries (steel, incineration, combustion of biofuels…). A catalytic treatment may be an advantageous route for the purification of such effluents [1–3].
The catalytic oxidation of VOCs to carbon dioxide and water has been identified as one of the most efficient ways to their destruction at low concentrations and to meet increasingly environmental regulations. The practical applications of the catalytic oxidation process require heating large amounts of gas containing low concentrations of VOCs to find the oxidation temperature. Therefore, highly active catalysts at low temperatures are required [4]. Moreover, the oxidation of hydrocarbons in the presence of NOx can be interesting both for automotive pollution control and in various industrial plants [2,3]. However, the studies are usually oriented to the complete NOx reduction in the presence of low amounts of light hydrocarbons [5–9] but not the inverse: complete oxidation of VOCs in the presence of NOx.
The use of zirconia as oxidation catalysts has been claimed for its thermal stability and specific area [4] and also for its selectivity for NOx reduction to N2 [10].
The introduction of alkali metals in catalysts developed for the oxidation of hydrocarbons in the presence of NOx is expected to provide interesting effects. In some cases, alkali metals are known to be activity promoters for the oxidation of hydrocarbons as well as for NOx reduction [10]. Previous works of our laboratory [1,11] presented this effect of alkali metals in the oxidation of soot.
The introduction of both alkali metal and transition metal (cobalt or copper) on ZrO2 are investigated and provided synergistic effects in the soot oxidation [11]. Moreover, cobalt and copper oxides are known to be active phases for VOCs oxidation [12–14] and also for NOx reduction [2,15]. Co and Cu were then also impregnated on the samples for improving the catalytic behaviours.
Therefore, the aim of this work is the study of the effect of alkali elements (Li, Na, K, Rb, Cs) on the physicochemical characteristics of zirconium oxide (ZrO2) and its catalytic properties in oxidation of toluene and/or carbonaceous particles and/or conversion of nitrogen oxides. The effects of Cs/Co and Cs/Cu ratios in the different catalytic tests were also studied.
2 Experimental
2.1 Catalysts synthesis
The zirconium oxide support was prepared by a precipitation method, adding dropwise an aqueous solution of zirconyl (IV) chloride to an ammonia solution under continuous stirring. The precipitate was filtered and washed to remove remaining chloride ions [16]. This solid was dried at 115 °C for 24 h and subsequently calcined under air flow (2 L h−1) at 300 °C for 4 h [1,4,11].
The alkali/ZrO2 systems were obtained by impregnation of aqueous alkali carbonates on a ZrO2 support, with an initial specific area of 201 m2 g−1.
Alkali–Co0.1/ZrO2 catalysts were prepared by co-impregnation of cobalt (CoCO3·nH2O) and an alkali carbonate onto ZrO2 solid. Alkali–Cu0.1/ZrO2 catalysts were prepared with the same method (using CuCO3·nCu(OH)2).
After drying, the samples were calcined under air flow (2 L h−1) at 600 °C for 4 h. The as-obtained solids will be denoted by alkalix–M0.1/ZrO2 where M is the symbol of the impregnated transition metal, x and 0.1 correspond respectively to alkali/Zr and M/ZrO2 atomic ratios.
2.2 Catalysts characterization
The chemical composition of the samples was determined by ICP–MS with a Varian 820 MS apparatus.
TG–DTA experiments were studied with a Netzsch STA 409 apparatus. About 30 mg of dried catalyst were loaded in an alumina crucible and heated from room temperature to 1000 °C (heating rate: 5 °C min−1) in air flow (75 mL min−1).
BET surface areas were measured by nitrogen adsorption at 77 K on a Qsurf M1 apparatus (Thermo Electron Corporation).
FTIR experiments were performed with pellets prepared by pressing about 200 mg of catalyst powder (1 wt.%) diluted in KBr. Measurements were recorded on a Bruker Equinox 55 FTIR spectrometer at ambient temperature in the 4000–400 cm−1 range. Spectra were recorded with a resolution of 2 cm−1 and by co-addition of 64 scans. In situ DRIFT spectra were recorded on the same apparatus equipped with accessories for the diffuse reflectance and a room environment stainless steel manufactured by Harrick Scientific Corporation. A double thermocouple system can regulate the temperature inside the chamber and near the sample. The gaseous flow passing through the chamber is controlled by mass flow controllers. The pretreatment (350 °C for 4 h) and the catalytic test have been performed under the same conditions (gas flow rates, temperature ramp), but with 30 mg of pure sample. All spectra are presented in absorbance after subtracting the spectrum of the reference sample (KBr powder).
XRD measurements were performed by a Bruker D8 Advance diffractometer using CuKα radiation (λ = 1.5406 Å) in the 2θ range 20–55°, with a step size of 0.02° and an integration time of 6 seconds.
Carbon black (CB) Printex U (from Evonik) was chosen as model for soot particles oxidation. Its chemical mass composition in percentage is: C = 92; H = 0.95; O = 4.4; N = 0.32 and S = 0.57. The specific area of the CB reaches 100 m2 g−1 [17]. The CB–catalyst mixture was performed and prepared in loose contact conditions [1,18]. Six percent of CB and 94% of catalyst were introduced in a small flask and were simply shaken for 20 minutes. The catalytic test towards CB oxidation was studied by TG–DTA measurements. About 30 mg of CB–catalyst mixture were loaded in an alumina crucible and heated from room temperature to 650 °C (heating rate: 5 °C min−1) in air flow (75 mL min−1). TG–DTA results will be used to determine catalytic performances and CB oxidation rate.
The oxidation of toluene was carried out in a continuous flow reactor with a fixed bed at atmospheric pressure. Before each test, the catalyst (100 mg) was dried in air (2 L h−1) at 500 °C (1 °C min−1) for 4 h in order to remove the hydroxyl groups, water or moisture adsorbed on the catalyst and thus to activate it [19]. The feed gas reactant mixture was made of 1000 ppm of toluene and 10% O2 in N2 (total flow rate: 100 mL min−1). After a stable feed, it passed through the catalyst and the temperature was increased from room temperature to 500 °C at a rate of 1 °C min−1. Toluene conversion was checked in parallel using a CP–4900 μGC (Varian).
The simultaneous oxidation of toluene and CB was studied, considering 1000 ppm of toluene and 10% O2 in N2 passed through CB–catalyst mixture (100 mg of catalyst + 6% of CB).
The effect of the presence of NOx (NO2 + NO) in the feed gas (900 ppm) was also studied for the both different oxidations. The analysis of combustion products (NOx, NO, NO2, CO and CO2) was obtained with Xentra 4900C continuous emissions analyzer (Servomex). Toluene conversion was checked using a CP–4900 μGC.
The volume hourly space velocity (VHSV), calculated at ambient temperature and atmospheric pressure, is 105,000 h−1. Since the work was always carried out in the same reaction conditions (same toluene and/or NOx flow and catalyst weight), the conversion values, which are directly comparable, were given.
3 Results and discussion
3.1 Effect of alkali metals on ZrO2
The theoretical alkali metal content of 0.15 ratio molar in the catalysts was almost confirmed by ICP–MS. Indeed, the experimental molar ratio of the alkali metal in the samples was found to be slightly lower: 0.129 ± 0.015 (Table 1).
Chemical composition of the catalysts calcined at 600 °C and specific areas of the catalysts before and after calcination at 600 °C.
| Catalyst | Chemical composition | BET specific area (m2 g−1) | |||
| Theoretical content (Alkali/ZrO2 and/or M/ZrO2, wt.%) | Experimental content (Alkali/ZrO2 and/or M/ZrO2, wt.%) | Corresponding experimental molar ratio | Before calcination | After calcination at 600 °C for 4 h | |
| ZrO2 | – | – | – | 201 | 84 |
| Li0.15/ZrO2 | 0.845 | 0.813 | 0.144 | 173 | 21 |
| Na0.15/ZrO2 | 2.819 | 2.433 | 0.129 | 156 | 34 |
| K0.15/ZrO2 | 4.809 | 3.660 | 0.114 | 134 | 34 |
| Rb0.15/ZrO2 | 10.250 | 8.351 | 0.122 | 107 | 27 |
| Cs0.15/ZrO2 | 16.235 | 14.210 | 0.131 | 86 | 21 |
| Cs0.15–Co0.1/ZrO2 | 16.235 (Cs) | 10.198 (Cs) | 0.094 (Cs) | 85 | 17 |
| 4.783 (Co) | 3.505 (Co) | 0.073 (Co) | |||
| Cs0.015–Co0.1/ZrO2 | 1.623 (Cs) | 1.346 (Cs) | 0.012 (Cs) | 204 | 68 |
| 4.783 (Co) | 4.229 (Co) | 0.088 (Co) | |||
| Co0.1/ZrO2 | 4.783 | 4.119 | 0.086 | 192 | 95 |
| Cs0.15–Cu0.1/ZrO2 | 16.235 (Cs) | 10.834 (Cs) | 0.100 (Cs) | 83 | 12 |
| 5.153 (Cu) | 3.797 (Cu) | 0.074 (Cu) | |||
| Cs0.015–Cu0.1/ZrO2 | 1.623 (Cs) | 1.260 (Cs) | 0.012 (Cs) | 196 | 96 |
| 5.153 (Cu) | 5.014 (Cu) | 0.097 (Cu) | |||
| Cu0.1/ZrO2 | 5.153 | 4.452 | 0.086 | 199 | 90 |
The specific area of the catalysts decreases with increasing temperature of calcination (Table 1). It decreases from 201 m2 g−1 for ZrO2 calcined at 300 °C to 84 m2 g−1 for the same sample calcined at 600 °C. For the impregnated alkali samples before calcination, the specific area decreases when the molar mass of the alkali metal increases. It goes from 173 m2 g−1 in the case of Li0.15/ZrO2 to 86 m2 g−1 in the case of Cs0.15/ZrO2. The decrease of specific area can be attributed to the reorganization within the network of zirconia reducing the porosity of the solid [20]. After calcination at 600 °C, the specific surface of Li0.15/ZrO2 is four times (21 m2 g−1) lower than the value of the support ZrO2 (84 m2 g−1). The addition of an alkali promoter leads to decrease markedly the specific surface of our catalysts. K and Na can migrate into the solid ZrO2 and stabilize the tetragonal phase. Na promotes the good crystallization of the solid [20]. Other alkali metals (Li, Rb, Cs) facilitate the transformation of the tetragonal phase to the monoclinic phase. Indeed, the monoclinic phase (formed with Li, Cs) has a smaller surface area compared to the tetragonal phase, and then, the specific surface of the samples with Li and Cs is smaller than the value of the sample with Na.
A XRD study of ZrO2 (calcined at 300 °C and 600 °C) and the different alkali0.15/ZrO2 catalysts calcined at 600 °C was carried out and the diffractograms obtained are displayed in Fig. 1. It shows that ZrO2 calcined at 300 °C is an amorphous phase. The monoclinic phase (JCPDS 65.1023) appears significantly in the case of ZrO2 calcined at 600 °C and Li0.15/ZrO2. For the tetragonal phase (JCPDS 50.1089), it is stabilized in the case of Na0.15/ZrO2, and less apparent in the case of K0.15/ZrO2. Indeed, according to Liu et al. [21], Na is able to diffuse inside the bulk of particles and stabilises the zirconia tetragonal phase. The tetragonal phase is predominant in the other catalysts. These results are in accordance with BET specific area results (Table 1).
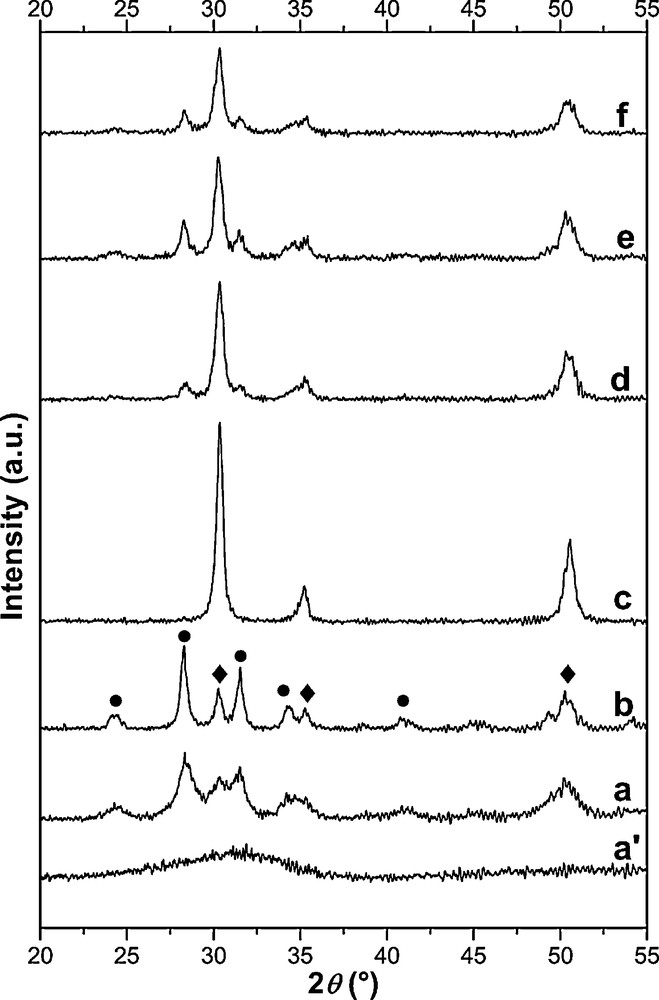
XRD patterns of ZrO2 (a’: calcined at 300 °C, a: calcined at 600 °C) and catalysts calcined at 600 °C (b: Li0.15/ZrO2, c: Na0.15/ZrO2, d: K0.15/ZrO2, e: Rb0.15/ZrO2, f: Cs0.15/ZrO2); (•) monoclinic phase; (♦) tetragonal phase.
DTA experiments (Fig. 2) of the different samples of alkali0.15/ZrO2 show the existence of exothermic peaks. These peaks are not accompanied by any significant mass loss (TG curves not shown). For the support calcined at 300 °C and the catalysts impregnated and dried at 115 °C, the first peak of crystallization of the tetragonal phase [16] is shifted to a higher temperature (428 °C in the case of the support and 433–600 °C in the case of promoted catalysts versus the molar mass of the alkali). Going from Li to Cs, the intensity of the first peak decreases; therefore the crystallization of amorphous ZrO2 to tetragonal phase is inhibited. For the same type of preparation, the delay of crystallization increases when the molar mass of the alkali increases. A peak appears at high temperature (910 °C) in the case of Na0.15/ZrO2. It corresponds to the crystallization to monoclinic phase [22]. Indeed, the stability of the tetragonal phase (Fig. 1) can accelerate the appearance of monoclinic phase at this temperature. This observation is consistent with the observation of Liu et al. [22]. Thus, the presence of alkali metals on ZrO2 calcined at 300 °C has an effect on the mode of crystallization of ZrO2 during the calcination of catalyst up to 600 °C.
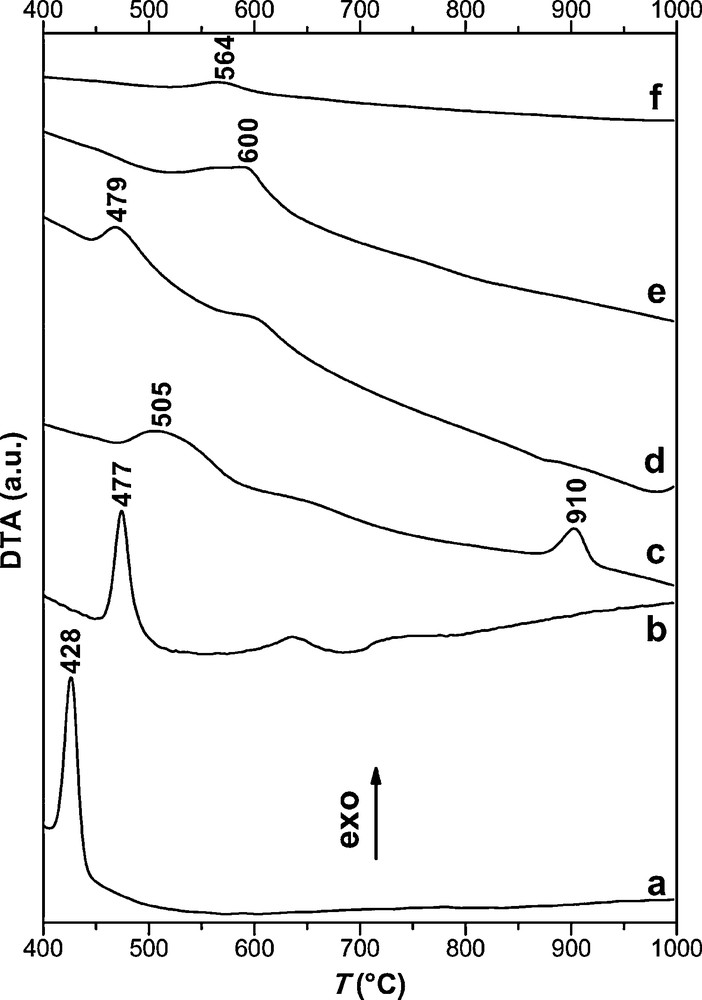
DTA curves of ZrO2 calcined at 300 °C (a: and dried solids, b: Li0.15/ZrO2, c: Na0.15/ZrO2, d: K0.15/ZrO2, e: Rb0.15/ZrO2, f: Cs0.15/ZrO2).
3.2 Impregnation of transition metals
The theoretical content of transition metals was confirmed by elemental analysis (Table 1). The molar ratio M/ZrO2 was between 0.073 and 0.097. When the transition metal was impregnated alone or with a low amount of Cs, the molar ratio was between 0.086 and 0.097 molar ratio. The Cs/ZrO2 molar ratio was 0.012. When the transition metal was impregnated with a high amount of Cs, the M/ZrO2 molar ratio decreased to 0.073–0.074 and the corresponding Cs/ZrO2 molar ratio was 0.094–0.1.
The impregnation of transition metal leads to materials with similar specific areas, comparable to that obtained from the support (in similar experimental conditions). The adding of a low amount of Cs leads also to the same observations, but in the case of the impregnation of Cu or Co with a high amount of Cs, the specific areas of corresponding catalysts are similar to those obtained from Cs0.15/ZrO2 (in similar experimental conditions).
A XRD study of Csx–M0.1/ZrO2 catalysts calcined at 600 °C was carried out and the obtained diffractograms are displayed in Fig. 3. For copper based catalysts (Fig. 3A), there is a stabilization of the tetragonal phase with all catalysts, but for the CuO phase (2θ = 38.8), CuO lines are observed (JCPDS 48.1484) in the presence of the alkali metal (Cs0.15–Cu0.1/ZrO2). The same phenomenon was observed by Lick et al. [23]. Thus, the alkali metal allows the formation of the CuO phase and should influence the Cu segregation, leading to observable crystals with the XRD technique [23]. For catalysts with cobalt (Fig. 3B), in the absence of Cs or in its presence with a low amount, there is a line (at 2θ = 37.09) corresponding to Co3O4 (JCPDS 42.1467). The absence of a Co oxide phase peak in the XRD pattern for the Cs0.15–Co0.1/ZrO2 sample shows that the oxide particles must be finely dispersed as amorphous or small sized (size < 3 nm) particles invisible by XRD [14]. Park et al. [24] demonstrated that the characteristic lines of Co3O4 are shown only with Na/Co ≤ 0.015 (atomic ratio) and for samples with Na/Co ≥ 0.03, characteristic peaks of β-Na0.6CoO2 increase with increasing Na content.
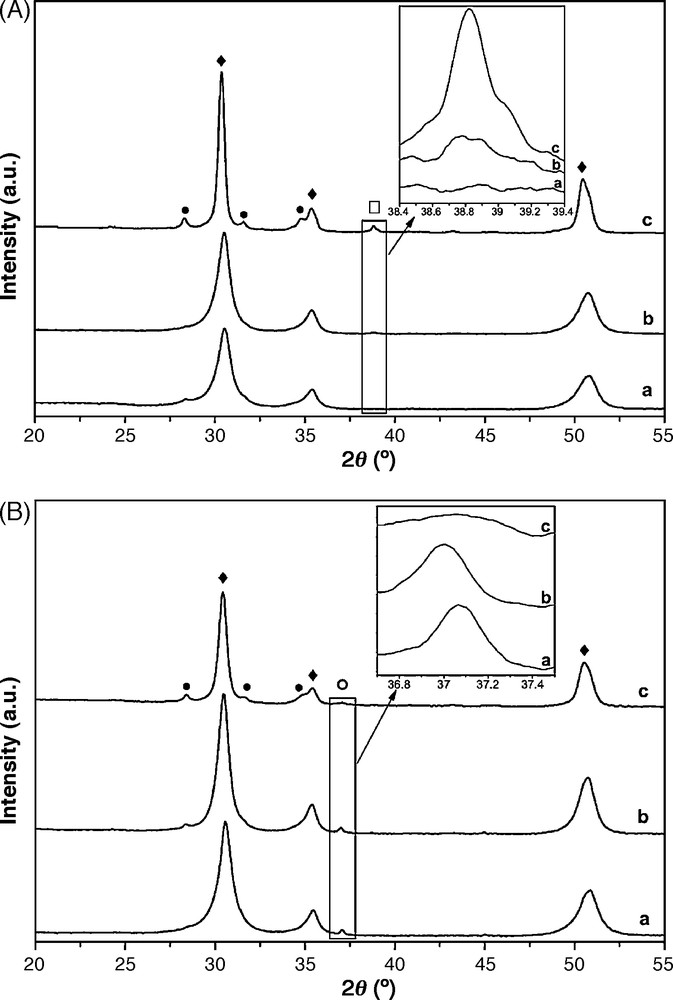
XRD patterns of Csx–M0.1ZrO2 (A: copper based catalysts, B: cobalt based catalysts) calcined at 600 °C (a: M0.1/ZrO2, b: Cs0.015–M0.1/ZrO2, c: Cs0.15–M0.1/ZrO2); (•) monoclinic phase; (♦) tetragonal phase, (□) CuO phase, (°) Co3O4 cubic phase.
3.3 Oxidation tests
3.3.1 Catalytic oxidation of carbon black
Catalytic performance in CB oxidation was determined by the use of DTA–TG curves. The maximum of the curve corresponds to the temperature of the highest rate of CB oxidation Tmax (Fig. 4). The oxidation of CB with ZrO2 alone is between 414 and 617 °C with a maximum CB oxidation rate (975 μg s−1 g−1CB) at Tmax = 575 °C. A shift to lower Tmax occurs in the presence of catalysts promoted with alkali, especially for Cs0.15/ZrO2 (Tmax = 355 °C). The oxidation rate goes from 1966 μg s−1 g−1CB with Li0.15/ZrO2 to 2516 μg s−1 g−1CB with Cs0.15/ZrO2. The higher performance of Cs-loaded catalyst for soot ignition cannot be explained by the specific area [25]. The results show that the use of alkali-impregnated catalyst is beneficial for the oxidation of CB, reaching the best results with the “heavier” alkali. The addition of transition metals (especially Cu), provided for the enhancement of toluene oxidation, is also interesting for the oxidation of CB. However, for Co based catalyst (Cs0.15–Co0.1/ZrO2), Tmax value is lower than for Cs0.15/ZrO2. For the catalyst impregnated by Cu and Cs, the CB oxidation rate reaches a maximum value of 2346 μg s−1 g−1CB and at Tmax (347 °C), slightly lower than that achieved with Cs0.15/ZrO2. These experiments suggest a synergistic effect between the alkali metal and Cu already observed in a previous work [11]. The alkali metal promoter interacts with the supported copper(II) species and favours their participation following a redox mechanism. It is assumed that the formation of CuO phase, under the form of large or small particles, is responsible for this activity enhancement, CuO species being more active than isolated copper and copper clusters for CB oxidation reaction [26]. The performance of CuO based catalysts in the CB oxidation has been also attributed to the high mobility of the copper ions during burn-off [27]. Moreover, it was shown in the study of Laversin et al. [11] that the alkali promoters are species that not only favour CB–catalyst contact, but also enhance the participation of active oxygen from copper oxide and oxide carrier to oxidize CB. Furthermore, in the case of Cs0.015–Cu0.1/ZrO2 and Cu0.1/ZrO2, the maximum CB oxidation rate is lower than those for the catalysts: Cs0.15/ZrO2 and Cs0.15–Cu0.1/ZrO2. Moreover, Tmax of these catalysts is also the highest among all the catalysts. This shows the contribution of Cs in the oxidation of carbonaceous particles. In fact, the presence of alkali metals favours contact between the catalyst and CB and enhances the catalyst's ability to release active oxygen species. Moreover, the presence of higher alkali content induces further activity improvement [11]. The XRD results (Fig. 3A) show that in the presence of high amount of Cs, the CuO phase appears on the XRD patterns. Tmax found for Cs0.15–Cu0.1/ZrO2 catalyst is lower than those of Cs0.015–Cu0.1/ZrO2 and Cu0.1/ZrO2 catalysts. This suggests that the activity can be associated mainly with Cs and with the wetting effect of the catalytic surface that this metal confers. Cs- and Cu-based catalysts will then be used in the others tests of CB oxidation.
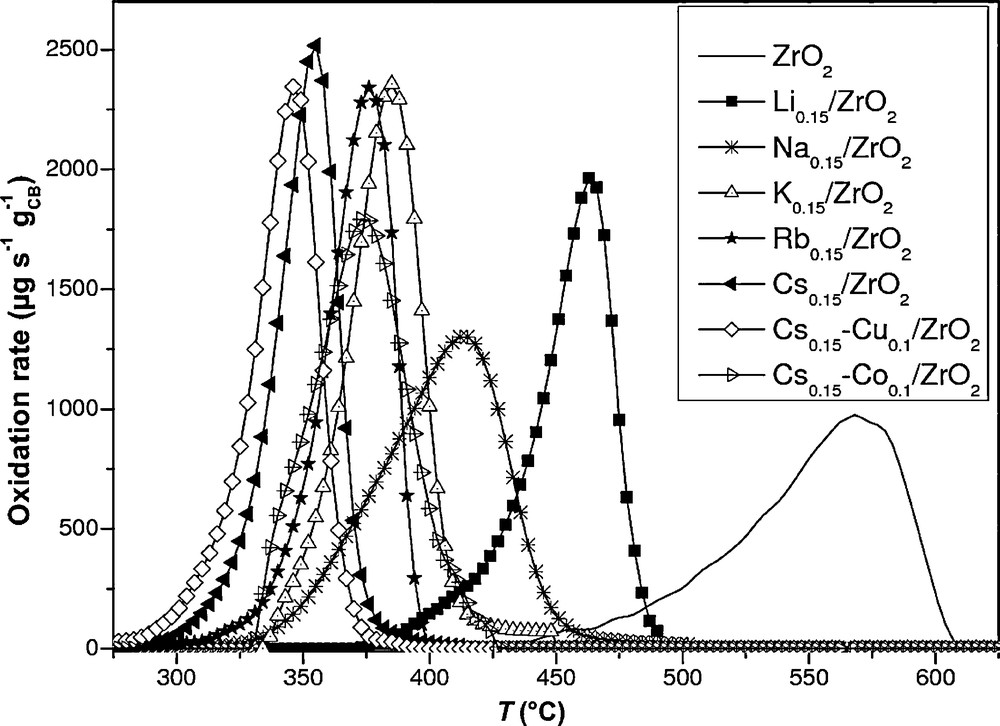
CB oxidation vs. reaction temperature.
3.3.2 Catalytic oxidation of toluene
The behaviour of the different catalysts for the toluene oxidation is presented in Fig. 5 and Table 2. For the catalysts promoted by alkali alone, the oxidation of toluene is only complete at 500 °C for the best catalyst Cs0.15/ZrO2. Cobalt or copper impregnation on Cs0.15/ZrO2 leads to powerful catalysts, especially for Cs0.15–Co0.1/ZrO2. Moreover, the solid with a low amount of Cs (Cs0.015–Co0.1/ZrO2) allows us to reach a T50%1 = 296 °C instead of T50% = 371 °C for Cs0.15–Co0.1/ZrO2 (Table 2). Moreover, cobalt impregnation alone on ZrO2 presents also a good performance, similar to Cs0.015–Co0.1/ZrO2 for toluene oxidation. Indeed, the presence of Co3O4 phase in Co based catalysts leads to improve toluene conversion to CO2 [13,14]. This Co3O4 phase is observable in our catalysts (Fig. 3B). The catalytic performance of Cs0.15–Co0.1/ZrO2 is lower than that of the same sample without, or with a low amount, of Cs. Therefore, the presence of high amount of alkali metal decreases the activity of the Co3O4 phase for the toluene oxidation. The good performance of cobalt-impregnated catalyst should be due to dispersed species of cobalt. Thus, it seems that the amount of reducible species plays a non-negligible role in the catalytic properties in oxidation reactions. The higher the amount of reducible species in the temperature range of catalytic activity is, the better the catalytic performance [19]. The good performance of Co based catalysts in comparison with Cs based catalysts for toluene oxidation could be due to the easier reducibility of Co3O4 compared to Cs2O. In fact, Co shows reduction peaks in the temperature range 100–400 °C [14,19], although Cs shows a maximum reduction temperature pointed out at 520 °C [1]. One reason for the lower performance of Cs high amount catalyst is the possible formation of a Cs–Co phase. Thus, the XRD results (Fig. 3B) show that the Co3O4 phase formation is diminished in the presence of high amount of Cs.
3.3.3 Catalytic removal of CB in the presence of toluene or NOx
3.3.3.1 Oxidation of carbon black in the presence of toluene
For comparison reasons, the tests of CB oxidation were all recorded in the same conditions by the use of a microreactor (Table 3). The ignition temperature Ti for CB oxidation was determined as light-off temperature, at which a significant evolution of CO2 started. In a previous study [28], Aouad et al. showed that the presence of propene in reactant gases flow was shown to be beneficial to CB elimination in the presence of RuxCe catalysts, knowing that the temperature of total oxidation of propene is lower than that of CB (NC 330, Evonik). In this work, the tests were performed with toluene and CB (Printex U). For all the catalysts, a decrease between 10–20 °C of Ti and Tmax is recorded for CB oxidation in the presence of toluene in comparison with simple CB oxidation. Thus, the presence of the hydrocarbon in the reactant gases mixtures is not only beneficial to the oxidation of a part of CB at low temperatures but also to the faster oxidation of CB [28]. The exothermic effect of the toluene oxidation and/or toluene adsorption on CB can explain this enhancement. However, the toluene oxidation seems not to be improved by the CB oxidation. The CB oxidation is enhanced in the presence of toluene but especially for the catalysts with a high amount of Cs (Cs0.15/ZrO2 and Cs0.15–Cu0.1/ZrO2). However, the catalyst Cs0.15–Cu0.1/ZrO2 is the best one. This can be attributed to the both participation of a high content of alkali metal and CuO phase.
CB oxidation and NOx conversion values.
| Catalyst | Tmax and (Ti) for CB oxidation (°C) | Maximum of NOx conversion (%) | ||
| CB + O2 | CB + O2 + toluene | CB + O2 + NO | ||
| Cs0.15/ZrO2 | 349 (275) | 325 (235) | 323 (270) | 22.6 (340–450 °C) |
| Cs0.15–Cu0.1/ZrO2 | 321 (268) | 308 (239) | 355 (255) | 25.9 (at 370 °C) |
| Cs0.015–Cu0.1/ZrO2 | 496 (362) | 451 (410) | 384 (291) | 22.6 (at 390 °C) |
| Cu0.1/ZrO2 | 498 (335) | 497 (329) | 405 (200) | 16.3 (at 410 °C) |
3.3.3.2 Oxidation of carbon black in the presence of NOx
The results concerning the CB oxidation in the presence of NOx are shown in Table 3 and Fig. 6. Dotted lines in the figure represent the NOx concentration in the gas feed in the presence of 10% O2, i.e., the points below and above this line mean the occurrence of NOx sorption/reduction and NOx desorption respectively. For the sample Cs0.15/ZrO2, Ti of CB oxidation in the presence of NOx is slightly enhanced compared to that in the test of CB oxidation alone, but lower than that in presence of toluene. The same phenomenon is recorded for the values of Tmax. An increase of NOx is also observable. Cs0.15/ZrO2 exhibits sorptive NOx, which is followed by a broad NOx desorption at higher temperatures. It gives rise to a sharp NOx desorption, which starts before the beginning of CO2 evolution. Unlike the case of the catalyst alone (not shown), very sharp and temporary desorption of NOx appears at 323 °C (also Tmax of CB oxidation). After this, the NOx concentration drops suddenly below the inlet level suggesting the occurrence of reactions between NO and CB [25] in the temperature range 323–450 °C. This is consistent with a larger area below the dotted line than that above the line (Fig. 6A). This phenomenon is in accordance with the observation of Ito et al. [25]. First, a sharp desorption of NOx should result from a very fast and local exotherm due to soot ignition. As described above, the temperature of soot ignition is lowered when a large amount of NO is pre-adsorbed on the catalysts. If soot would directly be reacted with desorbed NOx, the ignition must be observed after the beginning of NOx desorption. However, this is not the case. This leads to another important suggestion that soot ignition is presumably promoted by adsorbed NOx species [25]. For Cs0.15–Cu0.1/ZrO2, Tmax of CB oxidation is less good; it rises from 321 °C to 355 °C, but a conversion of NOx is detected at 370 °C, which is close to the maximum CB oxidation rate. The presence of NOx is unfavourable for toluene oxidation with this catalyst although this catalyst records the high NOx conversion. For Cs0.015–Cu0.1/ZrO2, Ti and Tmax of CB oxidation are considerably enhanced. A decrease of about 110° is observed for Tmax and 70° for Ti. A good NOx conversion is recorded with this catalyst. In the case of the catalyst Cu0.1/ZrO2, the CB oxidation is enhanced with a shift to a lower temperature in the presence of NOx (Table 3), but there is a low NOx conversion for Cu0.1/ZrO2 compared to Cs0.015–Cu0.1/ZrO2. As described on Fig. 6, the ignition temperature is lowered when a large amount of NOx is pre-adsorbed on these catalysts. If the CB directly reacts with desorbed NOx, the ignition must be observed after the beginning of NOx desorption. The addition of Cs not only activates CB ignition, but also increases NOx adsorption, which is also effective for CB ignition [25].
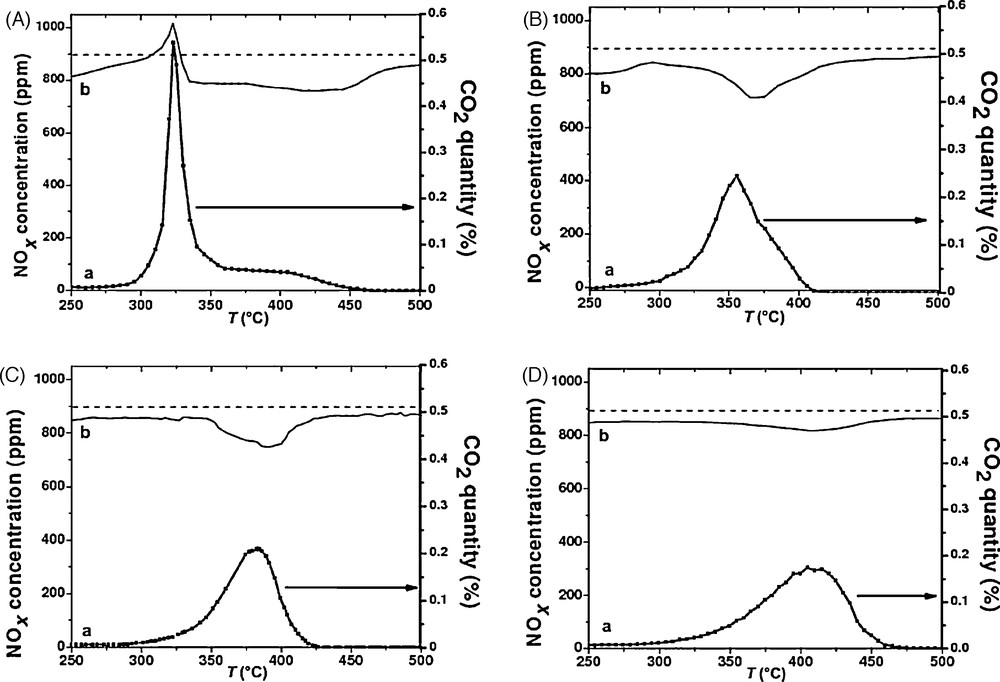
Oxidation of CB vs. reaction temperature with the catalysts (A: Cs0.15/ZrO2, B: Cs0.15–Cu0.1/ZrO2, C: Cs0.015–Cu0.1/ZrO2, D: Cu0.1/ZrO2) in the presence of NOx (a: CO2 quantity of CB oxidation, b: NOx concentration).
Fig. 8 shows the infrared spectra after CB oxidation tests without (spectra (a)) and in the presence of NOx (spectra (b)). The NOx sorption leads to the formation of bidentate and/or monodentate nitrates species on the surface. Two bands at 1380 and 1335 cm−1 are detected after tests in the presence of NOx for Cs0.15/ZrO2 and Cs0.15–Cu0.1/ZrO2. These bands are attributed to free NO3− groups (1380 cm−1) and to monodentate NO3− groups (1335 cm−1) [1]. Presence of bridged NO3− groups could not be excluded; their characteristic band (∼1620 cm−1) could be masked by adsorbed water band [1]. The amount of nitrate is large for the catalyst Cs0.15–Cu0.1/ZrO2 and lower for Cs0.15/ZrO2. For the samples Cs0.015–Cu0.1/ZrO2 and Cu0.1/ZrO2, there is not any significant adsorption. The large amount of nitrate adsorbed in the case of Cs0.15/ZrO2 and Cs0.15–Cu0.1/ZrO2 is due to the strong basicity of Cs. The oxidative adsorption of NO was also increased to form nitrate on the solid surface [25]. The use of a low amount of Cs impregnated with Cu, leads to a great shift of Ti and Tmax of CB oxidation to a low range of temperatures. The catalysts Csx–Cu0.1/ZrO2 possess the combination of NOx–oxidation activity and moderate basicity, yielding in significant synergism in the oxidative adsorption as to yield nitrite (NO2−) and/or nitrate (NO3−) on the surface. This explains the high NOx conversion in the case of these catalysts, compared to Cs0.15/ZrO2 or Cu0.1/ZrO2. The maximum NOx conversion corresponds to Tmax of CB oxidation (Table 3). Comparing the curves of NOx in the absence of CB (Fig. 7B) and in the presence of CB (Fig. 7C), the NOx conversion is just observed when a maximum of CB is oxidized at Tmax. This conversion is accompanied with a decrease of NO2 quantity (curve a). Thus, NO2 participates in the CB oxidation. This is explained by the need for NO2 as an intermediate for the catalyst to work efficiently [6], especially in the oxidation of CB [29]. The detailed mechanism of CB–NOx interaction is not clear at this stage, but this may be explained by considering adsorbed nitrate species as an oxidizing agent [25]. The oxidation of NO to the nitrate is supposed to proceed in two steps: [15]
| (1) |
| (2) |
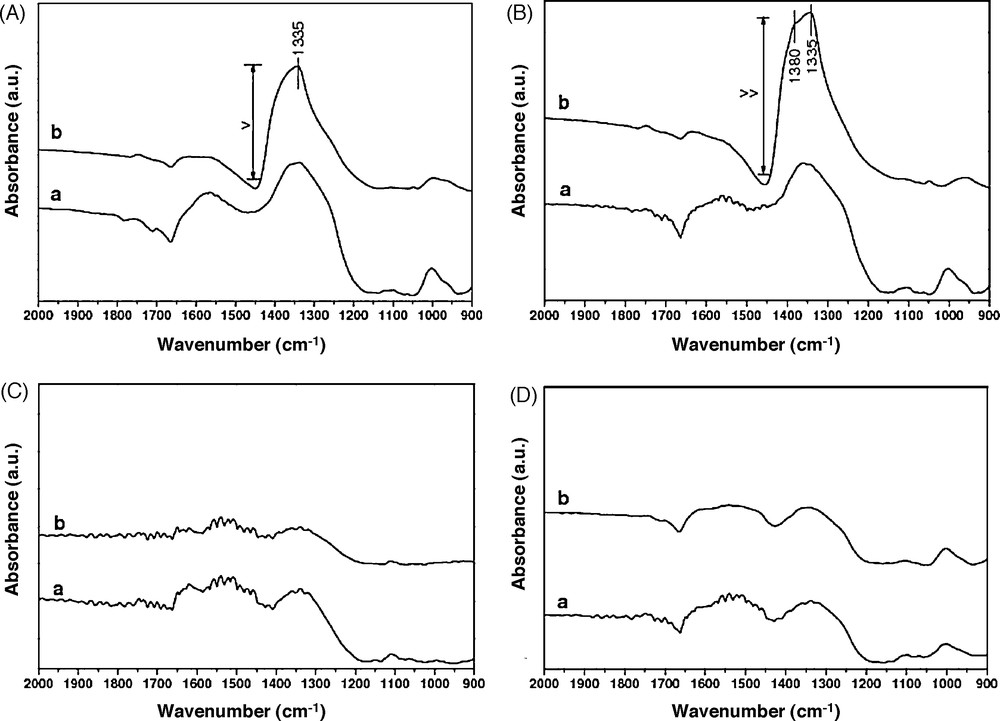
Infrared spectra of the catalysts (A: Cs0.15/ZrO2, B: Cs0.15–Cu0.1/ZrO2, C: Cs0.015–Cu0.1/ZrO2, D: Cu0.1/ZrO2) after test (a: CB + O2, b: CB + O2 + NO).
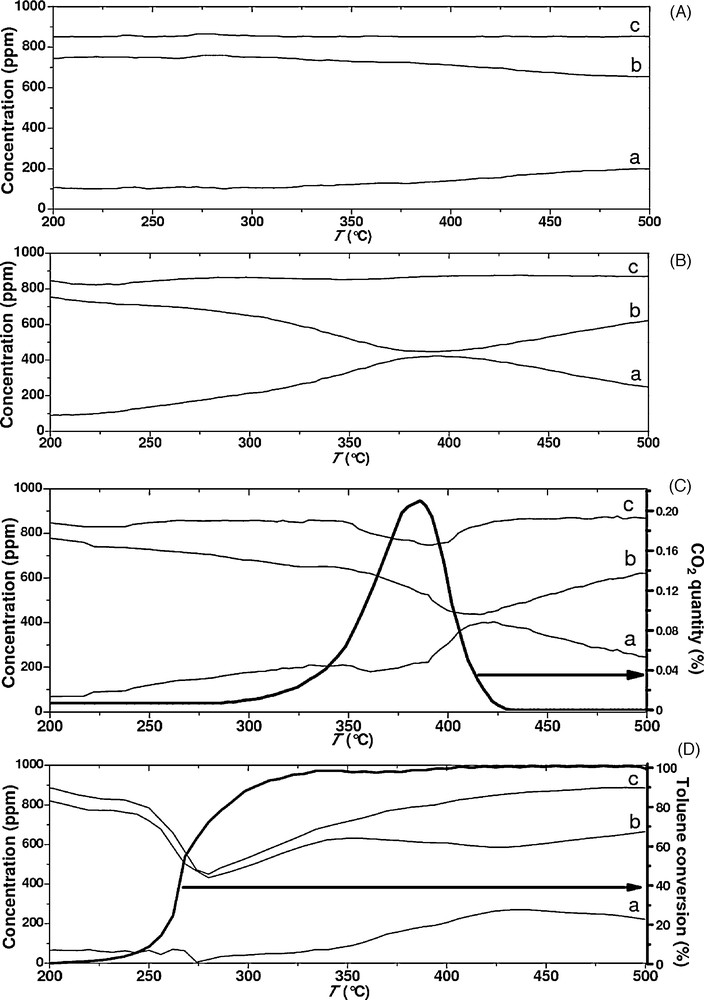
Effect of the presence of catalyst (Cs0.015–Cu0.1/ZrO2) and reducer (CB or toluene) in the conversion of NOx and the oxidation of NO (A: without catalyst, B: with catalyst, C: with catalyst in the presence of CB, D: with catalyst in the presence of toluene, a: NO2, b: NO, c: NOx).
3.3.4 Catalytic oxidation of toluene in the presence of CB or NOx
3.3.4.1 Oxidation of toluene in the presence of CB
The results of comparison between toluene oxidation alone and toluene oxidation in the presence of CB are summarized on Table 2. Ti of toluene oxidation is determined when a significant toluene conversion started. For Cs0.15/ZrO2, T50% of toluene conversion is less good in the presence of CB, but Ti is slightly enhanced. This is due to the oxidation of CB proceeding before that of toluene oxidation. The same result is obtained with Cs0.15–Co0.1/ZrO2. For Cs0.015–Co0.1/ZrO2 and Co0.1/ZrO2, T50% and Ti are both enhanced in the presence of CB. In this case, adsorption of toluene on CB should be important. The results show the interest of adding a low amount of alkali metal to cobalt in comparison with the simple catalyst Co0.1/ZrO2.
3.3.4.2 Oxidation of toluene in the presence of NOx
The oxidation of toluene in the presence of NOx is presented in Fig. 9 and Table 2. For Cs0.15/ZrO2, Ti and T50% of toluene oxidation decrease in the presence of NOx. The addition of Co to Cs0.15/ZrO2 enhances toluene oxidation, but not enough to reach the conversion obtained without NOx. The low NOx concentration observed at low temperatures (Table 2) should be due mainly to the adsorption of NOx species by the catalyst with a alkali amount, by the formation of nitrates [15]. The diminution of the amount of Cs leads to enhance the performance, greatly for NOx conversion and slightly for toluene oxidation compared to the same oxidation without presence of NOx. Co0.1/ZrO2 catalyst allows us to enhance NOx conversion in comparison with Cs0.015–Co0.1/ZrO2, but the oxidation of toluene is more efficient in the presence of NOx over the Cs0.015–Co0.1/ZrO2 catalyst. Moreover, on Fig. 9B (b), toluene oxidation increases when the concentration of NOx reaches its minimum. Therefore, the adding of a low amount of Cs to cobalt based catalysts leads to enhance toluene oxidation.
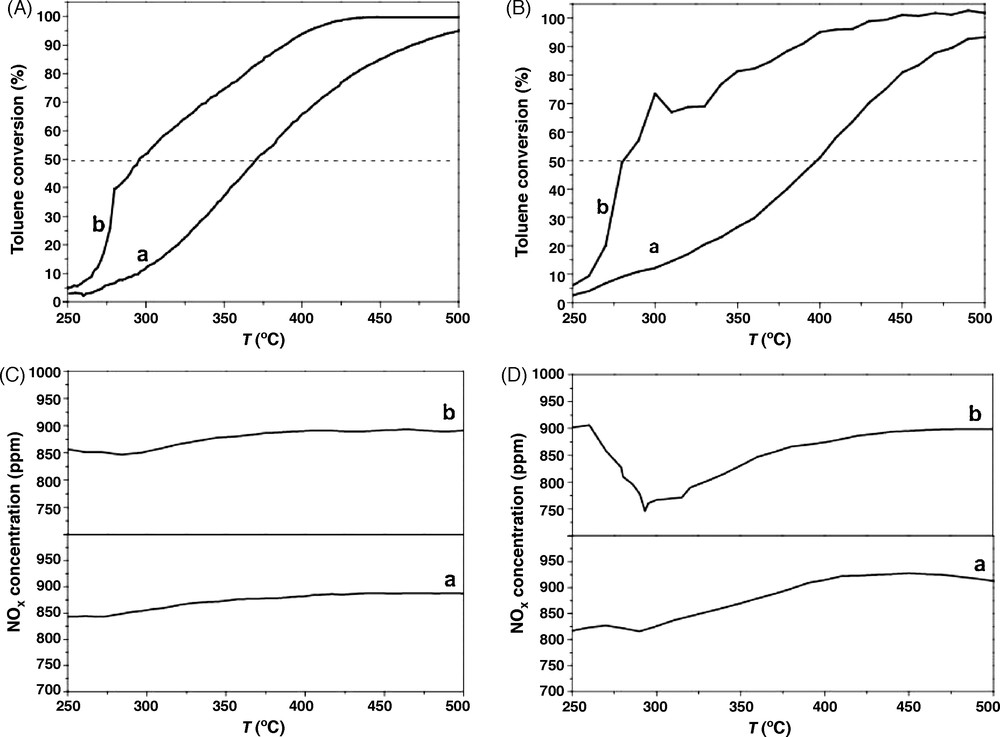
Toluene conversion and NOx concentration vs. reaction temperature on the catalysts (a: Cs0.15–Co0.1/ZrO2, b: Cs0.015–Co0.1/ZrO2) (A: toluene conversion without NOx, B: toluene conversion with NOx, C: NOx concentration without toluene, D: NOx concentration with toluene).
FTIR measurements for catalysts tested (Fig. 10) are carried after simple toluene oxidation at 500 °C (spectra (a)) and after oxidation of toluene in the presence of NOx (spectra (b)). For Cs based catalysts, a band at 1335 cm−1 corresponding to monodentate NO3− [1] is observed for the tests of toluene oxidation in the presence of NOx. So, the catalysts with the higher alkali content show the presence of a strong absorbance band ascribed to monodentate NO3− species (1335 cm−1). Therefore, NOx could be stored as nitrate species on catalysts with higher alkali content. This phenomenon is in good agreement with previous works revealing the strong interaction of NOx over alkali containing catalysts [1]. In this case, active oxidation sites of toluene could be occupied by NOx. This could explain the lower performance of Cs high amount catalysts. On the contrary, nitrite species (1265 cm−1) band showed in the case of Cs0.015–Co0.1/ZrO2 and Co0.1/ZrO2 catalysts demonstrates that the transition metals have the ability to form NO2 which is a stronger oxidizer than O2 [23].
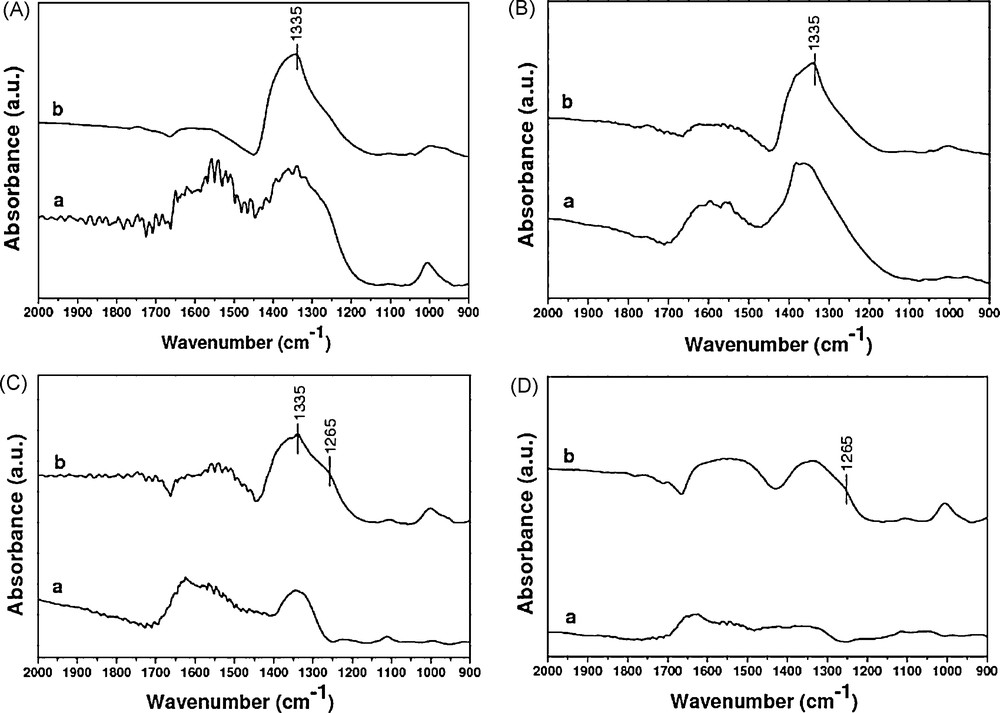
Infrared spectra of different catalysts (A: Cs0.15/ZrO2, B: Cs0.15–Co0.1/ZrO2, C: Cs0.015–Co0.1/ZrO2, D: Co0.1/ZrO2) after tests: (a: toluene + O2, b: toluene + O2 + NO).
3.4 Operando DRIFT study
Fig. 11 shows in situ FT–IR spectra of Cs0.015–Co0.1/ZrO2 at 350 °C. When introducing toluene and O2, there are bands at 1660 and 1425 cm−1 corresponding to substituted aromatic compounds, a band at 1305 cm−1 corresponding to skeletal C–C stretching vibration and a band at 1220 cm−1 corresponding to carbon single bonded oxygen stretching. In the presence of NO in the gas mixture (toluene + O2 + NO), the bands at 1660, 1425 and 1220 cm−1 are more intense and a band at 1540 cm−1 corresponding to R–NO2 appears. Thus, the toluene oxidation is observed by the strong bands for aromatic substitutes, especially for the experiment with NOx.
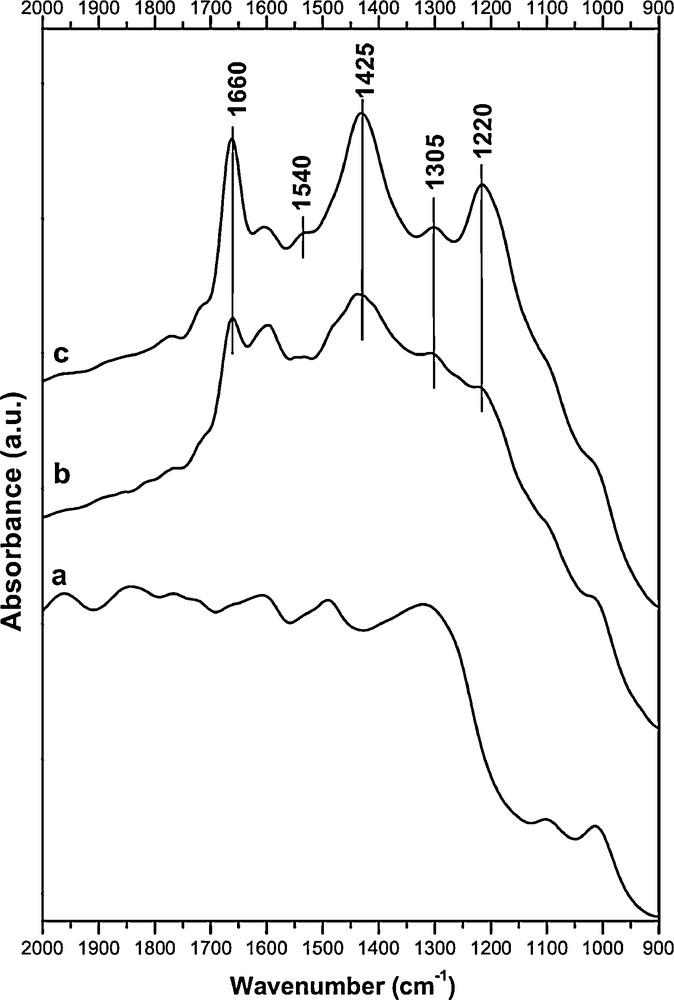
In situ infrared spectra of Cs0.015–Co0.1/ZrO2 at 350 °C (a: pre-treatment under air flow, b: toluene + O2, c: toluene + O2 + NO).
4 Conclusion
In this work, the catalytic performance of both oxidation of toluene and soot by alkali/ZrO2 is found to increase in the following order: Li0.15/ZrO2 ≈ Na0.15/ZrO2 < K0.15/ZrO2 < Rb0.15/ZrO2 < Cs0.15/ZrO2.
Catalysts impregnated by Co and Cs are active in the oxidation of toluene, whereas the catalysts based on Cu are more active in CB oxidation due to the active of CuO phase. Moreover, the contribution of cesium can enhance the participation of active oxygen from copper oxide to oxidize CB. In the case of the simultaneous oxidation of CB and toluene, the presence of VOCs enhances the oxidation of CB through the exothermic effect of VOCs oxidation. The catalyst impregnated with Cs and Cu is the most active one for CB oxidation alone or in the presence of toluene.
The presence of NOx is favourable for the oxidation of toluene and CB. For the CB oxidation, a significant decrease of CB oxidation temperature in the presence of NOx is obtained with Cs0.015–Cu0.1/ZrO2. Due to the strong basicity of Cs, the oxidative adsorption of NOx is increased to form nitrate on the solid surface. The CB combustion over such NOx-sorbing catalysts is further accelerated, suggesting the occurrence of the NOx–CB interaction on the catalyst surface. Adsorbed nitrate species are considered as oxidizing agents to promote CB ignition. The addition of Cs helps the activation of CB ignition, but also increases NOx adsorption, which is also effective for CB ignition.
For toluene oxidation, Cs0.015–Co0.1/ZrO2 is the best catalyst for toluene oxidation in the presence of CB or in the presence of NOx.
Thus, our catalysts show good performances in the both simple and simultaneous removal of VOCs or CB in the presence of NOx.
Acknowledgements
The “Nord-Pas de Calais” Region, the “Syndicat Mixte de la Côte d’Opale” and European Union via Interreg IV “Redugaz project” are gratefully acknowledged for financial support.
1 Temperature at which 50% of toluene is oxidized.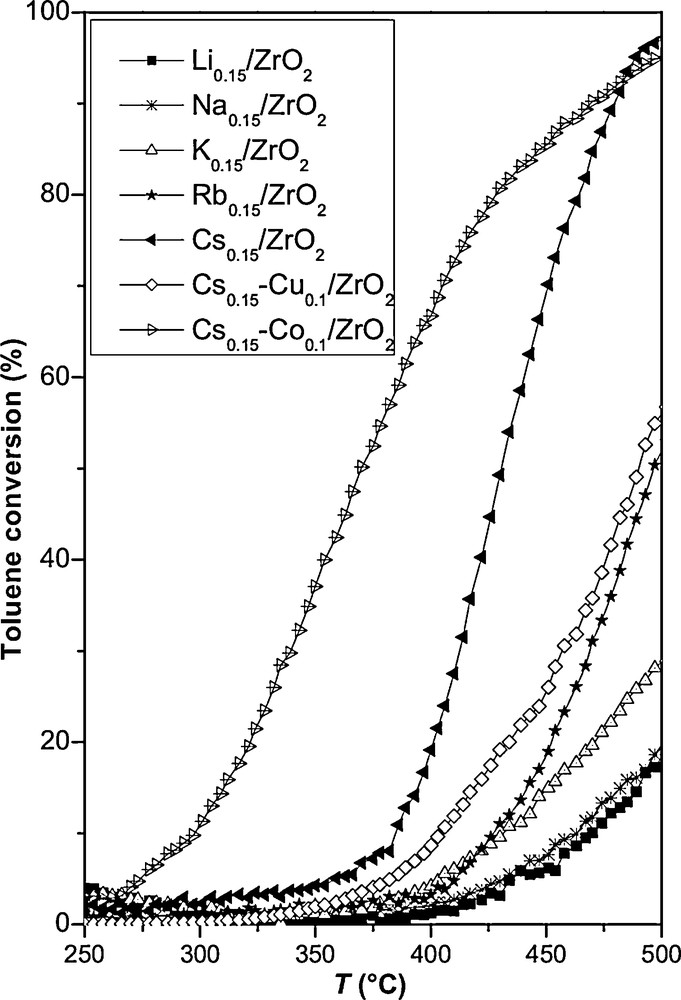
| Catalyst | T50 % and (Ti) values for toluene oxidation (°C) | Maximum of NOx conversion (%) | ||
| Toluene + O2 | Toluene + O2 + CB | Toluene + O2 + NO | ||
| Cs0.15/ZrO2 | 443 (283) | 451 (276) | 490 (308) | 14.9 (at 430 °C) |
| Cs0.15–Cu0.1/ZrO2 | 487 (300) | - | - | - |
| Cs0.15–Co0.1/ZrO2 | 371 (260) | 423 (220) | 399 (250) | Not observable |
| Cs0.015–Co0.1/ZrO2 | 296 (260) | 269 (208) | 279 (210) | 22.9 (at 293 °C) |
| Co0.1/ZrO2 | 294 (260) | 285 (193) | 285 (220) | 64.1 (at 300 °C) |