

1 Introduction
Since the first publications of Gedye and Giguere in 1986, the use of microwaves has progressively emerged as a popular non conventional heating source in the field of organic synthesis, largely due to frequent acceleration in reaction rates, improved yields and selectivities [1]. Nevertheless, application of microwaves in the area of carbohydrate chemistry is less documented [2]. Although research in this field is still in its infancy, recourse to microwave processes often provides, with remarkable yields and atom efficiency, new carbohydrate-based structures that are not easily available by any another means (or only via painstaking multi-step protocols). This review proposes therefore selected, recent and non exhaustive illustrations of the brilliant application of microwaves to promote famous carbohydrates “model” reactions.
2 Chemical transformations of monosaccharides
Common and largely-available sugars (i.e. sucrose, xylose and glucose) present several hydroxylated stereogenic centres which are difficult to chemically differentiate. Therefore, the regioselective derivatisation task of such structures is a very challenging purpose.
2.1 Regioselective hydroxyls protections/deprotections
One of the most thematic studies in sugar chemistry is thus the regioselective protection of carbohydrate hydroxyl functionalities, typically with acetic anhydride, aldehydes, acetyl, pivaloyl, or benzoyl halides, together with catalytic amounts of a Lewis acid or base [3]. Results under microwave irradiation indicate that a few minutes is usually enough to convert quantitatively the starting monosaccharide, under mild reaction conditions (i.e. without solvent or with reduced catalyst loadings), into its (partially or totally) O-protected counterpart (Fig. 1). The microwave strategy is convenient as higher yields are associated to minor side-product generation, an advantage over classical thermally-driven protocols. Moreover, microwaves allow an accurate control of the regioselectivity of the reaction, notably by an adequate tuning of operating conditions, which is difficultly attainable under “conventional” conditions.

Illustration of some hydroxyls protection/deprotection strategies under microwave conditions.
As an illustration of the usefulness of microwaves in hydroxyl protection, the formation of pivaloyl esters is of special relevence. Indeed, due to the steric hindrance of pivaloyl groups, these esterification reactions are typically slow under “classical” activation conditions. In this context, Oscarson et al. demonstrated that microwave dielectric heating was an interesting option for regioselective pivaloate formation under basic conditions (Fig. 2) [4]. Noteworthy was the observation that, even if the reaction occured in high yields after a few minutes in the presence of pyridine as the base, the desired adduct was isolated along with a side-product derived from the 5,6- to 3,5-acetal migration prior to acylation. Acetal migration, catalysed probably by the formed pyridinium chloride, was, however, completly restrained when using a polystyrene-supported DIEA or DMAP base instead of free pyridine.
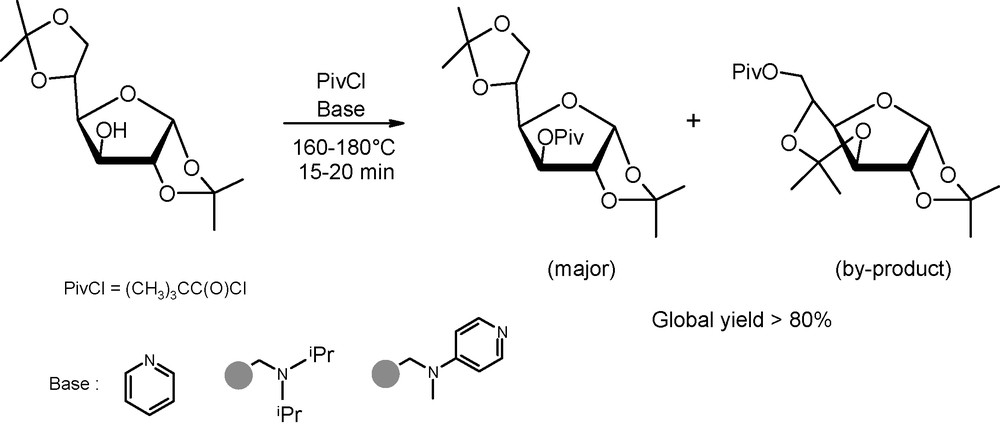
Pivaloylation of 1,2:5,6-di-O-isopropylidene-α-d-glucofuranose under basic conditions.
Even if often advantageous, cleaner and efficient, microwaves suffered, however, from several limitations, mainly due to the sensitivity of some carbohydrates (or derivatives) to elevated temperatures. In this context, Salanski and Queneau evaluated the mono-acetalisation of O-unprotected sucrose by citral under acidic conditions in dimethyl formamide (Fig. 3) [5]. Reaction at 100 °C under “conventional” oil-bath conditions and inert atmosphere, afforded the desired sucrose acetal in 83% yield after 2 min (vs. 59% in an open vessel). In parallel, cleavage of the glycosidic linkage occured when increasing reaction times beyond 5 min. When applying microwaves, under the same temperature and concentrations conditions, the yield reached only 42% after an identical runtime in open vessel conditions, together with increased amounts of hydrolysed adduct (26% after 10 min of microwave exposure), demonstrating that microwaves do not always improve the outcome of a reaction.

Mono-acetalisation of O-unprotected sucrose with citral dimethyl actetal in the presence of pyridinium p-toluenesulfonate (PPTS).
Regarding finally the selective deprotection studies in mild basic conditions (including the attractive application of Brönsted base impregnated onto alumina as the deprotective agent), the effect of microwaves is demonstrated to be highly decisive, as a drastic diminution of reaction times are encountered, without alteration of the initial anomeric configuration [6]. For instance, Ley reported on the possibility of microwave-assisted hydrolysis, on neutral alumina, of a set of pivaloyl protected sugars (Fig. 4) [7]. The regioselective deprotection occured selectively, cleanly and efficiently, without group migration or anomerisation, after only a few minutes of microwave heating at 75 °C. As a comparison, depivaloylation could not be achieved using an alternative “classical” heating source.
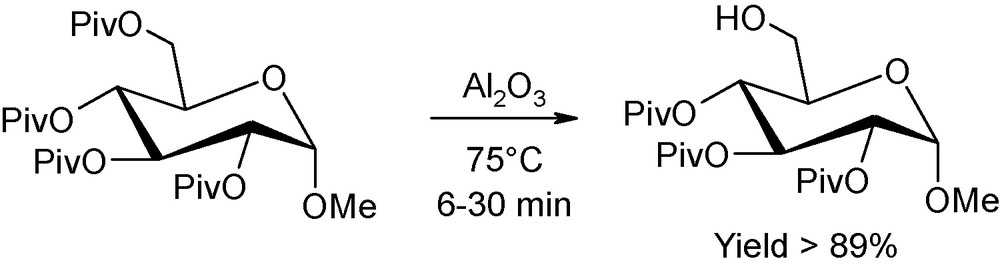
Regioselective microwave-promoted depivaloylation onto alumina.
2.2 Derivatisation at the anomeric position
2.2.1 Glycosylations
Beside hydroxyls protection/deprotection sequences, microwaves can successfully be employed in alkylation/glycosylation reactions [8]. Microwave-accelerated Fischer glycosylation, under acidic conditions, affords in good yields and in a single step, alkyl-chain glycosides, preferentially as the thermodynamic α-pyranoside isomer. When using conventional heat, a mixture of α, β-furanosides and α, β-pyranosides is systematically isolated, whilst classical Koenigs–Knorr and Helferich multi-step and time-consuming glycosylations of fatty alcohols provide otherwise the corresponding alkyl α, β-glucopyranosides (Fig. 5) [9].

Microwave-induced synthesis of alkyl O-glycosides via Fischer-type glycosylation.
From the literature, however, little information can be found about how microwaves could control the outcome of such glycosylation reactions: correlation between applied temperature and yields, impact of the microwave heating on the possible thermal decomposition of saccharides, control of the anomeric ratio. Kovensky et al. reported recently some answers in this field exploring the microwave-assisted glycosylation of peracetylated glucose catalysed by a Lewis acid in solvent-free conditions (Fig. 6) [8c] A set temperature of 115 °C was judged enough in order to prevent any decomposition of initial sugar by browning reaction. In these conditions, glycosylations of fatty alcohols were fast and high-yielding. In parallel, it was underlined that the physical nature (melting point) of the alcohol and its polarity impacted over yields and anomeric selctivities. Most polar alcohols provided the corresponding α-adduct in very short reaction times (less than 5 min), whilst long chain alkyl alcohols afforded preferentially the corresponding β-adduct. Although microwave heating increased the rate of glycosylation, it also resulted in yield reduction (degradation of the resulting glycoside) when exposure times were prolonged, especially with most polar alcohols.

Microwave-promoted glycosylation of peracetylated glucose.
Nowadays, the interest for well-defined monosubstituted glycosides is significant and is related to their particular physical properties suited for use as liquid crystals or as non toxic and biodegradable surfactants [10]. Moreover, these molecules play an important role as chemical synthons in the pharmaceutical sector [11]. For this reason, alkylation/glycosylation reactions are amongst the most extensively studied ones the field of carbohydrate chemistry under microwave conditions [12].
Novel glycosylation concepts have also emerged in the last decade. They concern the conjugation of an alcohol as the acceptor and either 4-pentenyl glycosides or methyl glycosides as the donors (Fig. 7). Usually, alkyl glycosides are not relevant as glycosyl donors, as they require harsh acidic conditions to break the glycosyl bonds, but 4-pentenyl and methyl-glycosides are proven to be suitable alternatives, in particular under microwave irradiation [13]. For instance, methyl-2,3,4,6-tetra-O-benzyl-β-d-glucopyranoside can be converted into octyl-2,3,4,6-tetra-O-benzyl-d-glucopyranoside (mixture of α and β anomers) with a 2-fold excess of n-octanol and using ytterbium triflate as the acid promoter. Under conventional conditions, the conversion yield does not exceed 46% after 1 h at 120 °C. At the opposite, yield reaches up to 72% after 10 min (or 88% after 30 min) for reactions performed, under exactly the same conditions (reaction scale, amount of reagents), in a microwave single-mode reactor.
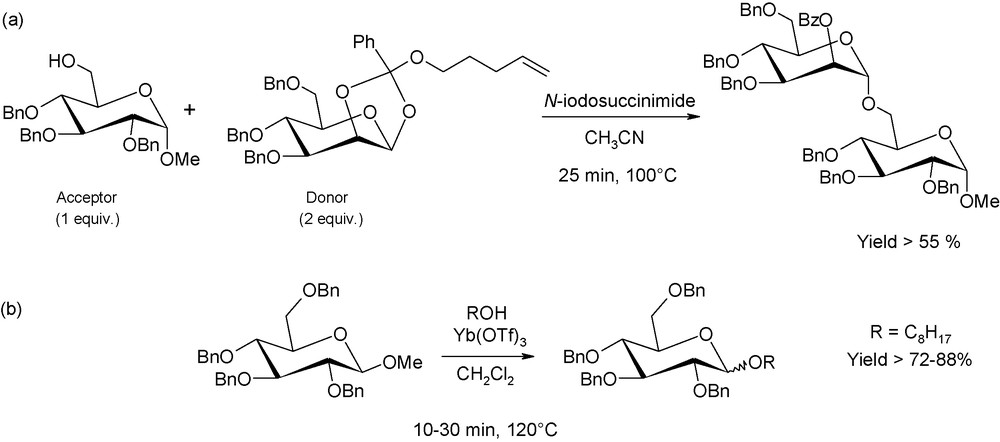
(a) Saccharide coupling using n-pentenyl glycosyl donors; (b) acetal-exchange type glycosylations involving methyl glycosides.
2.2.2 C-aryl glycosides production
The synthesis of C-aryl glycosides has also been evaluated under microwave heating. These molecules are indeed unique carbohydrates, with an aromatic cycle directly bound to the anomeric carbon, and exhibit diverse biological activities such as antibacterial, antitumor and enzyme inhibitory effects [14]. Lei et al. have described a new method for the microwave-assisted preparation of such anomerically pure C-aryl glycosides, involving a palladium-catalysed cross-coupling reaction between a perbenzylated pyranoid glycal and an aryl bromide (Fig. 8) [15]. This practical method was proven to be more rapid and stereospecific than the majority of other art-known techniques [16]. Indeed, only a few minutes at 170 °C was enough to achieve up to 75% yield of this Heck cross-coupling reaction between an aryl bromide and a 3-fold excess of pyranoid glycal, while prolonged reaction time (several days) and a large excess of glycal were necessary to obtain the desired adduct (α and β mixture) under thermally-driven conditions.

Microwave-induced synthesis of aryl C-glycosides.
2.2.3 β-Glycosylamine synthesis
Bejugam and Flitsch have reported on the efficient microwave-assisted production of glycosylamines from mono-, di- and trisaccharides (Fig. 9) [17]. These β-glycosylamines, suitable intermediates in the production of glycopeptides, [18] are classicaly prepared by treatment of a fully deprotected sugar with 40–50 equivalents of ammonium bicarbonate at room temperature for 6 days (Kochetkov amination) [19]. Results under microwave conditions were noticeable as all syntheses were complete within 90 min under relatively low reaction temperatures (about 40 °C) and reduced quantity of ammonium bicarbonate (5-fold excess), providing the corresponding glycosylamine adduct as the pure β anomer. Key to the success of the reaction was firstly use of anhydrous DMSO as the solvent and, secondly, work below 60 °C in order to prevent any significant dimerisation.

Microwave-assisted production of β-glycosylamines using ammonium bicarbonate.
Even if highly promising and fast, this microwave route does not compete with the “greener” protocol developed in 1995 by Lubineau et al. This approach, valuable for the straightforward obtention of glycosylamine from d-glucose, d-galactose, lactose, cellobiose and maltose consists in the simple treatment at 42 °C for 36 h of the saccharide with an aqueous solution of ammonia in the presence of one equivalent of ammonium hydrogen carbonate [20].
2.3 Other derivatisations
Beside anomeric centre derivatisation, microwaves have also been employed to induce the regioselective functionalisation of sugars with branched-chain or unsaturated alcohols. Some of these carbohydrate-based derivatives are constituents of antibiotics and nucleotides or are valuable starting materials for the production of biodegradable and biocompatible polymers [21]. In this context, Andrade et al. have proposed a multi-step microwave-assisted protocol, consisting of the selective protection of primary carbon positions followed by a one-step regioselective formylation. These sugar O-formates undergo then a Witting olefination in a microwave oven and in the absence of solvent to afford, in high yields and in short reaction times, valuable carbohydrate derivatives with vinyl ether-type chains selectively appended to the primary carbon positions (Fig. 10) [21c].
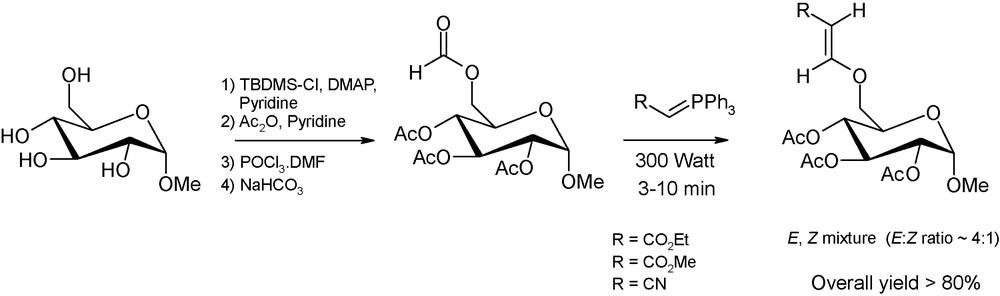
Microwave-promoted synthesis of glucose derivatives containing an unsaturated group. The first step is decribed in a previous work of Andrade et al. [22].
Interesting also is the straightforward approach reported by Yadav et al. for the obtention of imidazo[1,2-a]pyridine scaffolds from d-glucose and d-xylose (Fig. 11) [23]. These iminosugars are potent therapeutic agents for genetic and metabolic infections [24]. However, their “classical” synthesis is fastidious, time-consuming and required the manipulation of toxic solvents and strong Lewis acids as the promoters [25]. Microwaves are a powerful strategy for the direct one-step production of these important molecules as reactions are performed in solventless conditions, using milder acidic promoters, in high yields after only a few minutes of irradiation at 80 °C.
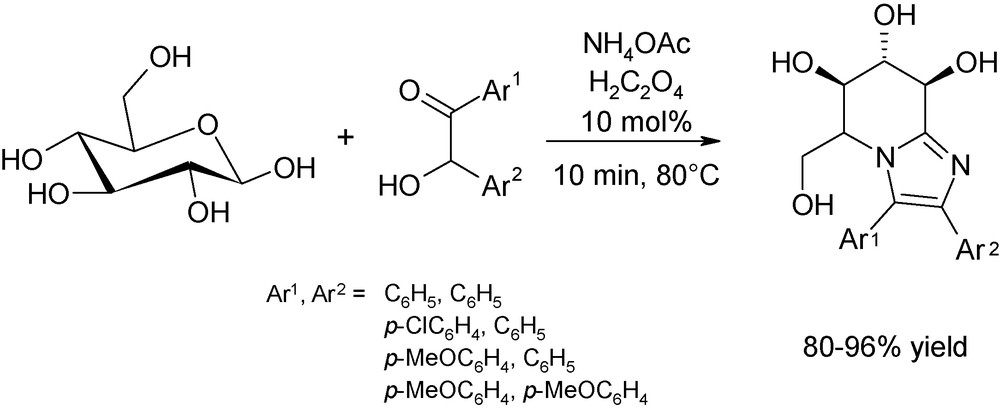
Quantitative microwave-assisted synthesis of iminosugar-bearing tetrahydroimidazo[1,2-a]pyridines from d-glucose catalysed by oxalic acid.
Microwaves have also been explored for a panel of monosaccharides related transformations, including mutarotations, [26] halogenations, [27] Tipson-Cohen protocol for the production of unsaturated pyranosides [28], or oxidations [29]. In general, faster and cleaner reactions are described, even if these specific reactions appear more as “laboratory curiosities”.
2.4 Microwave-assisted click chemistry
Copper(I)-catalysed azide-alkyne 1,3-dipolar cycloadditions (also referred as “click chemistry”) has progressively gained special attention in the field of carbohydrate chemistry [30]. This reaction is indeed a valuable synthetic tool for the preparation of a wide range of sugar-based architectures such as glycopeptides, glycopolymers, glycosylated biomolecules or immobilisation of carbohydrates onto solid surfaces (glycoarrays).
Even if most “click” reactions can be performed at room temperatures, there are instances where some “process intensification” are needed [31]. As an illustration, Lucas et al. reported in 2008 on the synthesis of a triazole-linked 3’-5’ dithymidine dimer, interesting molecule in biochemistry and genomic research, making use of this 1,3-dipolar cycloaddition (Fig. 12) [32]. Whilst coupling reaction between the azide derivative and the propargyl precursor affords 80% of the wanted product after 5 h at 80 °C in an oil bath, reaction under identical conditions (concentrations and temperature) provides the same isolated yield after only 3 min of microwave exposure.
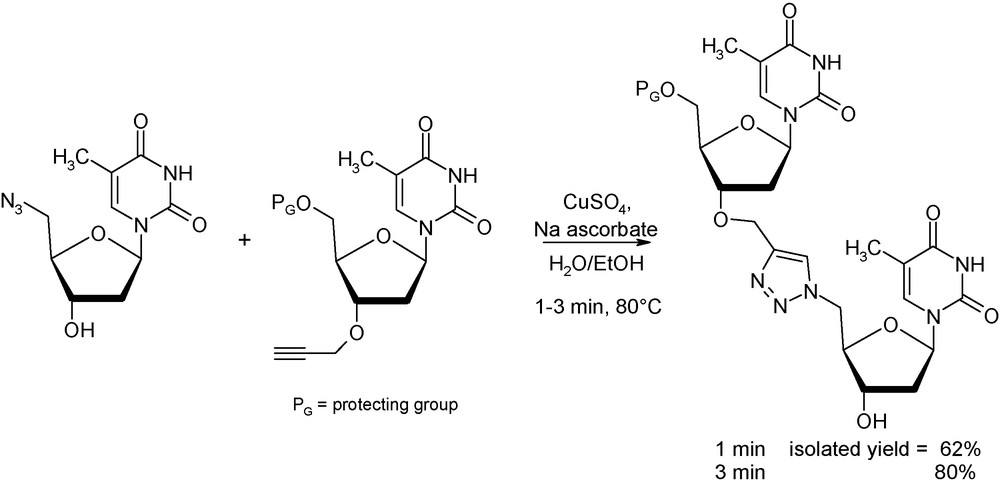
Microwave-assisted click chemistry between azido and propargyl precursors.
Using the same Huisgen 1,3-cycloaddition, Liskamp et al. reported on the brillant involvment of microwaves for the preparation of a panel of triazole glycodendrimers, up to the nonavalent level, starting from various (O-protected or O-unprotected) azido mono- or disaccharides and multivalent alkyne-linked dendrimers (Fig. 13). The protocol is highly selective and efficient, providing the desired triazole-linked glycodendrimers in up to 95% yield after an irradiation time of 20 min at 80 °C in sealed vessels [33].

Microwave-promoted synthesis of triazole-linked glycodendrimers by click chemistry.
2.5 Synergy between microwaves and heterogeneous catalysis
Nowadays, to the best of our knowledge, publications dealing with the involvement of microwaves in sugar chemistry are still limited. Nevertheless, new trends are observed and concern the ingenious synergic combination of heterogeneous catalysts and microwave activation, as a cleaner and more efficient methodology. For instance, this technique has recently been published for the production of new compounds arising from acidic sugars (Fig. 14). In this example, considerable alteration of the selectivity of the reaction is reported using inorganic acids loaded onto silica. Novel platform lactonic derivatives are thus isolated from d-glucuronic acid, whilst disubstituted α,β-pyranosidic and α,β-furanosidic molecules are recovered under “classical” oil-bath heating [34].
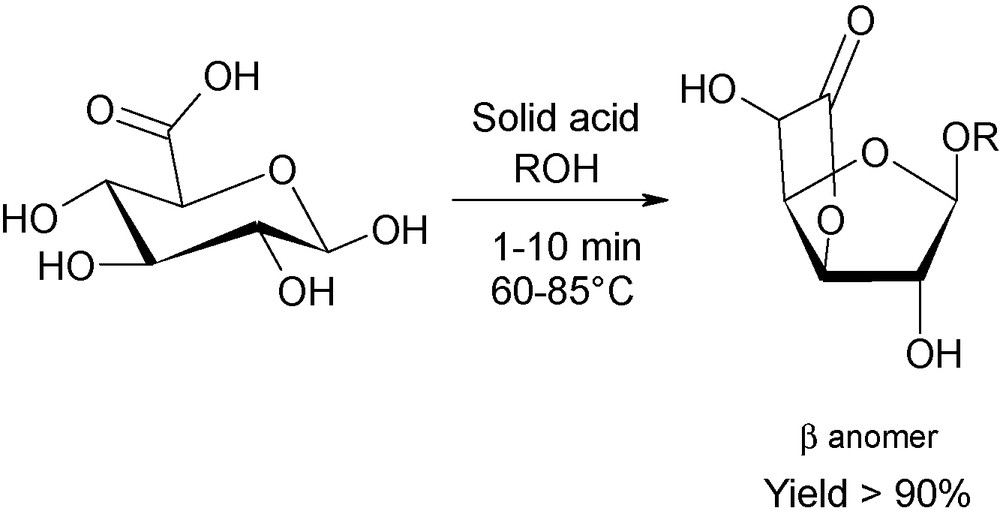
Microwave-induced lactonisation of d-glucuronic acid using supported acid catalysts.
Using the same strategy, the kinetically-favored disubstituted β-furanosidic isomer were isolated, in up to 85%, when O-unprotected d-galacturonic acid was treated with a 5-fold excess of alcohol after a microwave exposure time of 5 min at 85 °C, demonstrating the clear selectivity of microwave dielectric heating [35].
Noteworthy also is the production, catalysed by heterogeneous TiO2 and ZrO2 or ion-exchange resins, of 5-hydroxymethylfurfural (5-HMF), a valuable intermediate for pharmaceuticals, fine chemicals and furan-based polymers (Fig. 15) [36]. The dehydration reaction of glucose or fructose is performed either in water/organic media or in ionic liquid conditions. A comparison with conventional heating reveals that microwaves have a remarkable accelerating effect not only over monosaccharides conversions, but also on 5-HMF yields.
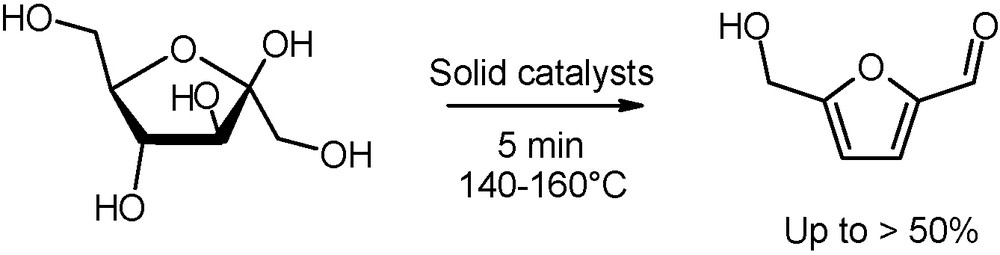
Microwave-assisted dehydration of fructose into 5-hydroxymethylfurfural.
3 Enzymatic modifications of monosaccharides
In parallel to chemical derivatisations of monosaccharides, it has been suggested that microwave activation enhances kinetics, yields and selectivities in enzymatic-mediated processes [37]. For instance, enzymatic (trans)glycosylations at 80–110 °C, using almond-β-glucosidase in dry media, and for reagents impregnated over alumina, demonstrates unprecedented results, with complete substrate conversion within 2–3 h [38]. These observations contrast with previous works, involving extended reaction times and large excess of alcohol. In the same manner, immobilised Candida antarctica lipase (Novozym 435) catalysed in satisfactory yields and purities the selective esterification of several sugars with dodecanoic acid under microwave irradiation [39].
4 Effect of microwave heating on polysaccharides
Microwave reactors have also been employed to promote either the functionalisation or the hydrolysis of polysaccharides. In particular, cellulose, the major component of lignocellulosic biomass, has been chosen as a focus.
4.1 Hydrolysis and pretreatment of cellulose
The conversion of cellulose into high added value materials is currently one of the most pursuits worldwide [40]. Cellulose is composed of d-glucose subunits joined together in long chains β-1,4 glucosidic bonds. Therefore, its enzymatic saccharification provides glucose that can readily be fermented to produce fuels ethanol or other intermediate chemicals [41]. The question of the microwave-assisted decomposition of cellulose into valuable by-products was evaluated for the first time in the 1980s. Allan et al. reported on the formation of,6-anhydro-β-d-glucopyranoside (levoglucosan), an interesting chiral synthon, when pyrolysing cellulose samples in a domestic microwave oven at 130 Watt for 5 min (Fig. 16) [42]. Yields in levoglucosan were moderate (about 40%), together with charred residues and water, whilst cellulose conversion was nearly quantitative.

Microwave-induced production of levoglucosan from cellulosic materials.
In 2007, Orozco published the dilute acid (chemical) hydrolysis of cellulose in a batch microwave reactor. The recourse to dilute phosphoric acid, at 150–200 °C for 2–10 min, led to the production of a mixture of simple monosaccharides (namely xylose, arabinose and glucose) whose respective yields could be modulated by an appropriate tuning of reaction conditions (i.e. acid concentration and microwave irradiation conditions) [43]. More recently, Zhang et al. have described the synergic application of microwaves and zeolites heterogeneous catalysts in ionic liquid to mediate the hydrolysis of cellulose into pure glucose. Compared with conventional heating mode, microwaves greatly enhance reducing sugars yields, and significantly reduce reaction times, while repressing side reactions such as dehydration and/or cross-polymerisation. As an illustration, a typical hydrolysis microwave-assisted hydrolysis of Avicel cellulose provides, after only 8 min at 240 Watt, 37% of glucose using HY zeolite as the acidic catalyst and 1-butyl-3-methylimidazolium chloride as the recyclable ionic liquid medium [44].
Microwaves have also emerged as an effective tool for the alkali pretreatment of cellulosic materials. The main purpose of such a pretreatment is to noticeably decrease contents of lignin and hemicellulose, reduce cellulose crystallinity and increase the porosity of the resulting materials. The microwave preliminary treatment has proven a beneficial effect, as it accelerates the subsequent enzymatic saccharification rates [45]. Compared to conventional alkali pretreatment, it is underlined that microwaves have an impact over weight loss and composition of the resulting cellulosic-containing material. For instance, wheat straw pretreatment at 700 Watt, in 1% NaOH aqueous solution, induces a starting material weight loss of 48.4% and a composition of 79.6% cellulose, 5.7% lignin and 7.8% hemicellulose after 25 min, compared to a weight loss of 44.7% and a composition of 73.5% cellulose, 7.2% lignin and 11.2% hemicellulose after 60 min of conventional heating. As a conclusion, microwave pretreatment is very efficient as it contributes to remove more lignin and hemicellulose after a shorter reaction time than other conventional alkali treatment.
4.2 Derivatisation of cellulose
Functionalisation of cellulose has attracted considerable attention since the 1930s. Indeed, by introduction of various functional groups onto the main polysaccharide chain, additional physico-chemical properties can be defined. Phosphorilated microcrystalline cellulose presents promising properties, such as high adsorption of serum protein, flame retardancy and corrosion inhibition. The microwave-assisted cellulose phosphorylation was devised in 2002 by Besson using urea as the swelling solvent (Fig. 17) [46]. This environmentally friendly procedure is efficient, even without cellulose pretreatment, and leads to monosubstituted phosphorus acid esters of cellulose with various degrees of substitution of the hydroxyl moities (0.2–2.8). Moreover, considerable improvements of this degree of substitution can be achieved in comparison with conventional heating protocols.
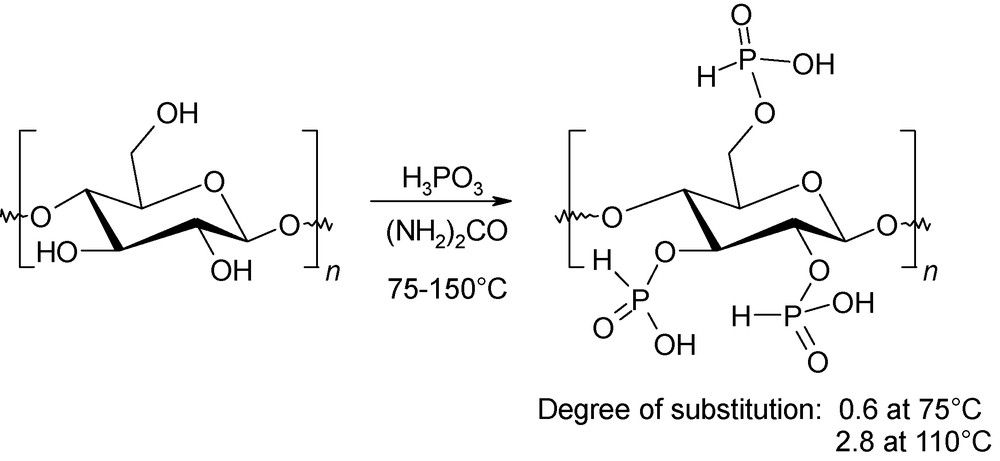
Microwave-induced phosphorylation of cellulose.
Microwave-induced acylation or esterification of cellulose to provide novel biodegradable biopolymers are also both extensively studied topics (Fig. 18). The first attempts at heterogeneous acylation of cellulose with short-chain carboxylic acids, under microwave conditions, were reported in 1999 by Gourson et al. [47]. Acylation of cellulose, in solventless conditions, by dodecanoyl chloride and with addition of 4-dimethylaminopyridine as the catalyst, afforded in 30% yield the corresponding esterified cellulose after 10–20 min of irradiation in a domestic microwave oven (750 W). Compared to conventional heat, reaction times were substantially reduced from several hours to a few minutes.

Acylation or (trans)esterification of cellulose induced by microwave irradiation.
Nevertheless, publications on the dissolution and the microwave-assisted chemical derivatisation, by acylation, of cellulose in binary solvent systems, such as N,N-dimethylacetamide/lithium chloride, have opened new vistas for cellulose functionalisation [48]. Microwave-assisted production of fatty esters of cellulose in such a binary solvent system results in a dramatic drop in reaction time as 1 min is enough to convert quantitatively the starting cellulose material into its corresponding long-chain carboxylic esters. In the same vein, ionic liquids appear as convenient media for the microwave-assisted functionalisation of microcrystalline cellulose by anhydrides [49].
Finally, the possibility of obtaining cellulose long-chain esters, via esterification or transesterification processes, has been studying under microwave heating. For instance, production of cellulose stearate, by transesterification of methylstearate with low-molecular weight crystalline cellulose, is achieved in solventless conditions using p-toluenesulfonic acid as the acid catalyst. Typically, a short period of time (10 min) at 90 °C affords cellulose stearate with a degree of esterification three times higher than the one estimated under conventional heating conditions. Duration of the reaction, as power of the microwave heating, is established to impact upon the degree of substitution [50].
5 Scale-up of microwave-promoted reactions
All examples reported here above deal with reactions performed generally at laboratory scale (several milligrams per run) using devoted single-mode microwave instruments. For microwave-assisted synthesis of carbohydrates derivatives to become a widely accepted industrial technology, there is a need to develop techniques for larger production (ideally beyond the Kg-scale).
As inferred above, glycosylation reactions are probably the most extensively studied in the field of microwave-assisted carbohydrate chemistry. An interesting contribution was presented by Loupy in 2000 who devised the use of a batch microwave platform for the glycosylation of decanol with peracetyl derivatives up to several hundred grams per run (Fig. 19). Yields were comparable to those obtained under identical conditions (temperature, reaction time, reagents ratio) in laboratory-scale experiments (1–3 gr-scale) [51].
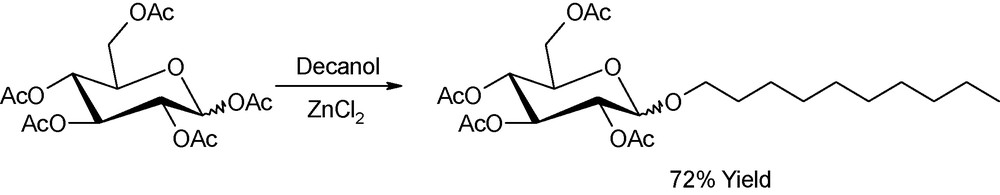
Glycosylation of decanol with peracetylated glucose.
Nuchter et al. accomplished also a valuable scaling-up of microwave-assisted Fischer glycosylations up to the kilogram-scale with an improvement in economic efficiency [52]. In batch reactions, several monosaccharides (i.e. glucose, mannose, galactose and butyl glucose) reacted with a 3–30-fold excess of alcohol (methanol, ethanol, butanol or octanol) in the presence of a catalytic amount of homogeneous acid. Yields were nearly quantitative after 20–60 min of microwave exposure at temperatures ranging between 60 and 140 °C. In further experiments, scale-up of Fischer glycosylations of aliphatic alcohol with glucose or derivatives were also investigated using continuous flow methods. Practically, the premixed educts were circulated by a pump through the microwave reactor, which was preheated to 110–140 °C and 12–16 bars by microwave irradiation. Under these conditions, yields of the corresponding adducts were quantitative after a residence time of 5–10 min within the microwave cavity.
6 Conclusions
Microwave-heating was demonstrated to be an useful method to assist manipulations on mono- and polysaccharides. Short reaction times are usually associated to improved yields and selectivities, under milder reaction conditions, compared to classical thermally-driven processes.
Acknowledgements
This work has been carried out in the framework of the “Technose” Excellence Programme supported by the “Région Wallonne” (Belgium). The authors are grateful to the “Région Wallonne” for its financial support.