

1 Introduction
Carbohydrates are among the most common organic molecules and play a central role in all biological systems; therefore, their chemistry and synthesis are of fundamental interest. Due to the easy access of many carbohydrates, they have been extensively investigated throughout the time of modern chemistry. In the first years, the main interests were the physical properties as well as simple reactions in order to determine the structure of monosaccharides. Later, the focus moved more and more to the assembly of carbohydrates to oligosaccharides, which has been much refined and optimized during the years since the pioneering work a century ago. Sophisticated methods have been developed and the complexity of prepared oligosaccharides has increased tremendously during the last three decades. Even though nearly all types of glycosidic bond can be made today, some are still troublesome and far from standardized methods can be used – Hans Paulsen's statement1 [1] from 1982 is still valid today!
With the appearance of more glycosylation methodologies and new techniques, both in synthesis and analysis, there has been an increasing interest in understanding the effects and parameters important for the reactivity and selectivity of the glycosylation reaction [2]. Several “concepts” and explanations have appeared in the last three decades. The main focus has been on the effects taking place in ordinary donor systems, e.g., D-glucose in the 4C1 conformation. It is well understood that having an ester protective group on O-2 gives neighbor group participation, which results preferentially in the 1,2-trans product, i.e., β-glucoside. The effect of anchimeric assistance has also been shown to apply in carbohydrate chemistry; therefore, an increase in reactivity of 2-acyl donors having a 1,2-trans relationship are normally more reactive than their 1,2-cis counterparts [3].
It was early recognized that it is not only the substituent on O-2 that is important for the reactivity of the donor, but all substituents play a role. Generally, it has been observed that having more electron withdrawing groups (such as ester protective groups) resulted in a decrease in the reactivity (higher activation temperature), whereas less electron withdrawing groups increased the relative reactivity of the donor. This observation was put into a concept by Fraser-Reid [4] – the so called “armed-disarmed” concept, where ether protective groups are considered to be arming and acyl groups disarming (Scheme 1). This concepts has later been expanded to cover conformational restriction – torsional disarming by using tethering protective groups such as acetals, dispiroketals, etc. still with the focus on the 4C1 conformation (for the common D-sugars). The better understanding and control of glycosyl donors had a great influence on carbohydrate chemistry and resulted in a number of new methods, such as “one pot” oligosaccharide synthesis [2,5].
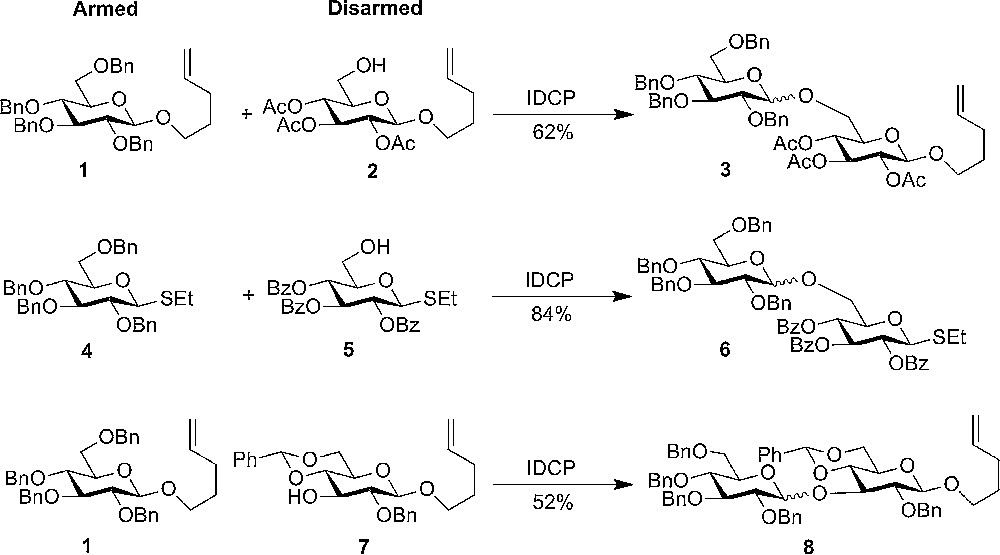
“Armed disarmed” concept - electron withdrawing groups are reducing the reactivity.
Recently, with the development of computational chemistry, the study of the glycosylation mechanism has gained renewed interest, and with the support of ingenious experimental work, the importance of conformation of the transition state (TS) has been shown to be crucial for the selectivity and stability of the oxocarbenium ion. Whitfield and coworkers related the stereoselectivity in the glycosylation with the conformation of the oxocarbenium ion, with the lowest energy conformation giving the major product [6]. Two families of conformers can be discerned, by restriction of the donor with e.g., a 1,3-dioxalane protective group, one of the conformer families is favorized, and therefore the selectivity increases. These theoretical studies can be used to explain experimental observed selectivities or to design face discriminated glycosyl donors. Internal hydrogen bonding as well as hydrogen bonding with the incoming acceptor was also shown to be of importance for the outcome of the glycosylations [7].
Woerpel and coworkers have also studied the influence of conformations on the facial selectivity in glycosylations by experiments, spectroscopy and computational methods [8]. By comparing the conformation of dioxocarbenium ions, which could be analyzed by NMR, with theoretical values of the related oxocarbenium ion, they observed a strong conformational preference for having alkoxy substituents pseudo-axially when having the half chair oxocarbenium ion due to electrostatic interactions. On the other hand, when having alkyl substituents, the preferred half chair conformation had equatorial substituents due to steric reasons (Fig. 1) [9].
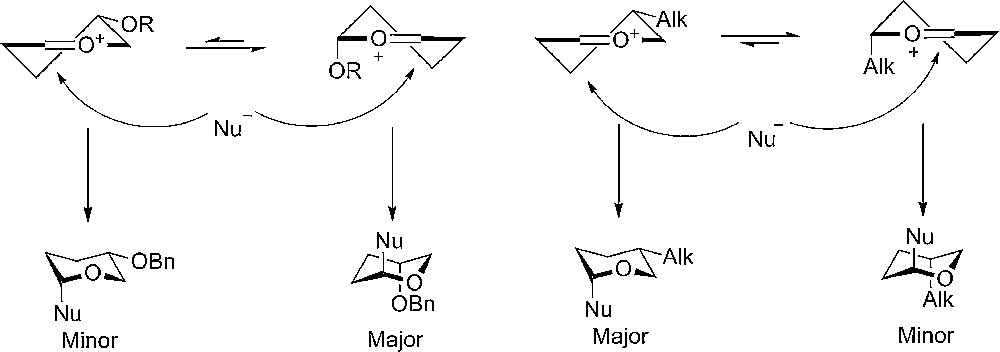
Pyran rings having a C3 or C4 alkoxy substituent prefer to adopt an axial rich half-chair conformation due to stereo electronic effects, which leads to the 1,4-trans product. Alkyl substituent leads to an equatorial rich half-chair conformation due to steric effects.
The established conformational preferences for a number of substrates could be used to predict the stereochemical outcome of reactions at the anomeric center and the theory could be confirmed by experiments.
Another approach to the analysis of conformational substituent effects, in connection with stabilization of a positive charge, was introduced by Bols and coworkers, who used piperidines to study the substituents influence of on the building-up of positive charge in a ring system. From pKa-measurements on the piperidonium ions, the effects from a wide variety of substituents could be quantified and a linear relation between base strength and the electron withdrawing capacity of the substituent could be established. Furthermore, it was clearly shown that an axial substituent, such as a hydroxyl group, was significantly less electron withdrawing than the equatorial counterpart (Fig. 2) [10].
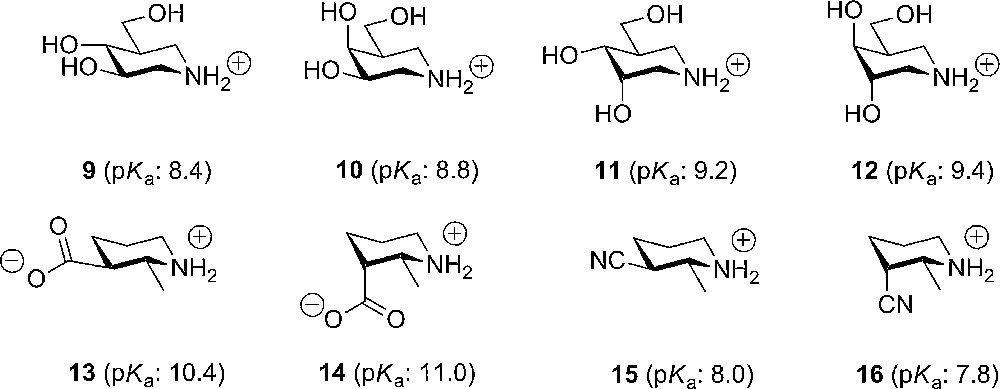
Some examples of the influence of stereo electronic effects on the pKa value in piperidines.
The observed effects were studied further in carbohydrates by measuring the hydrolysis of methyl pyranosides. Again, the conclusion was clear; when having more axial groups, a faster hydrolysis and hence a more stabilized oxocarbenium ion was the result. Further studies suggested that positively charged glycosylation intermediates attempt to adopt a conformation with a maximal number of axial substituents in order to gain stabilization. From the results obtained in the work on substituent effects in piperidines and pyranosides, it was proposed that axial rich glycosyl donors would be superior in reactivity and could alter the selectivity. Indeed, this was the case, as will be discussed later in this review (Fig. 3).
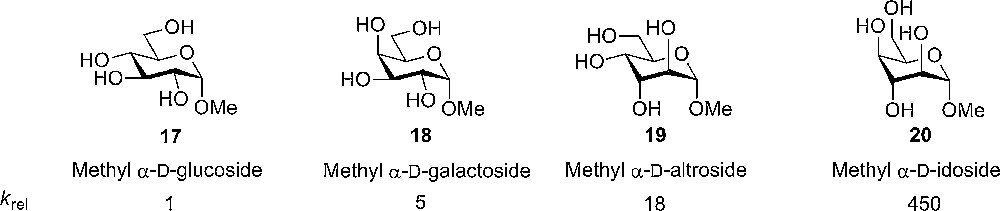
Hydrolysis of methyl glycosides with different configurations – more axial substituents gives faster hydrolysis.
In this review, we will focus on the reactions and properties of carbohydrates having unusual axial rich conformations, that is, conformations distinct from the ground state lowest energy conformation, i.e., as an example, the 4C1 conformation for D-glucose. The review is divided into three main chapters. The first concerns axial rich anhydrosugars, which are the earliest examples of “flipped” conformations and the most studied group of compounds in regard of synthesis and reactivity. The second chapter deals with the use of difunctional protective groups to tether substituents on the carbohydrate together and thereby induce a ring flip to a more axial rich conformation. The last chapter will focus on conformational changes induced by the substituent pattern – especially the use of bulky protective groups. This is a relative new methodology to flip the ring, but nevertheless a very promising way to enhance reactivity and selectivity in the reaction at the anomeric center.
2 Anhydrosugars
Anhydrosugars are widely used in carbohydrate chemistry as versatile intermediates. They are often stable and can be selectively activated when needed. The uses of anhydrosugars date back to the very early days of carbohydrate chemistry and have been extensively studied and reviewed [11]. One interesting feature of anhydrides is the conformational changes that are necessary for their synthesis and the difference in reactivity between the different derivatives. When introducing an anhydride in the sugar molecule, a bicyclic or tricyclic skeleton appears composed of oxirane, oxetane, oxolane and oxatane rings (3,4,5 and 6 membered oxocyclic rings, respectively). The oxirane (epoxide) is probably the most commonly used anhydro derivative due to its easy introduction and high reactivity. The conformation is preferentially a half-chair; this can, in many cases, be regarded as an axial rich conformation that should be part of this review. However, due to the substantial amount of work and reviews in this area, the focus will mainly be on flipped conformations, such as 1,6-anhydro and 3,6-anhydrosugars as well as the unusual such as 1,4- and 2,6-anhydrosugars. The use in glycosylation reactions with the reactivity and selectivity as main areas of interests will be presented.
A field where anhydrosugars have been extensively studied is in ring opening polymerizations. The unique properties of anomeric anhydrosugars, i.e., strained high energy conformations and at the same time activation of the anomeric center and protection/activation of the acceptor side make them very interesting starting materials. Since the first work appeared on 1,4-anhydro glucose derivatives to synthesize cellulose, many examples have appeared and reviewed [12].
In connection with this review, a few observations made in polymerization of glucosanes (1,6-anhydro) are worth pointing in order to understand some of the parameters important for the reactivity of the sugars. In the polymerization of levoglucosane derivatives containing azide groups in the 2,3 or 4 position, it was noticed that the exchange of a benzyl ether with an azido group decreased the reactivity. The decrease was found to be strongest when having a 2-azido substitution and smallest when having the 3-azido derivative (Scheme 2) [13]. The disarming effects of azido-groups are well known from glycosylation chemistry. From the polymerization of 1,6-anhydro derivatives, a difference in the relative reactivity of benzylated 1,6-anhydrohexoses has been established. Interestingly, the following decrease in reactivity was observed: manno > gluco > galacto > allo > altro [14] with the three first sugars being of similar reactivity, whereas allo only polymerizes slowly and altro does not polymerize at all under usual reaction conditions. In the unflipped 4C1 conformation, the opposite trend in reactivity is seen – but overall it makes sense that the more axial substituents give more reactive sugars. The substituent on C2 affects the anomeric reactivity more and in more ways that the rest of the substituents due to close contact with the anomeric center.
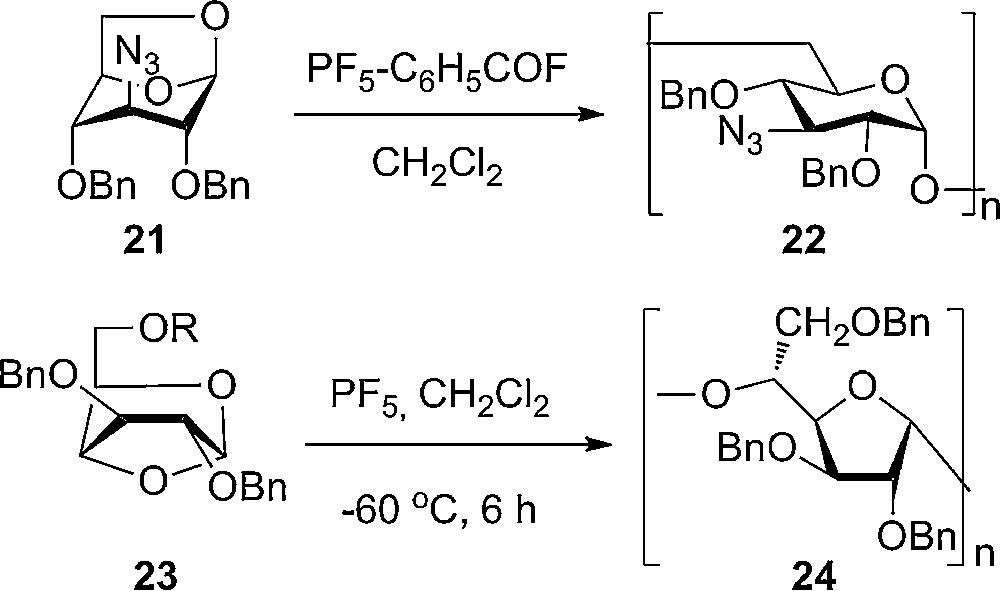
Ring opening polymerization of 1,6- and 1,4- anhydrosugars to give (1-6)-glucan derivatives and (1-5)-glucofuranan.
It has been suggested that the main driving force in these reactions is the release of strain by the ring opening, but due to the lower reactivity observed with gluco compared with manno, this cannot be the only explanation. Other factors such as the ease of conformational changes during the reaction could also influence the reactivity [15].
Polymerization of 1,4-anhydroglucose derivatives was first described by Micheel and co-workers [16] to give 1,4-α/β-glucopyranosidic polymers; later it was found to be 1,5-furanan [17]. The protective group pattern in the monomers was shown to play an important role for the ring opening polymerization (Scheme 2) [18]. Except for the polymerization reactions, 1,4-anhydro- and 1,4-anhydro-glucopyranosides have not been used in glycosylation reactions and studies of the reactivity in comparison with usual sugars have not appeared.
The increased reactivity of having an anhydro compound has been known for many years – one of the first to realize the unique properties was Haworth in the early 1940s [19]. Haworth observed that methyl 3,6-anhydro-2,4-dimethyl-α-D-glucopyranoside 25 was more reactive towards acid than the corresponding β 26 and when treated with acidic methanol it was converted into the more stable β-configuration (Scheme 3). In aqueous acid the pyrane ring opened to release strain to give the open chain aldehyde 27 and not the expected hemi-acetal. Non-aqueous hydrolysis experiments of unprotected methyl-3,6-α-D-glycopyranosides 28 rearranged to the less strained furanoside 29.
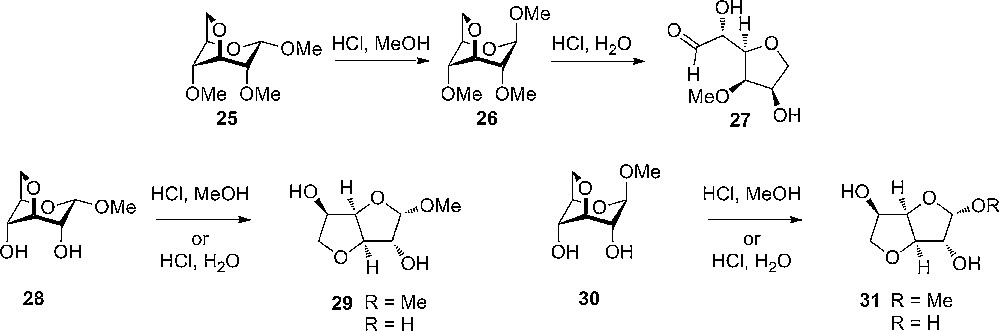
Haworth's hydrolysis of various 3,6-anhydro-glucopyranosides to give open form with protected substrate and furanosides with unprotected methyl glucosides.
Since then, several reports on the reactivity of anhydrosugars have appeared in the literature. One example is the observed acid sensitivity of 1’,2;3,6;3’,6’-trianhydrosucrose 32, which hydrolyze about 200 times faster than sucrose itself (Scheme 4) [20]. Another is the study by Foster et al., who continued the work by Haworth and coworkers, on the reactivity of 3,6-anhydrosugars. He invented an empirical model – so called i-values – that could predict the relative stability of the anhydrosugars. The greater the i-value of a molecule, the greater is its molecular strain and the lower its stability. It is, however, clear today that the reactivity differences seen for these molecules are caused by different electronic effects, not sterical effects [10]. From the work on 3,6-anhydrosugars, it was further more noticed that 3,6-anhydro-2-deoxy galactoside 34 hydrolyzes much faster that the corresponding 2-deoxygalactoside and the 3,6-anhydro-galactoside.
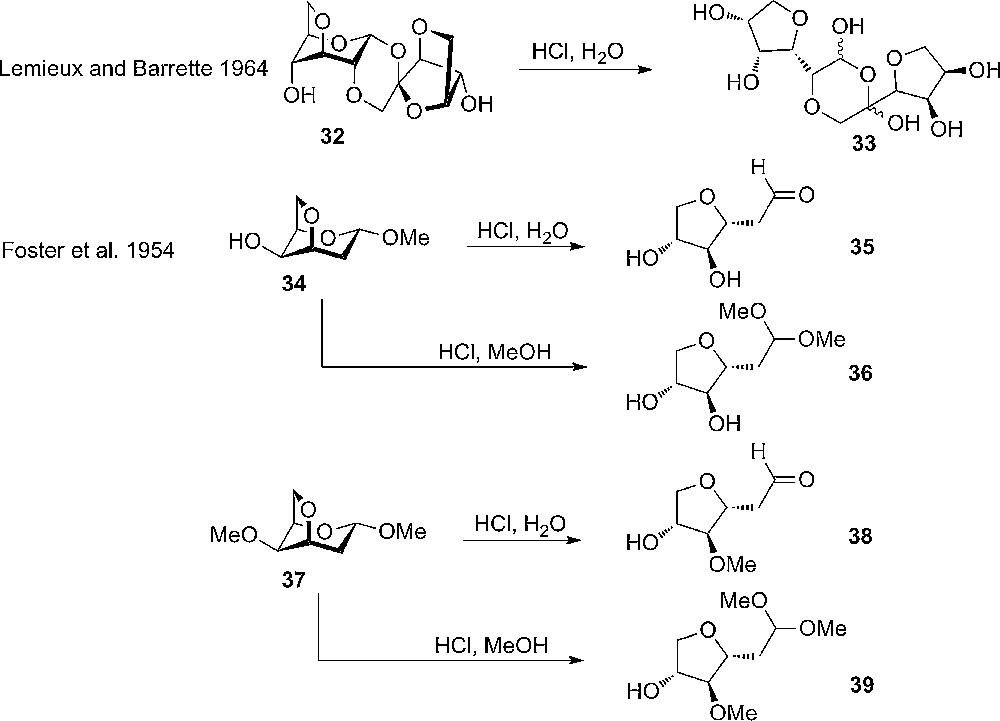
Early studies on the reactivity of different 3,6-anhydrosugars by Lemieux and Barrette and Foster et al.
Also, a difference between anomers of 3,6-anhydromannosides was observed – with the β being more reactive towards hydrolysis than the α. The difference between anomers were found to be opposite for glucosides, where the α hydrolyzed faster. The explanation for the diversity in reactivity was explained by strain in the different substrates [21].
A more recent example is the hydrolysis of agarose 40 (Scheme 5), a natural occurring polymer of the (1-3)-β-D-Galp-(1-4)-α-D-Galp-An- repeating unit. Upon reductive hydrolysis conditions, only cleavage of the flipped 3,6-anhydro sugar is observed, resulting in the reduced disaccharide 41 and tetrasaccharide 42 as major products.
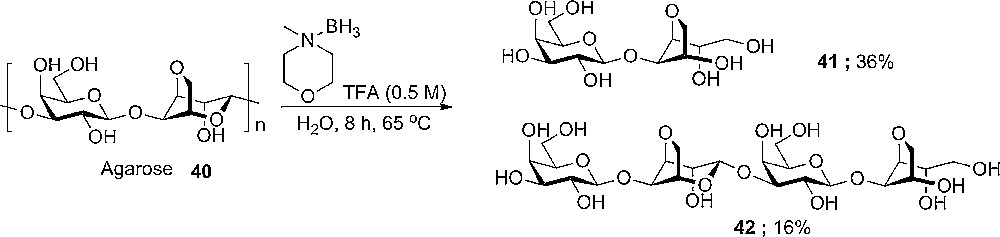
Hydrolysis of agarose 40 to give well defined di-and tetra-saccharide substructures.
A study to quantify the influence of the conformation on the reactivity was first accomplished by Bols and co-workers, who investigated the effects on glycoside reactivity by studying the hydrolysis of a selection of 3,6-anhydroglucosides. The reactivity was quantified by reaction kinetics and it was found that methyl 3,6-anhydro-β-D-glucosides 45, which adopts the 1C4 conformation, hydrolyses about 200 times faster than the methyl glucosides 43 and 44 in the usual 4C1 conformation (Table 1). Interestingly, it was found that methyl 3,6-anhydro-β-D-galactoside 47 was less reactive than the corresponding methyl galactoside 46. This observation was explained by the fact that the anhydrogalactoside adopts the B1,4 conformation and not the axial rich 1C4 (Scheme 6).
First order rate constants for acidic hydrolysis of glycosides. Data for 9, 10 and 12 from [22].
| Compound | k × 105 s−1 (60 °C) | Relative hydrolysis rate (2M HCl, 60 °C) |
| 0.708 | 1 | |
| 1.26 | 1.8 | |
| 316 | 446 | |
| 5.13 | 7.2 | |
| 2.71 | 3.8 |
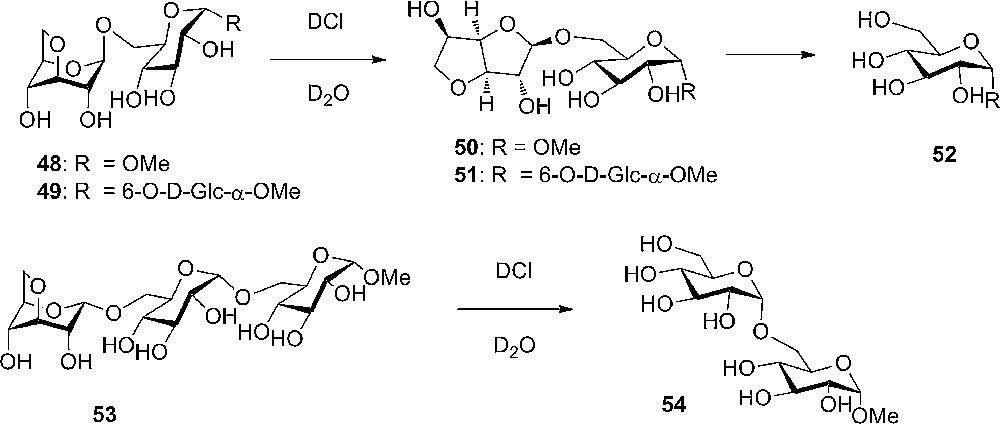
Hydrolysis of a di- and a tri-saccharide containing the 3,6-anhydro sugar unit.
When looking at linear oligosaccharides containing an anhydro-sugar unit, it was found that the hydrolysis selectively took place at this residue – again underlining the greater reactivity of axial rich carbohydrates. As Haworth described for the hydrolysis of the methyl 3,6-anhydro-α-D-glucoside 28 (Scheme 3), the less strained and hence more stable 3,6-anhydro furanosides are intermediates in the hydrolysis reactions (Scheme 6). Surprisingly, the same trend was not found in cyclic oligosaccharides such as β-cyclodextrin with one anhydro-sugar unit. The cyclodextrin breaks randomly to give an open chain, which is fast further hydrolyzed at the expected anhydro sugar to give a mixture of oligosaccharides of different length.
During the synthesis of the trisaccharide 53, containing the 3,6-anhydro-α-D-glucopyranoside, pentenyl 3,6-anhydro-2,4-di-O-benzyl-D-glucopyranoside (55α and 55β) were prepared and used in glycosylation reaction with the primary acceptor methyl 2,3,4-tri-O-benzyl-α-D-glucopyranoside (Scheme 7). Surprisingly, it was observed that the reaction was very selective towards the β-product 57 and not the desired α anomer. The selectivity was independent of the anomeric configuration of the donor used. The explanation for this remarkable selectivity could be the anomeric effect.
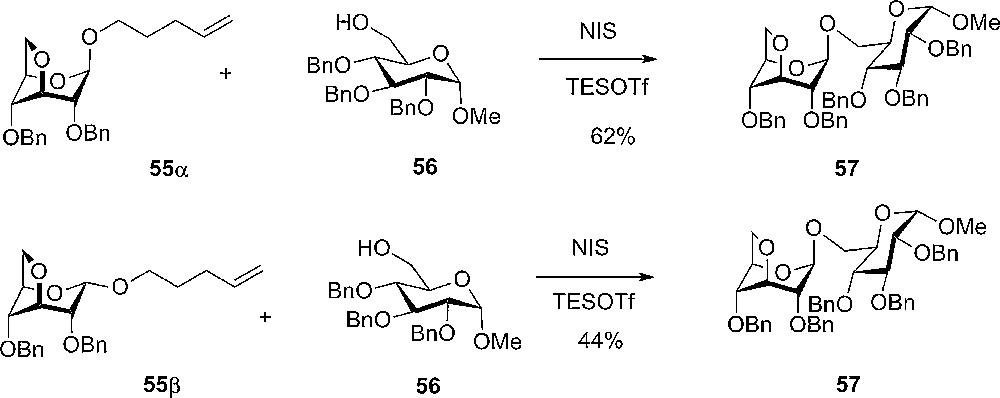
Glycosylation with 3,6-anhydro donors.
Bochkov and Kalinevitch describe the first use of 3,6-anhydro-α-D-galactopyranose-1,2-(methyl orthoacetate) 58 as a glycosyl donor in the synthesis of disaccharides containing the anhydro sugar (Scheme 8). From NMR studies, it was suggested that the donor adopts the B1,4 conformation rather than a 1C4. In the synthesis, it was noticed that the donor was considerable more reactive than comparable orthoesters of other sugars, as well as only one tenth of the catalyst HgBr2 was needed. The reaction provided 70% of the desired β-product 60 [23].
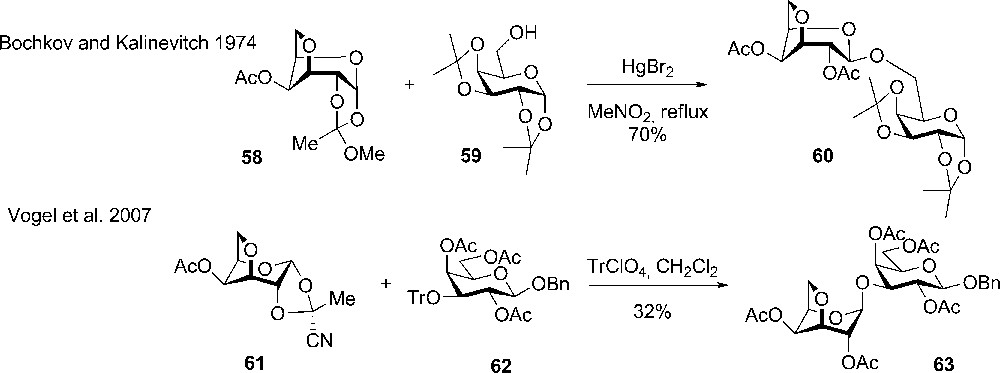
The use of 3,6-anhydro sugar ortho esters in glycosylation reactions.
Later, a similar approach was taken up by Vogel et al. who prepared 4-O-acetyl-3,6-anhydro-1,2-O (1-cyanoethylidene)-α-D-galactopyranose 61 and used it a galactosyl donor (Scheme 8). It was found from crystallography that the anhydro compound adopted a flipped 1C4 conformation [24]. In the coupling with 3-O-tritylated acceptors (rhanmo and galacto) an excellent β-selectivity was observed but the yields were modest (30 to 60%). Similar yields and high selectivity were obtained with 3,4-di-O-acetyl-1,2-O-(1-cyanoethylidene)-6-O-tosyl-α-D-galactopyranose (unflipped conformation), however, difference in reactivity between the two conformations was not investigated.
Recently, van der Marel and coworkers studied glycosylation with conformationally restricted uronic acids and found that galactono-3,6-lactones 64 are reactive and highly α-selective donors (Scheme 9) [25]. Similarly, the 2,6 and 3,6 lactones of mannuronic were prepared, but to this date no studies of them as glycosyldonors have appeared [26].
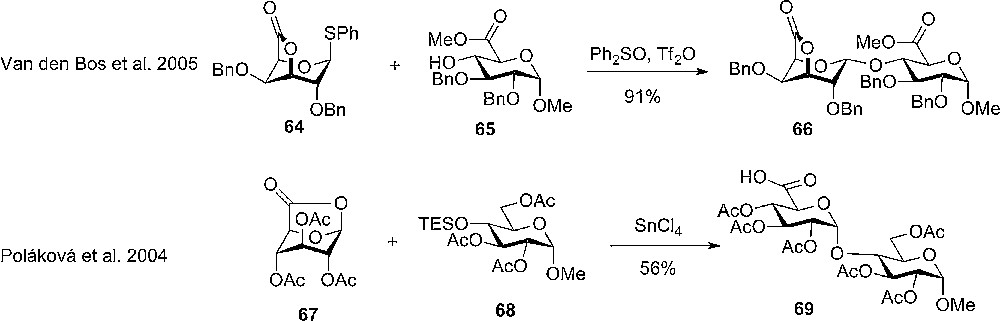
The use of conformationally restricted axial rich uronic acid glycosyl donors.
Another approach to the stereoselective synthesis of uronic acids was introduced by Murphy and coworkers, who studied the glycosylation reactions with inverted glucuronic donors (Scheme 9). In the SnCl4 mediated coupling with silyl ether acceptors (68), an increase in yield compared with usual unflipped donor was observed. The selectivity was further more excellent towards the α-anomer and it seemed like neighbor participation from the 2-O-acetyl did not play a role for the selectivity in the glycosylation [27].
The reactivity of 2,6-anhydro pyranosides was first studied by Hudson, who looked at the hydrolysis of methyl 2,6-anhydro-α-D-altroside 70, which was found to be less acid sensitive compared with the 3,6-anhydro derivatives of methyl-α-D-galactopyranoside and methyl α-D-glucopyranoside. Due to the formation of 1,6-anhydro-D-altrose 71 when hydrolyzing methyl α-D-altrose the rate of hydrolysis could not be measured and compared to the 2,6-anhydro-derivative, it was, however, stated that the compound hydrolyzed significant faster than methyl α-D-glucopyranoside [28]. Methyl 2,6-anhydro-3,4-di-O-α-D-mannoside 72 has also been prepared, but to the best of our knowledge the reactivity has not been studied [29] (Fig. 4).
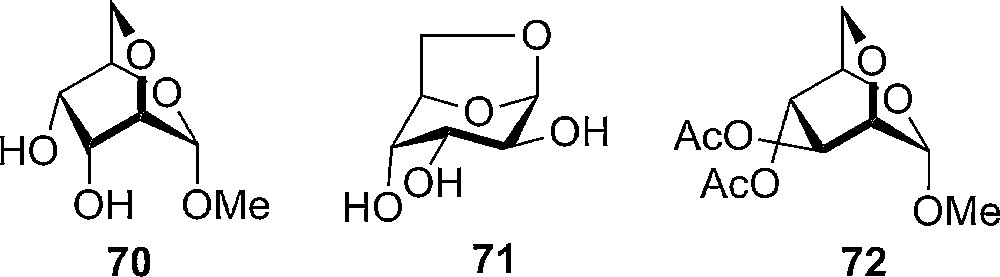
Anhydro sugars prepared by Horton et al. and Eades et al. respectively [30].
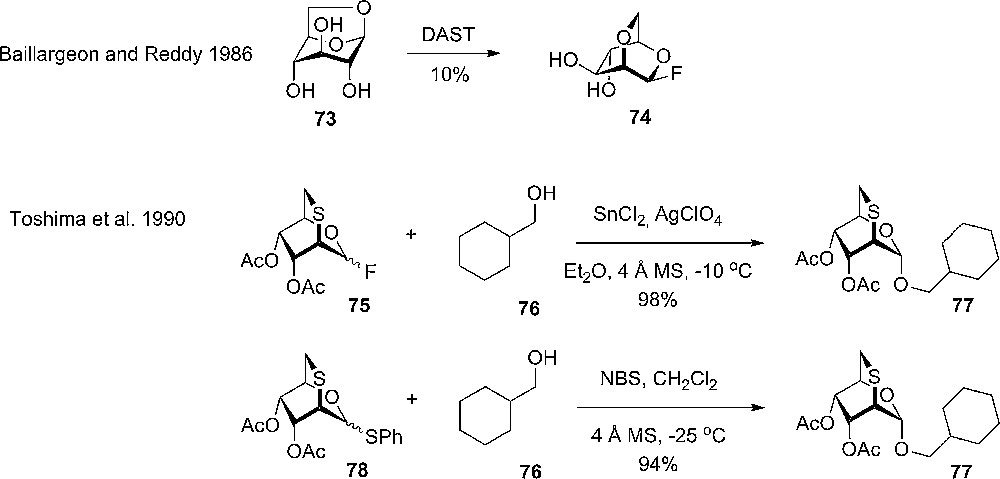
The application of 2,6-anhydro pyranosides and similar compounds in glycosylation reactions has been investigated and some interesting features of this class of donors have been discovered. The first report on the synthesis of a potential 2,6-anhydro donor came from Baillargeon and Reddy, who upon DAST mediated fluorination of 1,6-anhydrosugars observed the formation of a side-product – 2,6-anhydro-β-D-hexopyranosyl fluorides, due to a 1,2 migration (Scheme 10). The potential donors, one of them being 74, were analyzed by NMR and crystallography to confirm the structure and conformation, but were not used in glycosylations [30].
Early work on 2,6-anhydro-2-thio glycosyl donors.
Tatsuta and coworkers prepared and used 2,6-anhydro-2-thio glycopyranosyl fluoride 75 in glycosylation reactions. With the sulfide bridge, the compounds are easily converted into 2,6- deoxysugards, which are difficult to synthesize by using conventional donors. The glycosylation reactions of either the thioglycoside 78 or the glycosyl fluoride 75 proceed smoothly at low temperature and turned out to be very α-selective and to give high yields with simple acceptors (76). The influence of solvents in the reactions was limited and only with donor 79 having dichloromethane as solvent and slightly higher temperature (−10 °C instead of −40 °C) the β-anomer 80 was the sole product (Scheme 11) [31].
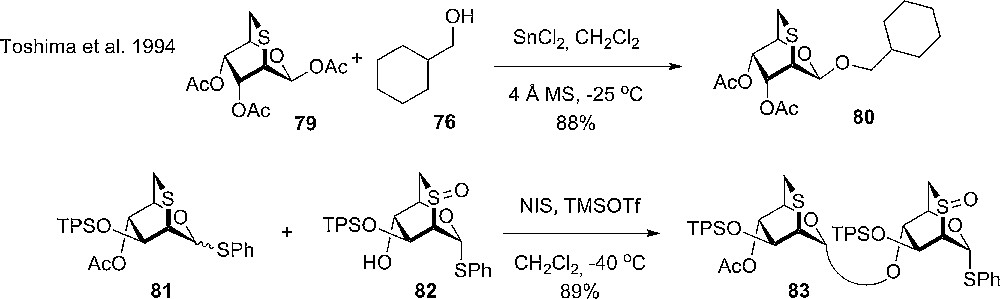
β-Selective glycosylation with 2,6-anhydro-2-thio glycosyl donor and disarming of a thioglycoside donor by oxidizing the ring sulphur.
When comparing the 2,6-thioanhydro donor with the usual 2,6-di-deoxy donor, it was found to be significantly more reactive and to provide an excellent α-selectivity, the usual donor gave high yields at higher temperature, but without selectivity. Again, no solvent effect nor anomeric configuration of the donor was observed to influence the selectivity of the donors (thioglycoside and glycosyl fluoride) in glycosylations. Interestingly, it was observed that having an anomeric acetate 79, altered the selectivity and a strong dependence on the solvent was observed, with ethereal solvents giving rise to α-selectivity and MeCN, toluene and CH2Cl2 giving excellent β-selectivity (Scheme 11). The outcome of the reactions was explained with the α-anomer being the kinetic product due to the repulsive electronic interaction between the incoming alcohol and the ring sulphur. The β-anomer, on the other hand, is the thermodynamic product due to 1,3-diaxial interactions with the 3 and 4 substituents in the sugar ring. Under the glycosylation conditions, the α- can anomerize to the more stabile β-anomer 80. Another feature with the new donor was illustrated with the oxidation of the ring sulphur to give the sulfinyl and sulfonyl respectively. The reactivity of the derivatives is reduced considerably, and will not be activated under the standard conditions. The oxidation state of the sulphur can then be reduced again when the donor functionality is needed (Scheme 11) [32].
The efficacy of the 2,6-anhydro donors were underlined with the synthesis of the natural products erythromycin A and olivomycin A trisaccharide [33,34].
The opening of 1,6-anhydrosugars by nucleophiles is a well known reaction in carbohydrate chemistry and the results that have emerged in this area have been reviewed elsewhere2 – the use of 1,6-anhydrothiols on the other hand is a relative small niche in carbohydrate chemistry [36] and most of the studies, regarding glycosylations, have been made by Stick and coworkers. The first report on glycosylation with thiolevoglucosane came already in the early eighties from Lundt and Skelbæk-Pedersen who were able to isolate a crystalline sulfonium salt 84 from the reaction with the thiolevoglucosane with triethyloxonium tetrafluoroborate, which could then react with nucleophiles to give O- or S- glucosides as well as substituted 1,2-O-alkylidenes depending on the nucleophiles used (Scheme 12) [37].
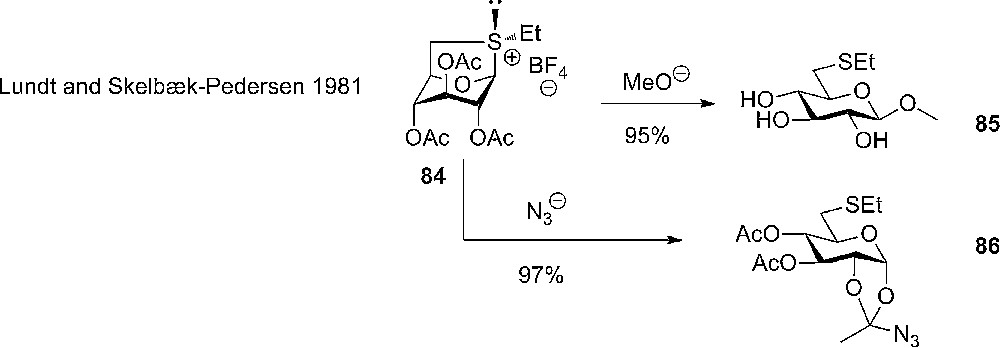
The use of crystalline sulfonium salt as donor in glycosylation with simple acceptors.
Stick investigated the use of 1,6-epithio- (87) and 1,6-episeleno-β-D-glucopyranose (88) as potential donors, which could be used in the synthesis of 6-deoxy containing oligosaccharides (Scheme 13). It turned out that migration of acetyl protective groups to the acceptor was a major problem and first when changing to the more stable benzoyl effective donors were obtained. Reasonable yields could be obtained in the glycosylations but only the seleno derivatives could easily be reduced to the desired 6-deoxy sugars [38].
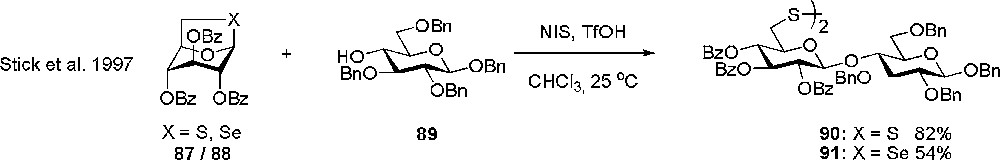
Glycosylation with 1,6-epithio and 1,6-episeleno-β-D-glucopyranose.
In the studies of the synthesis of β-acarbose the scope and limitation of the 3,4-anhydro-1,6-dideoxy-1,6-episeleno-β-D-glucose and the corresponding thio were evaluated. Again, the seleno based donor was superior compared with the sulphur, but decomposition of the donor became a problem [39]. The described donors could be oxidized to give sulfoxides and sulfones but none of them were particular efficient as glycosyl donors [40].
The reaction of anhydrosugars goes further than classical glycosylation chemistry. An example is the use of 3,6-anhydro bridged ulosyl bromide 92 as an intermediate in reductive cleavage as a route to the enolate, which upon trapping with an aldehyde 93 gives the aldol product 94 (C-glycoside).
The flipped conformation did not affect the anomeric selectivity, which was exclusively α, but the diastereoselectivity at the new stereo-center was improved compared with the use of a usual gluculosyl bromide.
3 Axial rich donors by tethering two or more functional groups together
In this chapter, the restriction of carbohydrates by tethering two functional groups together will be discussed. The use of bifunctional protective groups to tether functionalities together is used extensively in carbohydrate chemistry. Protective groups, such as benzylidene, isopropylidene, dispiroketals, silylenes, etc., are standard tools in the regioselective protection of carbohydrate building blocks. The influence on glycosylation selectivity and reactivity has been studied and used in the design of complex oligosaccharide synthesis. Taking into account the popularity of the protective groups it is striking that almost all examples in the literature concern pyranoses/pyranosides in the most common equatorial rich conformation – 4C1 for the most common D-sugars. The use of tethering protective groups to change the conformation of the sugar has been studied in some cases, such as for 1,2-orthoester (and similar compounds) but relatively little is known about the connection between conformation, reactivity and selectivity of the axial rich tethered compounds. This chapter will give an overview of the different types of tethering, their analysis and properties with the focus on reactions at the anomeric center (Scheme 14).
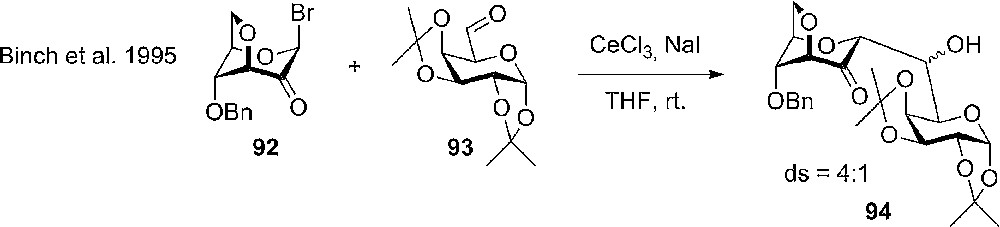
Reductive cleavage as a route to carbohydrate enolates and the application in C-glycoside synthesis.
3.1 Orthoesters and alkylidene tethering groups
One of the first reports about the conformations and properties of orthoesters describes some hydrolysis experiments made on α-D-glucopyranose 1,2-(ethyl orthoacetate)-triacetate. There is, however, not a direct comparison with similar compounds and hence no conclusion about reactivity. However, it was interestingly suggested that the orthoesters exist in another conformation than the expected 4C1 – this suggestion was build on the obtained optical rotation data [41].
The use of orthoesters as glycosyl donors dates back to Kochetkovs work from the early 1960s (Scheme 15) [42].
The first use of orthoesters as glycosyl donor by Kochetkov et al. [42].
Even though it was one of the only alternatives to the Koenigs-Knorr glycosylation, it was not used very much for the next decades. Recently, the orthoesters as glycosyl donors have gained renewed interest and many improvements to the method have been published in the last decade.3 There are, however still only a few examples, where the conformation of the donor has been thoroughly investigated and used to study the influence on reactivity and selectivity in glycosylation reactions.
In 1996, Makatsubo and coworkers investigated the use of the axial rich 3,6-di-O-benzyl-1,2,4-orthopivalate 98 as the monomer in cationic ring-opening polymerization in the chemical synthesis of cellulose (Scheme 16). By using triphenyl carbenium tetrafluoroborate as promoter, fully protected cellulose 99 could be obtained and after global deprotection the synthetic polymer showed excellent analytical similarity with a natural sample [44]. The importance of substituent effects was also studied. Both the 3-O-benzyl and the 1,2,4-orthopivaloyl groups are indispensable for the synthesis of (1-4)-β-pyranan. The substituent on C6 was found to influence the reaction as well, with less electron withdrawing groups (ethers) and electron donating groups (hydrogen) giving the best results due to electronic and not steric effects [45].
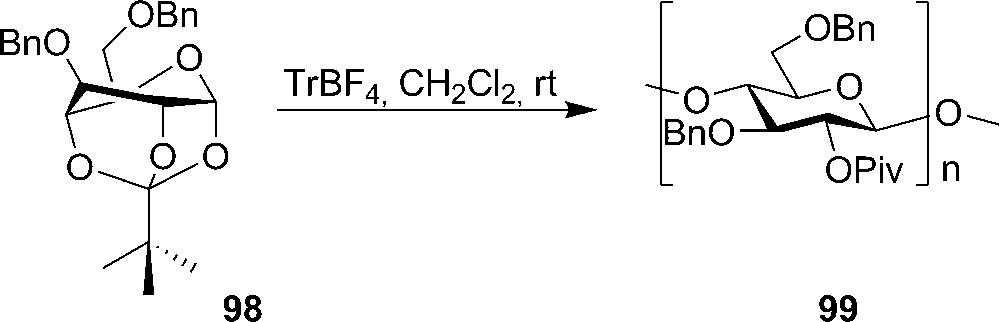
Cationic ring-opening polymerization of 3,6-Di-O-benzyl-α-D-glucose-1,2,4-orthopivalate in the chemical synthesis of cellulose.
Wong and coworker demonstrated the use of the 1,2,6-orthoester of mannose 100 as a potential glycosyl donor (Scheme 17). The orthoester was easily obtained from the unprotected 1,2-orthoester and could be further protected by benzylation of the 3-OH and 4-OH. In glycosylation with a simple primary alcohol in large excess, reasonable yields could be obtained, whereas primary alcohol sugar derivatives gave lower yields because the further reaction of 102 (as acceptor) was a side reaction [46].
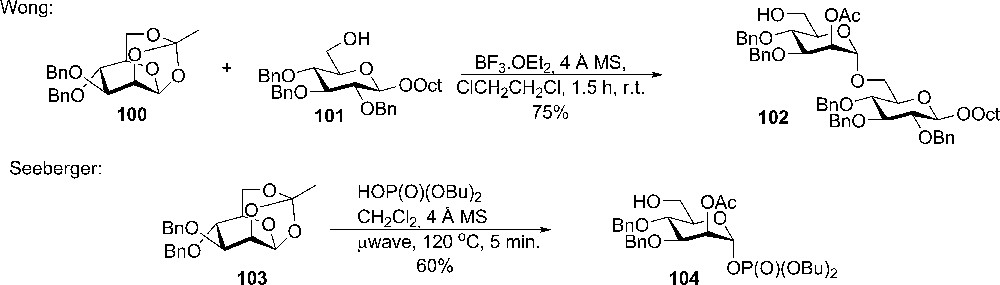
The use of 1,2,5-orthoester of mannose in glycosylation reaction by Wong et al. and in introduction of phosphate by Liu et al. [47].
Recently, pyranoid tricyclic orthoesters gained some interest again with the Seebergers group, who used the orthoesters as versatile starting points for the preparation of glycosyl phosphates, which were used in automated synthesis of lipomannan backbone α(1-6) hexamannoside (Scheme 17). It was noticed during the work that the tricyclic orthoester 103 was less reactive towards dibutyl-phosphate compared with the mannosyl 1,2-orthoester [47].
At the same time as Kochetkov's [42] pioneering work on orthoesters, several groups were interested in the analysis of the structure of orthoesters, which could now be analysed both in solution by NMR spectroscopy and in the solid state by X-ray crystallography. Coxon and Hall [47a] analyzed 1,2-alkylidene α-D-glucopyranosides by NMR and concluded that the pyranose ring essentially adopts a skewed boat conformation based on the dihedral angels obtained from the insertion of the measured coupling constants in the Karplus equation (Fig. 5) [48]. A further experimental point was the observation of a long range coupling (“W”) between H2 and H4.
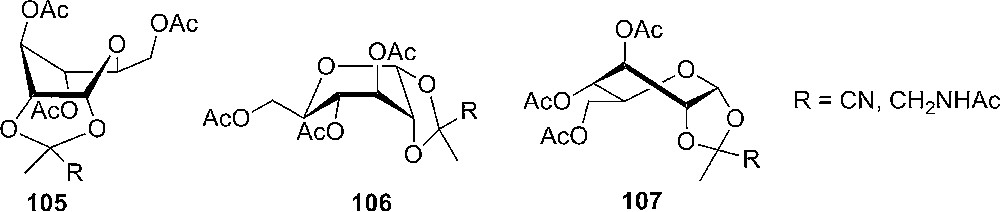
Conformations of 1,2-O-alkylidenede-α-D-glucopyranoses in solution suggested by Coxon and Hall [47a].
The following studies of the preparation and conformation of 1,2-O-aminoisopropylidene 108 and 1,2-O-(1-alkoxyethylidene) α-D-glucopyranose suggested that the conformation was rather a distorted, flattened chair more than a skewed boat and that the substitution changes (introduction of more bulky groups) in the orthoester did not change the conformation of the sugar (Fig. 6). This was furthermore justified by X-ray crystallography [49].
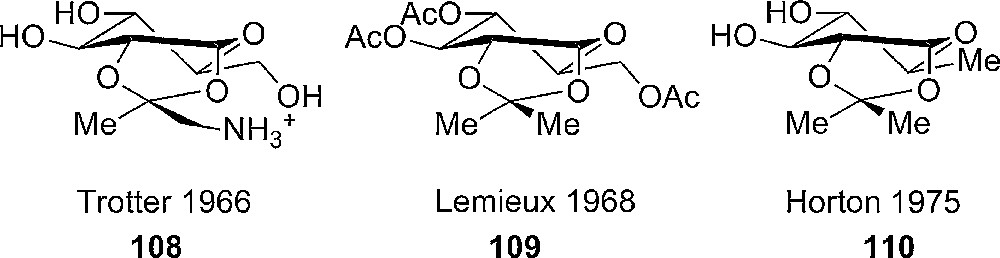
Compounds and conformations analyzed by Trotter et al. [49c], Lemieux et al. [49b] and Horton et al. [50] respectively – all suggested being in a distorted 4C1-chair.
With the use of paramagnetic shift reagents similar conclusions were drawn from the spectroscopic data isopropylidenated pyranose derivatives [50]. In the early studies on the conformation of pyranose rings having a fused 1,2-cis-cyclic acetal or orthoacetate ring (109, 110), a general trend is difficult to see, and there is controversy between the conclusions (Fig. 6) [51].
With the work of Foces-Foces and coworkers the conclusion became more clear and from X-ray crystallography a set of criteria, based on torsional angles to predict the conformation could be obtained (Fig. 7) [52].
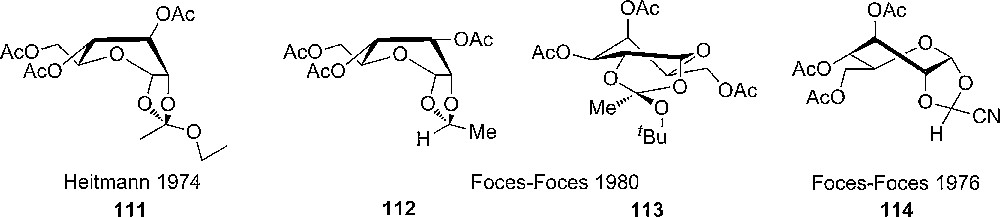
Suggested structures of different 1,2-cis tethered sugars.
From the analysis by NMR spectroscopy and computational methods of several sugar derivatives containing a 1,2-di-O- fused ring, some general trends could be observed. 1,2-O-alkylidene-α-D-allose and D-glucose derivatives have a 3S5 (skew boat) average conformation in solution, whereas the D-galacto derivative 113 has a distorted (4C1) chair conformation [53]. It was shown that the configuration at C7 (the dioxolane center carbon) strongly influences the conformation of the D-glucopyranoses; endo-methyl and phenyl substituents give rise to a 2S5 skew conformation, whereas the exo-methyl and phenyl have a flattened 4C1 major solution conformation. These results, obtained from NMR studies, are consistent with obtained crystal structures. Similarly, it was concluded from the analytical results that the pyranoid ring in the corresponding galacto-derivatives is unaffected of the C7-configuration [54]. The importance of the substitution pattern on C7 (in the dioxolane ring) was further demonstrated by studying the solid and solution phase structure of 1,2-O-(S)-(1-aminomethylethylidene)-α-D- and 1,2-O-(R)-(1-tert-butoxyethylidene)-α-D-glucopyranose, which showed to have a distorted 4C1 conformation and not the skew boat conformation [55]. Similar studies have been performed on D-xylo-, L-lyxo- and L-arabino-pyranosides [56].
With the conformational information in hand, the reactivity of acetylated exo- and endo-cyanoethylidene derivatives (115, 116, 119, 120) could be studied in glycosylation reactions (Scheme 18).4 The glycosylation of acetylated 1-(endo-cyano)ethylidine derivatives having D-gluco 119 (4C1), D-xylo 120 (OS2) and D-galacto (4C1) stereochemistry with either a primary or a secondary tritylated acceptor was faster than the glycosylation with the corresponding exo-derivatives 115 and 116. All the glycosylations with the acceptor 117 gave similar yields in the range of 54 to 65%. The difference in reactivity was explained with steric hindrance around the anomeric center, which is present in the exo-configurations and absent in the endo-configuration [58].
Thio-ortoesters are another interesting class of pyranoid rings fused with a dioxolane ring and they were introduced in carbohydrate chemistry by Magnussons in 1976 [59]. Even though their striking similarities with 1,2-alkylidenes and 1,2-orthoesters, the conformational information about these compounds is very limited. Kochetkov used 1,2-thio orthoesters 121 in glycosylations, but no improvements compared with unrestricted donors were observed and no relation between conformation and reactivity has been reported (Scheme 19) [60].
Kochetkov's synthesis of a disaccharide using a 1,2-O-thioorthoester as the donor.
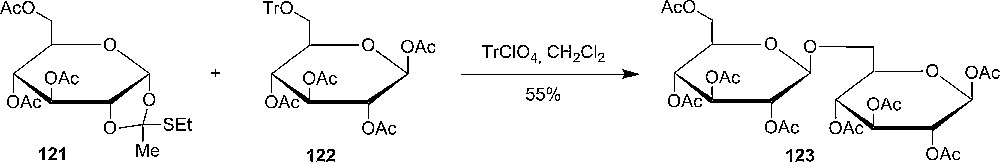
Van Boom and coworkers showed that the reactivity of 1,2-thioorthoesters 124 was higher in competition glycosylation reactions with a similar ethylthioglycoside 125, but with the limited number of reactions no clear conclusion could be drawn (Scheme 20) [61].
Van Boom's iodonium promoted glycosylation of 1,2-O-thioorthoesters in the presence of a thio-glucoside.
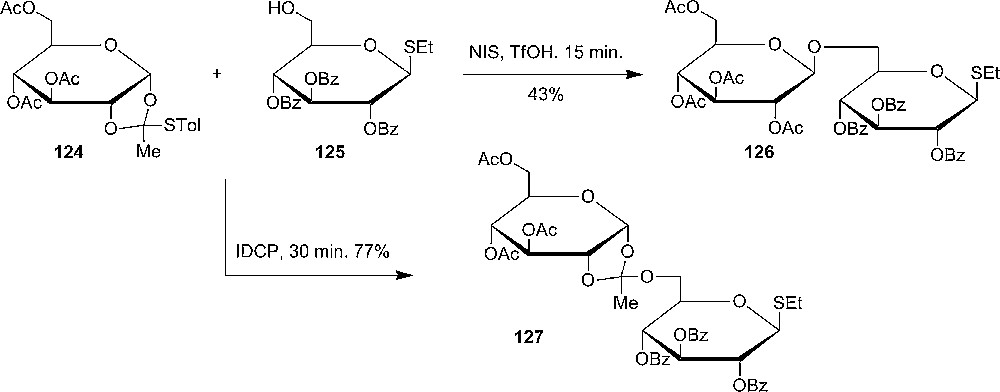
In 2006, Sivapriya and Chandrasekaran prepared and studied conformationally locked tricyclic 1,2,6-thio-orthoester, which was prepared from per-O-acetyl-6-O-tosyl-α-D-mannosyl bromide 128 using tetrathiomolybdate as the sulfur-transfer reagent. X-ray crystallography confirmed the skew boat like conformation of the product 130 (Scheme 21). Glycosylation reactions with the thio-orthoester could be performed with mild conditions using N-iodosuccinimide without using additional activators and resulted in reasonable yields of 132 at 0 °C (Scheme 21) [62].
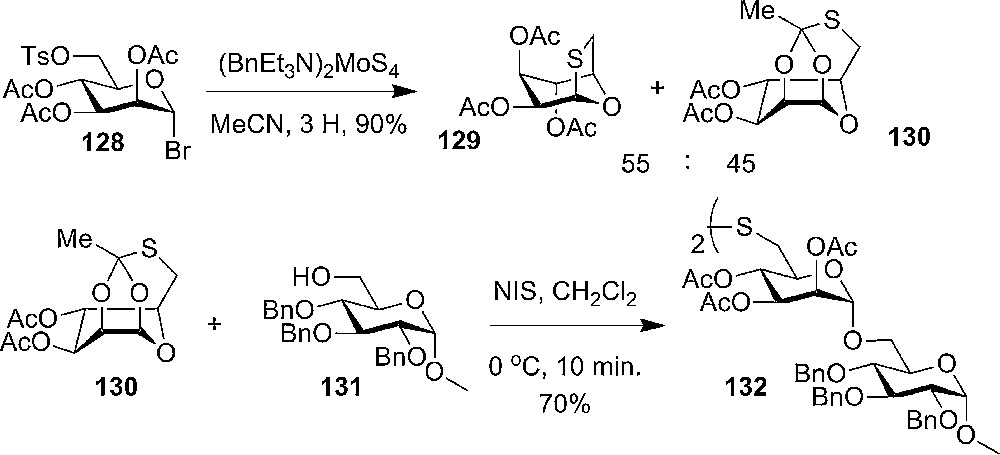
Chandrasekaran's synthesis of 1,2-O-6-S-thioorthoester of mannose and its use in glycosylation with a primary acceptor.
Recently, the interest in N-acetylhexosaminidases inhibitors has led to careful investigations of the conformation of N-acetylthiazolines [63] and N-acetyloxazolines,5 which are potential glycosyl donors [65]. It has been found that these systems are flexible and can adopt several conformations in solution – resembling a small library of inhibitor candidates. The influence on the reactivity at the anomeric center has so far not been investigated.
3.2 Alternative tethering methods
Presently, a few other methods of tethering pyranoid sugar rings into an axial rich conformation have been developed and used in carbohydrate chemistry.
Boric esters are one example. It has been known for decades that boronic acids react with carbohydrates and already in 1964 Ferrier et al. could isolate 2,4-phenylboronates of methyl α,β-D-xylopyranoside 137 (the all axial 1C4 conformation) as a crystalline compounds (Scheme 22). The use of boric esters as a 2,4-protective group was shortly investigated but low yielding [66].
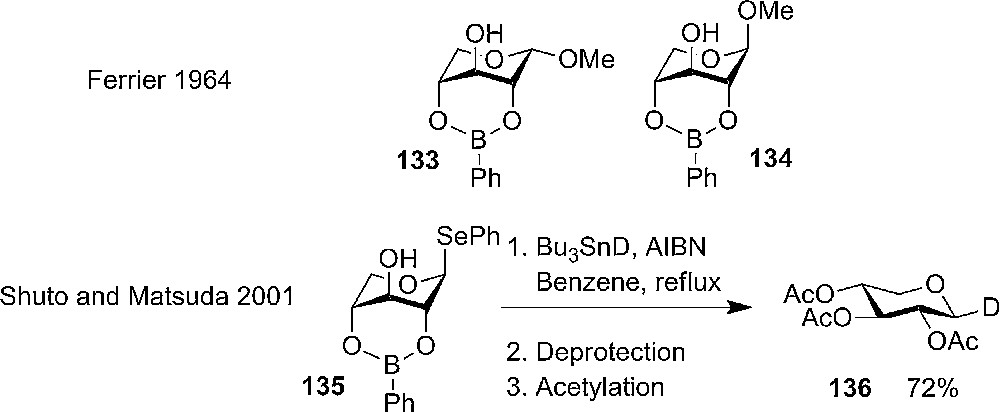
Ferrier's synthesis and analysis of methyl xylopyranoside 2,4-O-phenylboronate and the use of phenylboronate in stereo selective radical reaction by Abe et al. [72].
The early observation by Ferrier has not found much use in the first many years, but recently the boric ester was used as a 2,4-tethering protective group in order to investigate the influence of conformation in radical reactions on phenyl 1-seleno-β-D-xylopyranoside 2,4-cyclic-phenylboronate 135 (Scheme 22) [67].6
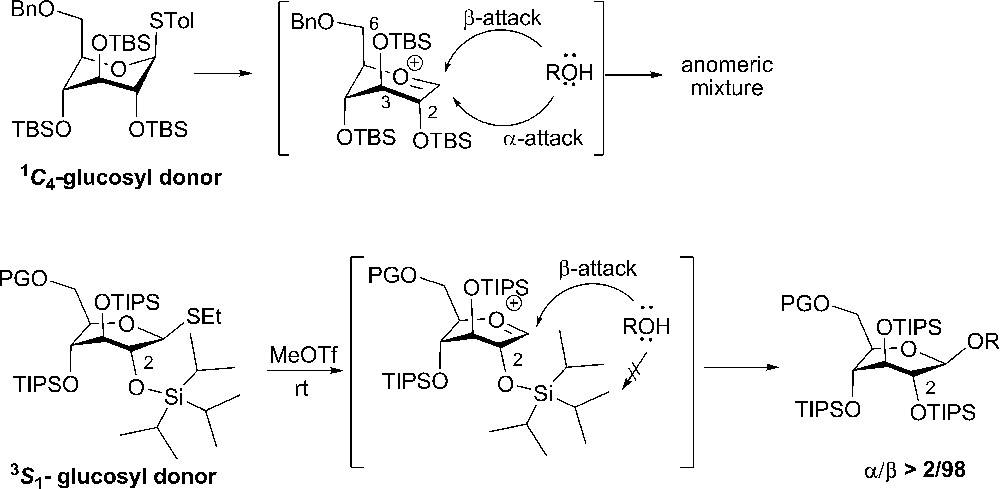
The use of dialkylsilylenes as tethering protective groups in axial rich glycosyl donors was demonstrated by Bols and coworkers, who prepared three different thioglycoside donor types: a 2,4-tethered 143 with a 1C4 conformation [68]; a 3,6-tethered 141 also in the all axial 1C4 conformation [69]; and finally a 4,6-tethered 137 locked in the all equatorial 4C1 conformations (Scheme 23) [70].
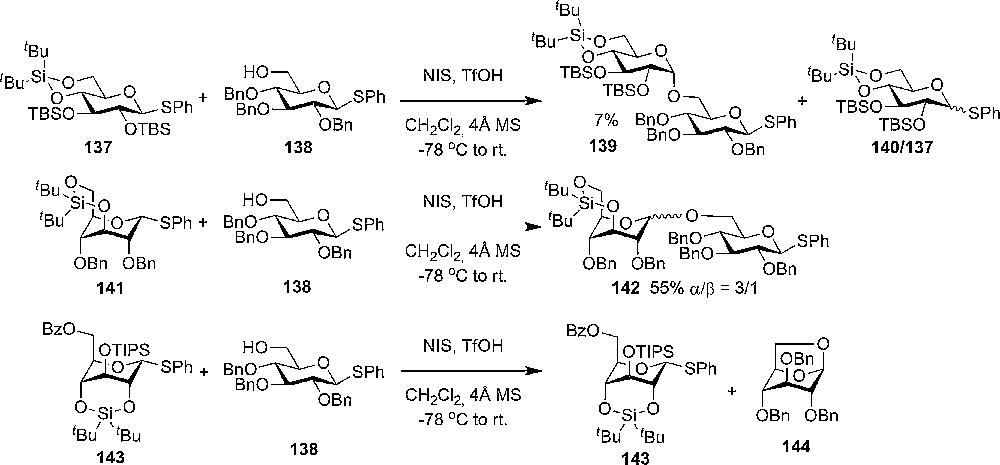
Glycosylations with bridged glucosyl donors restricted to the 4C1 and 1C4 conformations, respectively.
Upon comparison of the three donors in competition glycosylations with an armed glycosyl donor 138 with an acceptor side, it was found that the 3,6-tethered donor 141 was very reactive. It was readily activated at −78 °C, with NIS TfOH as the promoter system, and gave a reasonable yield of the disaccharide donor and selectivity towards the α product (55%, α/β = 3:1). The result clearly shows the enhanced reactivity by changing the conformation and that the disarming effect of tethering (torsional disarming) was overcome by the stereochemical effects. From the competition experiment the donor can be classified as “super armed”. Glycosylation between the all axial 2,4-tethered donor 143 and the armed donor 138 with a primary acceptor side results, surprisingly, not in the disaccharide donor, but in the isolation of unreacted 2,4-tethered donor and perbenzylated levoglucosane. In this case it seems like a conformational strain closer to the anomeric position gives rise to a decrease in reactivity, despite the stereoelectronical favorable all axial conformation. Further investigations with these types of donors have to be performed in order to clearly understand the different effects.
As expected from the stereoelectronic effect of having a locked all equatorial donor 137, almost no disaccharide was observed when using a steric hindered secondary donor/acceptor. A very small amount of disaccharide 139 could be isolated when having a primary acceptor 138 (7% β) (Scheme 23). These experiments show the importance of having axial substituents in order to increase reactivity of the donors. The effect of having silyl protective groups alone is minor, while torsional disarming by tethering 3,6-O together is no problem, the favorable stereo electronic effect from having an all axial conformation, when tethering 2,4-O together, can be overruled by steric strain (torsional disarming).
Yamada and coworkers have investigated alternatives to the silylene based tethering of glycosyl donors to give the desired all axial or axial rich conformation. To overcome some of the disadvantages of having bulky silyl protective groups i.e. restriction, silyl migration and difficulties in the synthesis, due to steric strain, 2,4-O-butenyl-2-ene was studied as a tethering protective group. The cyclic protective group was introduced from the 2,4-di-O-allyl precursor with a Grubbs first generation catalyst to give the 2,4-tethered intermediate, which was transferred to the desired product 145 in additional three steps. The bridged 1-O- acetyl-glucopyranose 145 (Fig. 8) was obtained in overall 8 steps from the known ethylthio-3,6-di-O-benzyl-β-D-glucopyranoside. By NMR studies the axial rich conformation was confirmed to be 3S1 skew boat. An important aspect of the new bridged glucopyranoside is the easy deprotection of the butenyl-2-ene by palladium mediated double bond migration and cleavage [71]. As far as we know, the properties regarding reactivity and selectivity of the new bicyclic glycopyranose have not yet been studied.
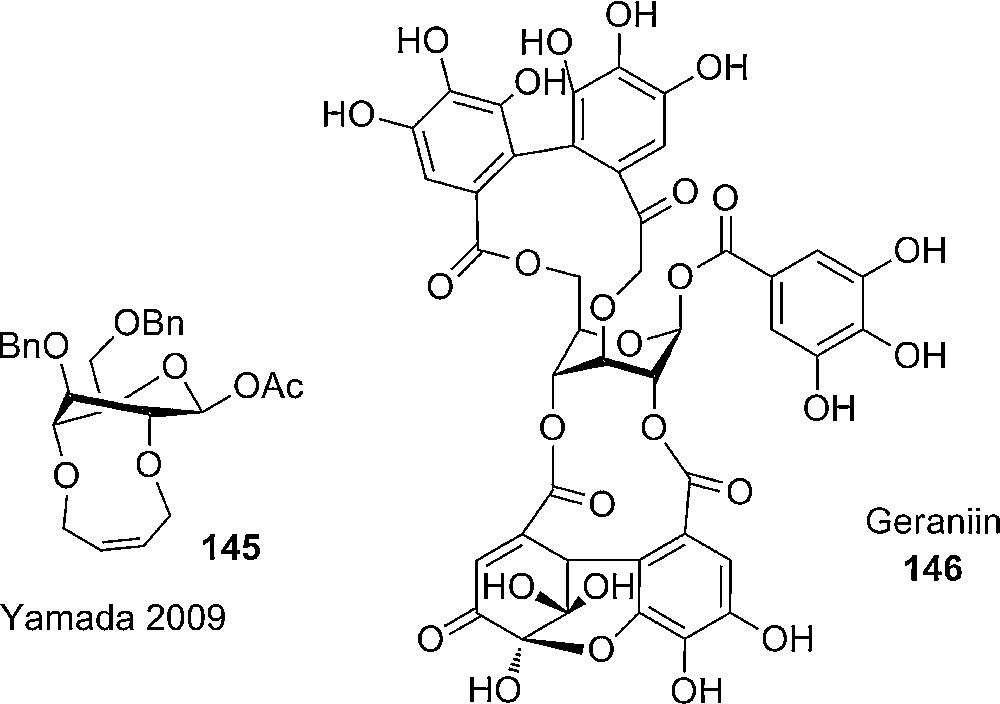
Yamada's 2,4-O-[(Z)-2-butenylene]-bridged glucopyranose and the natural axial rich glucopyranose derivative.
Interestingly 2,4- and 3,6-tethering of glucopyranosides are found in nature e.g. in corilagin and geraniin 146, which could be an inspiration for further developments in this field (Fig. 8) [72].
With the aid of carbocyclic annulations a new glucose derivative has been prepared by Mehta et al. (Fig. 9). The all axial racemic glucose derivative 147 was obtained by a de novo synthesis from tetralin. The trans-decaline structures represent an interesting model compound to investigate the locked axial rich conformations in the means of glycosidase inhibition and stereo electronic effects.
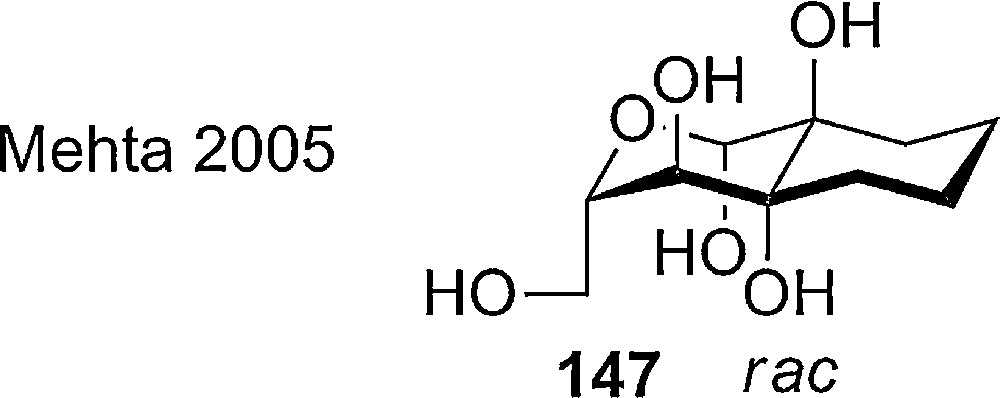
Annulation as a method to lock pyranoses in the axial rich conformation by Mehta.
Due to the lengthy racemic synthesis and the non removable bridge in this type of compound, the use in oligosaccharide synthesis is very limited.
4 Axial rich donors due to bulky substituent effect
4.1 Conformational studies of different alkyl and silyl - ether protected donors
Although the axial-rich conformations of pyranose are not so common and, in general, demand bulky substituents to force the ring flipping, there are several examples in literature where the conformation with more axial substituents is the most stable. Lemieux and Morgan reported pyridinium salt 148 that is forced to a 1C4 conformation wherein the pyridinium group has an equatorial orientation due to the attractions from the charge and the lone pair of the ring oxygen (Fig. 10) [73]. Compound 148 is highly reactive compared with its β-D-gluco diastereoisomer. The anomeric effect has been suggested to introduce a ring flip to an all axial conformation [74]. The α-D-idopyranose pentaacetate 149 that was characterized by 1H-NMR [75], and crystal structure [76] having the acetoxymethyl group equatorial and the four acetoxy groups axial (Fig. 10).
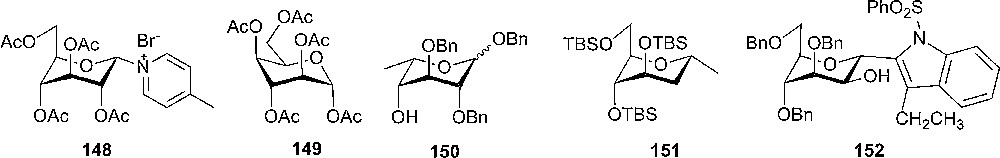
Donors in unusual conformations.
An interesting study of conformational changes of some anomeric O-benzylated D-gluco-, L-Ido- and hex-4-enopyranoside derivatives has been made by Kiss and Arnold [77]. They observed that a C-5 aliphatic substituent has a strong tendency to assume an equatorial position, pushing the benzyloxy groups into the axial position (Fig. 10, compound 150). Later, Tius and Busch-Petersen discovered that the 5-alken-1-ol having tri-TBS protecting groups will form only compound 151 during an oxymercuration reaction (Scheme 24). Compound 151 has the chair conformation with the silyloxy and silyloxymethyl groups axial and methyl equatorial, and is the minimum energy structure (Fig. 10) [78].
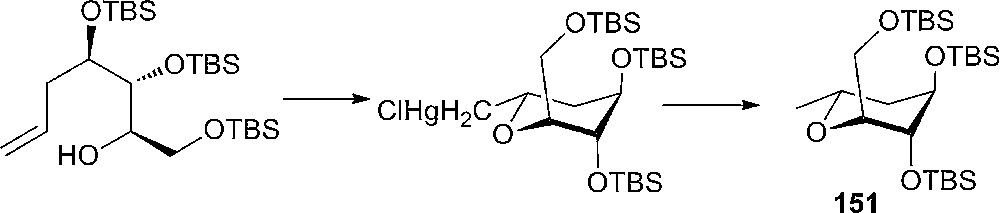
Oxymercuration of 5-alken-1-ols.
Manno-derivative 152 was used as intermediate in the total synthesis of a new class of glycoprotein identified in human RNase [79], An epoxide opening reaction of 1,2-anhydro-3,4,6-benzyloxy-mannose by a lithiated indole derivative gave 63% yield and 95:5 selectivity towards the α-stereoisomer 152. It was clearly observed that 152 adopts the 1C4-conformation with the tryptophan moiety equatorial orientated, presumably because of its bulkiness, as well as the absence of the anomeric effect [79].
The most common ones are with bulky O-silyl protecting groups [80]. The latter are widely used, owing to their ease of selective introduction and removal under mild conditions. An efficient method for preparation of fully O-silylated xylose and glucose derivatives has been reported by Shuto and coworkers using NaH and TIPSOTf or TBSOTf in THF. All products were conformationally restricted in an unusual 1C4-form and the yields were up to 85% [81].
4.2 Radicalic C-glycosylations
In 1999 Shuto and coworkers reported a stereoselective method for the synthesis of C-glycosides, compounds with unique biological activities, via a radical cyclization reaction of a vinylsilyl group as temporary connecting tether [82]. When the reaction was performed on a 2-O-dimethylvinylsilyl ether of 3,4,6-tri-O-benzyl-1-phenylselenyl-β-D-glucose 53, a mixture of products was obtained (Scheme 25) with different ratios, depending on the reaction conditions. It was suggested that the radical intermediate adopt a 4C1-conformation which facilitates an undesirable epimerization at the 5-position and/or an elimination of the benzyloxy group at the 4-position to give mainly the compounds 156 and/or 157. When the reaction was performed on substrates containing bulky silyl-protecting groups, the expected α-C-glucosides were obtained in good yields. The radical intermediate in this case was in a flipped 1C4-conformation due to steric repulsion between the adjacent bulky protecting groups at the 3- and 4-positions [83,84], therefore giving selectively the α-C-glucosides 160 and 161 after oxidation and protection steps (Scheme 25).
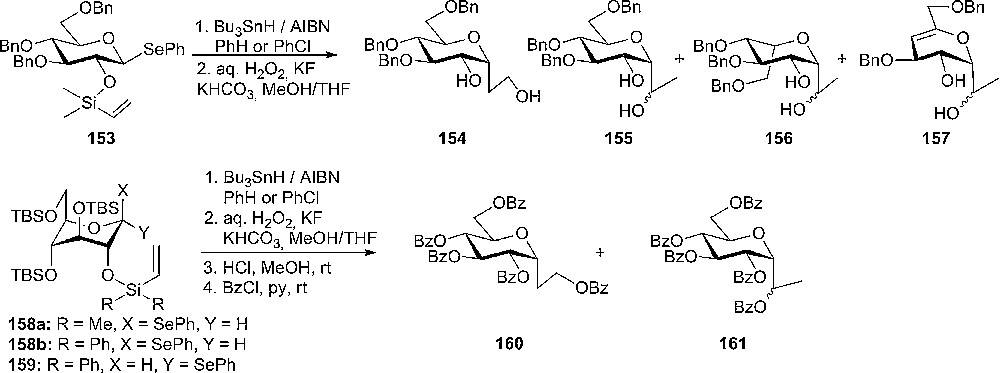
Radical C-glycosylation with vinylsilyl ether donors.
The change in conformation due to bulky protective groups has been reported also by Jackson and coworkers [84] when they studied the reaction of thiophenol with glucal epoxides. The main product was α-phenylthioglycoside having an unusual 1C4-conformation in the crystal structure. The bulky silyl ether groups are axial oriented and the conformation appear to be stabilized by a hydrogen bond between the C-2 hydroxy proton and O-1 [84].
Changing to an allylsilyl tether as protection group for the 2-position, the stereoselectivity of the radical cyclization changed, in favor of an equatorial-attack (Scheme 26). For tri-O-benzyl protected substrate 153b the stereoselectivity was up to α:β = 1:4.1, while the TBS-protected substrate 164 gave, under the same conditions, the desired α-C-glucoside 165 as the sole product in 75% yield [85]. Thus, the course of the radical cyclization was efficiently controlled by the change in conformation of the pyranose ring from 4C1 to 1C4-form. The method was successfully used for the synthesis of 3,7-anhydro-D-glycero-D-ido-octitol-1,5,6-trisphosphate 166, a potential IP3-receptor ligand which can be useful for proving the mechanism of IP3-mediated Ca2+ signaling pathway [86].
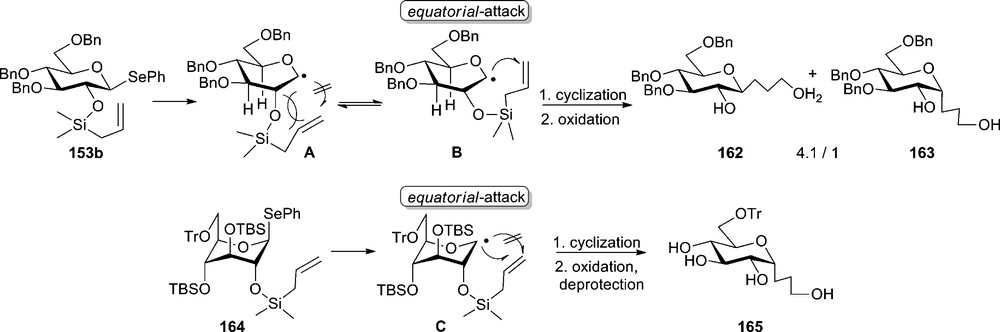
Radical C-glycosylation with allylsilyl tether.
The concept was further extended to other conformationally-restricted substrates and theoretical studies confirmed the influence of the anomeric effect over the stereoselectivity of these reactions. The pyranose ring conformation was restricted to its 4C1-form by using 2,3- or 3,4-O-cyclic-diketal protecting groups and to 1C4-form by using 2,4-O-cyclic-phenylboronate 135 and 2,3,4-tris-O-TIPS groups [67]. Radical deuteration and radical C-glycosylation reactions were investigated on the restricted xylose derivatives and compared with a conformationally unrestricted xylose substrate. Thus, the stereoselectivity was significantly increased by conformational restrictions. By NMR-studies, it was concluded that the radical intermediates formed in the reaction are in conformations similar to their substrates. When using the 4C1-form, a high selectivity for α-products (α/β = 97:3 – 85:15) was observed, whereas the 1C4-restricted gave selectively the β-products (α/β = 1:99 – 0:100). By looking at the stability of the radical intermediates in the transition state, it was established that the dominant factor controlling the stereoselectivity of these reactions is not steric hindrance but the kinetic anomeric effect that causes the highly stereoselective axial attack on the radical intermediate. This effect was previously observed by Giese and coworkers [87,88]. The radical deuteration reaction was further studied on 2,3- and 3,4-O-cyclic diketals of 1-phenylseleno-D-glucose derivatives that are restricted in 4C1-conformation, and on different O-silyl protected glucose derivatives locked in a 1C4-conformation, and compared with an unrestricted glucose derivative. Thus, the stereoselectivity was significantly improved by conformational restrictions, the 4C1-form giving good stereoselectivity for the α-product while the 4C1-form gave the β-product. It is noteworthy that 1C4-restricted compounds having a tetrahedral carbon substituent in the 5-position (–CH2OTIPS or –CH2OH), together with a 2-OTIPS protecting group, are too steric hindered both on α- and on β-sides, so the kinetic anomeric effect favoring the β-face attack is completely blocked [89]. The stereoselective radical cyclization reaction found applications in the synthesis of novel IP3 receptor ligands with α-C-glycosidic structure having different C-chain lengths at the anomeric position [90]. β-C-Glucosides can be efficiently synthesized also by starting from 6-O-allyldimetylsilyl ethers of the 1C4-restricted substrates via radical cyclization reactions [91]. As an application of the method, β-C-glucoside trisphosphates having a C1, C2 and C3 side chains have been synthesized using the conformational restriction strategy. Potential interactions of these compounds with the IP3 receptor binding site were subsequently explored [92].
It has been observed that for aryl C-olivosides the conformation is mainly dictated by the preference of the aryl-anomeric group to be equatorial; therefore the β-olivosides in a 4C1-conformation are thermodynamically more stable than their α-anomers [83]. The situation changes when the hydroxyls from 3- and 4-positions were protected with bulky t-butyldiphenylsilyl (TBDPS) groups that prefers to be antiperiplanar to each other in order to avoid a gauche interaction. In this case, the pyranoside ring is flipped to a 4C1-conformation. 3,4-OTBDPS-protected α-olivosides are very stable towards anomerization under Lewis acid conditions and in glycosylation reactions they form stereoselective α-aryl C-olivosides as sole products (Scheme 27) [83].
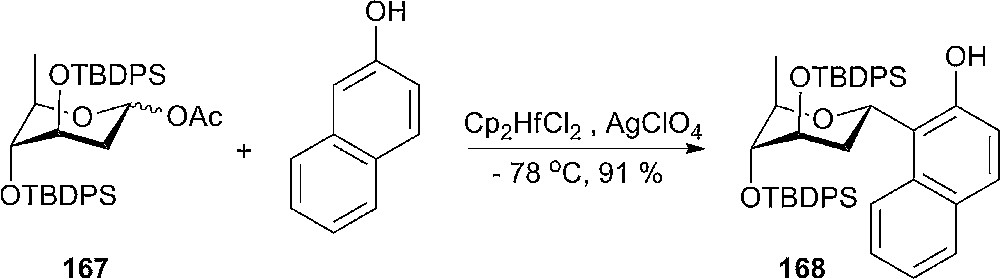
Glysosylation of α–olivosides.
The steric repulsion between adjacent bulky silyl protecting groups was a useful tool in the synthesis of the nucleoside antibiotic Herbicidin B [93]. The strategy involves an aldol-type C-glycosylation reaction promoted by samarium diiodide (SmI2) as a key step in the reaction (Scheme 28).
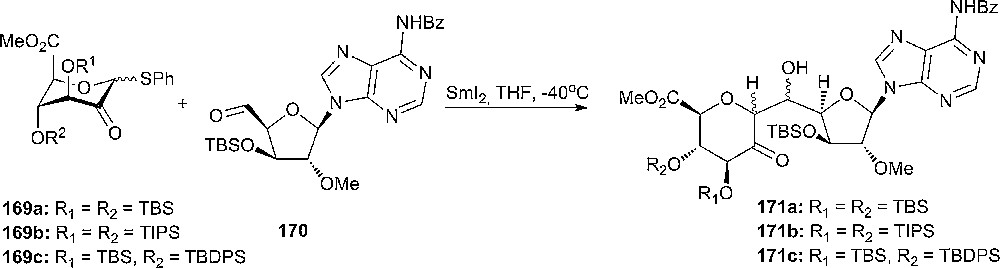
An aldol-type C-glycosylation reaction promoted by samarium diiodide.
Different silyl protected phenylthiouloses, 169a–c, have been prepared in order to ensure a high selectivity in the aldol-C-glycosylation reaction. It was observed that two vicinal TBS protecting groups are not bulky enough to flip the conformation in donor 169a, while the other donors adopted the 1C4-form with the substituents at the 3- and 4-positions being axial. SmI2-promoted aldol reaction with phenylthiouloses 169a-c and aldehyde 170 gave predominantly the desired 6’-α-aldol products 171a–c that were further used in the synthesis of herbicidin B [93].
Total synthesis of ravidomycin 174, an amino sugar congener of the gilvocarcin antibiotics, required a β-selective glycosylation step. Thus, the authors exploited the effect of large silyl protection group at the 3-position of the glycosyl donor 172. Consequently, the L-sugar 172 adopted the 1C4 (L) conformation [94] and under glycosylation conditions the oxonium intermediate underwent an equatorial C-glycoside formation to the desired β-anomer 173 in 83% yield (Scheme 29) [95]. Sterical hindrance undoubtedly plays an important role in forcing formation of the equatorial isomer.
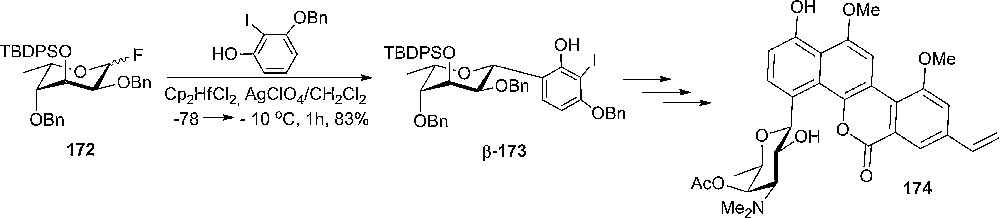
C-glycoside formation to the desired β-anomer 173.
As a continuation of the work with C-glycosylation reactions, Shuto and coworkers reported a method for controlling the α/β-stereoselectivity in the Lewis acid promoted C-glycosylations [96]. This is useful with smaller C-groups, such as allyl, where sterical hindrance does not dictate the equatorial product.
The stereoselectivity of the C-glycosylation can be influenced by controlling the kinetic anomeric effect based on the conformational restriction of the donors. Thus, the conformational restricted 4C1 donors gave increased α-selectivity, whereas the 1C4-restricted substrates completely reversed the stereoselectivity towards β-products. Anomeric allylations were performed on three types of donors: unrestricted 175, 4C1-restricted 176, 1C4-restricted 177 in order to compare their stereoselectivity (Scheme 30) [96].
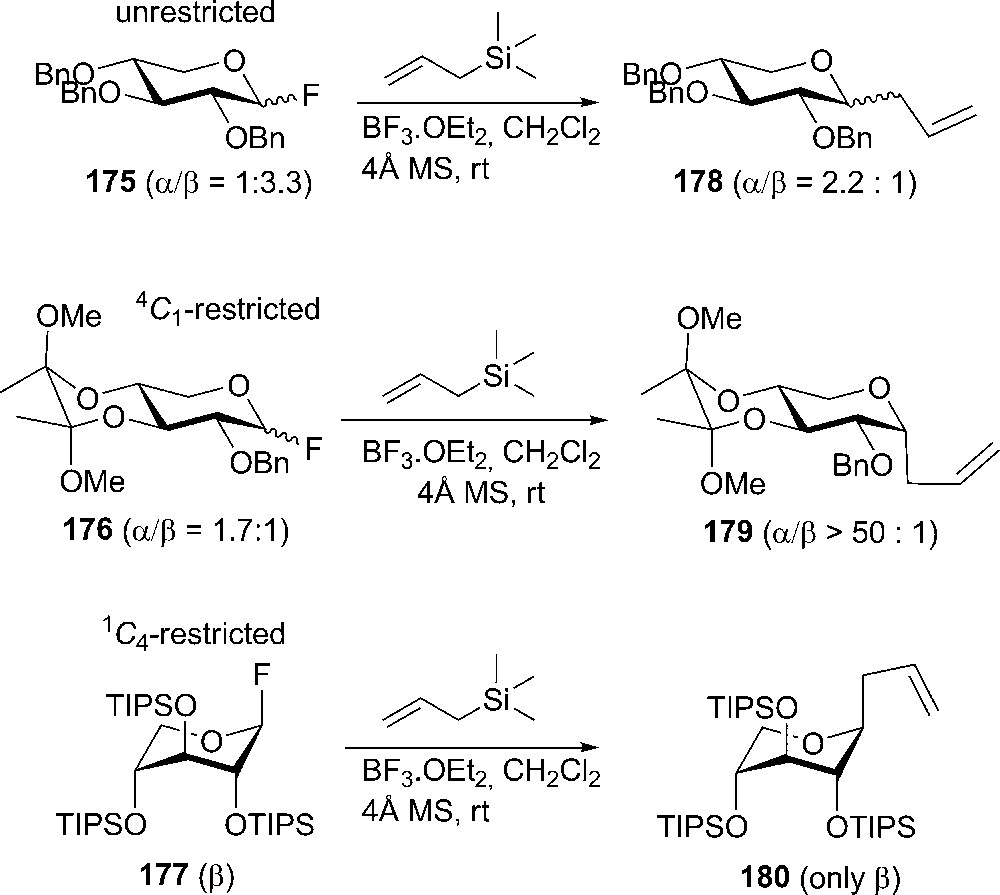
Lewis acid promoted C-glycosylation.
4.3 O-Glycosylations with donors in unusual conformation
Stereoselectivity of glycosylation reactions of D-glucal derivatives with PhSCl and PhSeCl has been studied. Depending on the substituents, the D-glucal derivatives exist preferably in 4H5 or 5H4 conformations (Fig. 11). The greatest stereoselectivity was observed with those glycals that exist preferably in the inverted 5H4 conformation since the diastereofacial selectivity is more pronounced than for the glycols in 4H5 conformation. In 5H4 form, the addition of PhSCl from the upper (β) face is more hindered (two axial substituents) than the addition from the bottom (α) face (Fig. 11) [97].
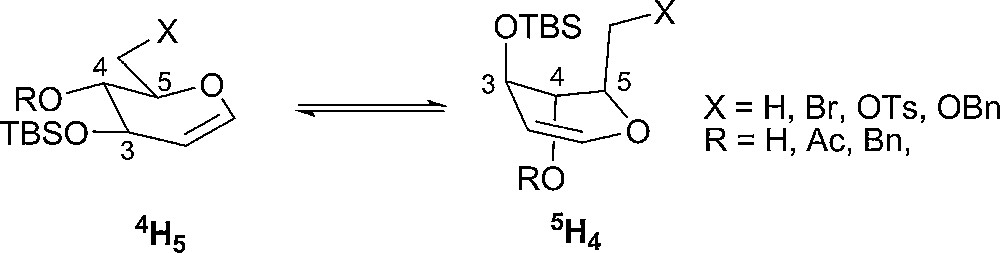
Conformations of D-glucal derivatives.
The authors concluded that the stereoselectivity of these reactions highly depends on the presence of an electronegative heteroatom substituent at position-6, as well as on the functionality at position-4, though the influence of the OTBS protecting group in the change of conformation is not discussed here. Some of the addition products were studied as glycosyl donors [98].
Stereoselective glycosylation of a family of 6-deoxy-3,4-silylated-1,2-glycals has been carried out under acid catalysis at room temperature (Scheme 31) [99]. The products could subsequently, by a endo-selective alkynol cycloisomerization reaction, be converted to disaccharides. D-Ribo-glycal 181 afforded the β-glycoside 186 very selective (β:α = 96:4) and the L-lyxo-glycal 182 formed complete setereoselective the α-glycoside 187. In both cases, the mechanism consists of an addition of the alcohol anti to the C3-substituent. On the other hand, the glycosylation of D-arabino- and L-xylo-glycals (183 and 184) proceeds with slower rates, hence Ph3P-HBr catalysis was used instead of CSA. A decrease in anomeric stereoselectivity was observed and it was suggested that thermodynamic equilibration at the anomeric center was responsible for the selectivity.
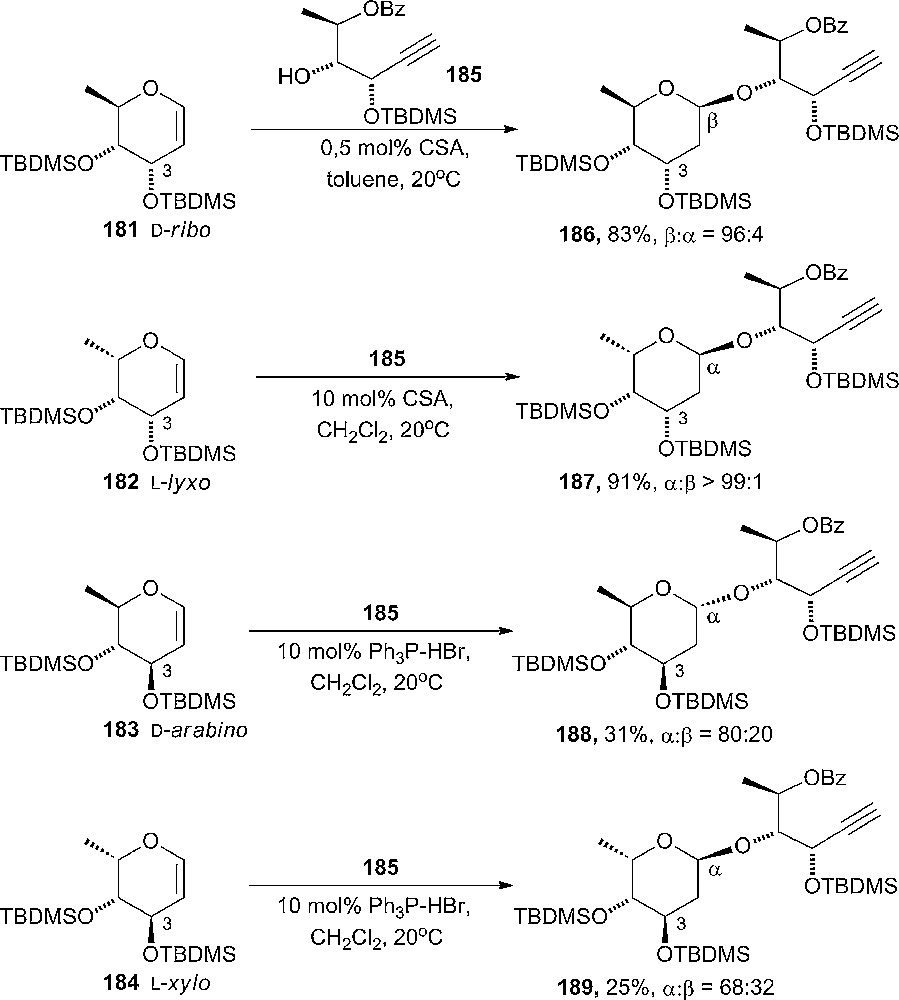
Stereoselective glycosylation of a family of 6-deoxy-3,4-silylated-1,2-glycals.
Donors in unusual conformations have been used in the synthesis of a tetragalactose moiety of the glycosyl phosphatidyl inositol anchor of the variant surface glycoprotein of Trypanasoma brucei [100]. Donor 192 was obtained as a mixture of two diastereomeric sulfoxides S:R in 1.8:1 ratio by oxidation of donor 191 and used as donor in highly selective glycosylation reactions (Scheme 32).
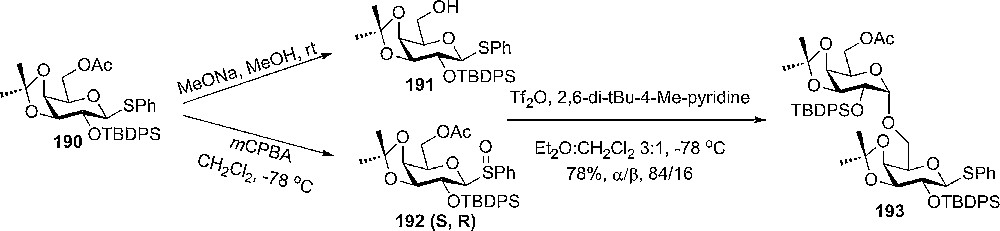
α-selective glycosylation with donors 191 and 192.
The high selectivity can be related to the conformation of compound 191 and also to the tendency of the sulfoxides method to give α-selectivity when the donor has a nonparticipating group in the 2-position [100]. The conformation of 190 has been studied by 1H-NMR data that showed that the combination of locking the pyranose ring with the isopropylidene group, together with introduction of a bulky OTBDPS group at the 2-position, results in a skew boat conformation.
Glycosylations with sulfoxides 194a or 194b having bulky silyl protecting groups gave a 1:1 mixture of α:β-glycosides 196. The donors were in a 3S1 skewed boat conformation (Scheme 33) as seen by 1H-NMR [101].
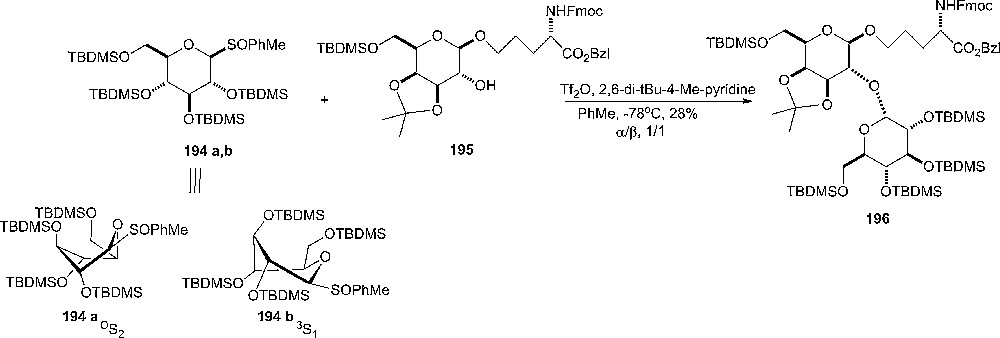
Glycosylation with donors in a 3S1 skewed boat conformations.
Impressive β-stereoselectivity in glycosylation reactions have been achieved using 2-deoxy-2-iodo-glucopyranosyl acetate donors and TMS-OTf or TBS-OTf as the promoter. The β-disaccharides were obtained in 62–92% yield and the selectivity varies from pure β-products to 9:1 β:α-disaccharides in the worst case (Scheme 34) [102].
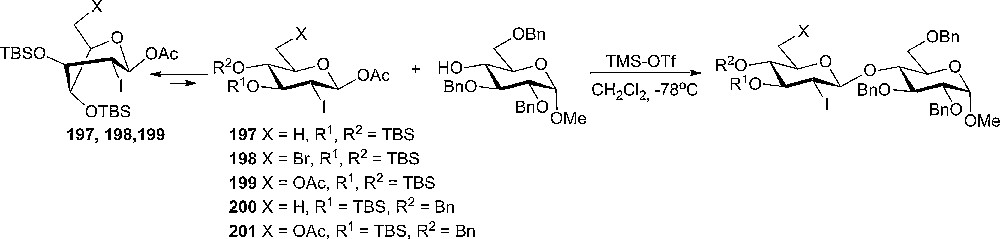
β-Selective glycosylations using 2-deoxy-2-iodo-glucopyranosyl acetate donors.
Donors 197–199 with silyl ether protecting groups in 3- and 4-positions were more reactive than donors 200 and 201, probably due to their twist-boat conformations, as determined by 1H-NMR analysis. Donors 200 and 201 adopt the usual 4C1 chair conformation. The influence of 2-iodo substituent as a stereodirecting group was not certified [102]. The method was extended to 2-deoxy-2-iodo-α-mannopyranosyl acetates and 2-deoxy-2-iodo-α-talopyranosyl acetate having OTBS protecting groups at 3- and/or 4-positions. The TMS-OTf or TBS-OTf promoted glycosylation reactions of these donors afforded exclusively the α-glycosides in 77–98% yield [103]. Roush and coworkers reported later the synthesis of 2-deoxy-β-glycosides using 2-deoxy-2-iodo-β-glucopyranosyl fluorides as mild and highly stereoselective glycosyl donors together with β-hydroxy ketones as acceptors [104]. Further experiments with 2-deoxy-β-glucopyranosyl fluorides showed a high β-stereoselectivity of the glycosylation reaction with a variety of acceptors. Therefore, the group applied the method in the total synthesis of formamicin, a cytotoxic plecomacrolide natural product [105].
The study continued by investigating the glycosylation reactions of 2-deoxy-2-iodo-glucopyranosyl trichloroacetimidates as donors [106]. In general, it was noted that the reactivity and the selectivity was depending on the conformational preference of the donor ground state. Conformationally constrained glycosyl imidates 202a and 202b afforded glycosylation with high β-selectivity (Scheme 35), the origin of which might be the conformationally inverted oxonium ion 203 or 204 intermediates. The high β-selectivity of the glycosylations with donors 202a,b was explained as a result of a favorable β-attack over the twist boat oxonium ion intermediates. The nucleophilic approach from the α-face is sterically unfavorable due to the pseudoaxial orientation of the iodine substituent. The SN2-like pathway was discounted since the products, stereochemistry was independent of the starting donor configuration [106].
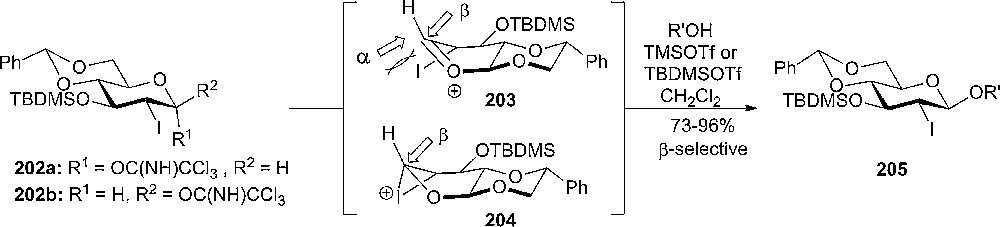
Conformationally constrained glycosyl imidates gave high β-selectivity in glycosylation.
Yamada and coworkers reported a flipping of the naturally stable ring conformation of L-rhamnose (1C4) and D-mannose (4C1) by the introduction of a TBS group at the 3-position and a TPS group at the 4-position of the sugar [107]. They also observed that having di-O-TBS groups is not sufficient for changing to axial rich conformation; therefore it was necessary to introduce the more bulky TPS group in order to prepare flipped rhamnoside 206 and D-mannose derivatives 207 and 208 (Fig. 12).
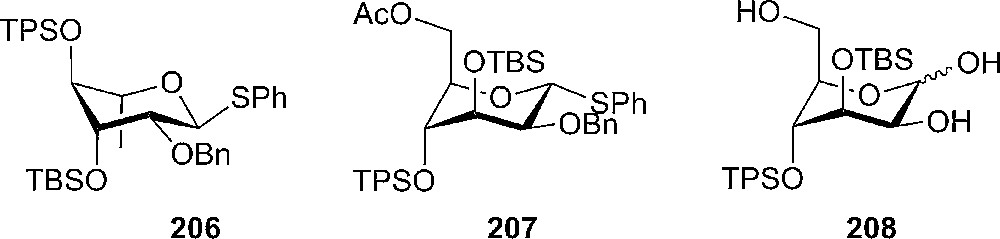
Flipped rhamnoside 206 and D-mannose derivatives 207 and 208.
Rhamnosylation reactions with L-rhamnosyl donor 206 in strained 4C1-conformation and NBS as the promoter, were performed with some β-selectivity [108]. The diastereoselectivity at the anomeric center can be influenced by selecting the solvent and the rhamnosyl acceptor. In ethereal solvents, the β-product formation was increased [109]. The corresponding trichloroacetimidate of rhamnosyl derivative 206 was tested in glycosylation reactions with cyclohexylmethanol as the acceptor [109]. The highest β-selectivity (27:73 210α:210β) was obtained in n-pentane and a bulky Lewis acid (9-BBNOTf) as catalyst at −95 °C. The rhamnosyl donor 209 has the 4C1 ring conformation that would diminish the general high α-selectivity of the rhamnosylation reactions (Scheme 36).
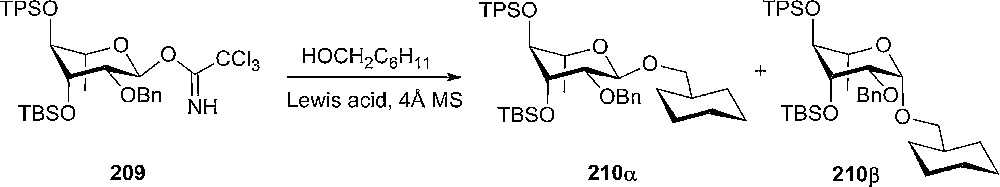
High α-selective rhamnosylation reaction.
During their study regarding the conformational changes in six-member rings, Yamada and coworkers observed the ring inversion into the axial-rich chair in myo-inositol derivatives when having two TBDMS, TIPS or TBDPS groups into the 3,4- or 4,5-positions [110]. The same ring flipping was noticed with D-glucose derivatives 213–216 and 218. Among all those compounds (Fig. 13), donor 214 could be isolated and exists in the full-axial chair conformation without a bridge structure. It is noteworthy to mention that the conformation of thioglucosides was more affected by the configuration of the anomeric center than by the variation of the silyl protecting groups [111].
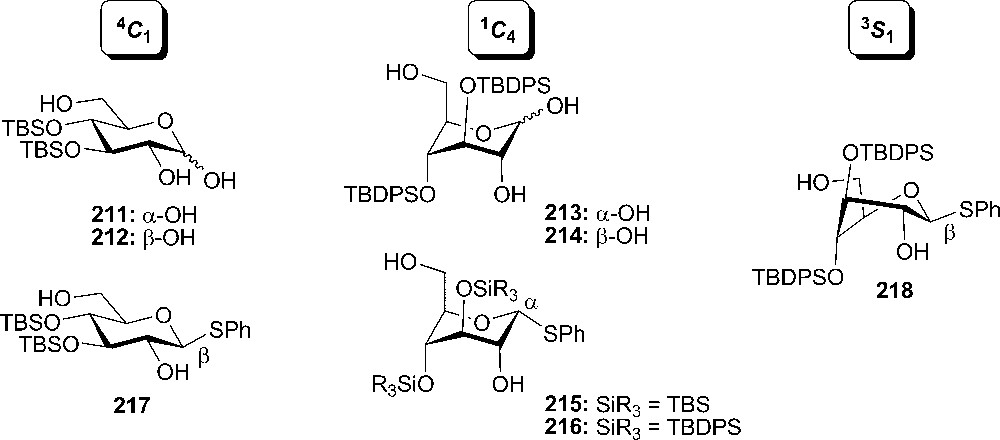
Different conformations of silylether protected donors.
The same group reported the first ring inversion of D-glucopyranoses into the axial-rich 1C4 form, induced by the silyloxy/silyloxy repulsions at the 2- and 3-positions [112]. When small TBS protecting groups were used, the repulsions were too small to flip the pyranose ring; therefore donor 219 has the major isomer α-219 in the equatorial-rich chair form, while the minor form β-219 has an axial-rich conformation (Fig. 14). A clear inversion is observed when more bulky silyl groups (TBDPS) are engaged and in this situation the anomeric stereochemistry does not have any effect [112].
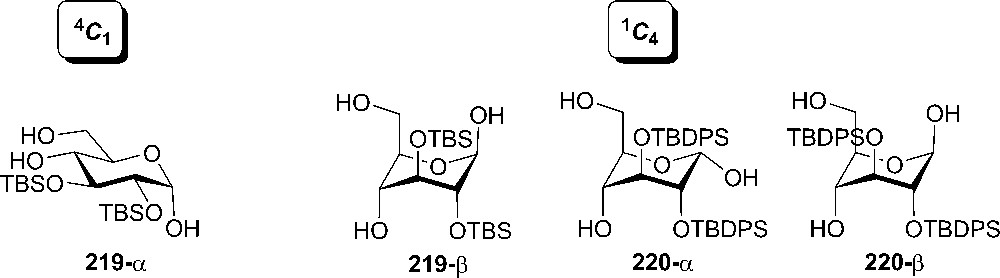
Bulky silyl groups flips donor conformation to axial-rich form.
Employing the donor conformational flip strategy, Kaneko and coworkers found a novel approach for the stereoselective synthesis of GM-237354, a potential antifungal agent [113]. They used 2-deoxy-2-iodo-glycopyranosyl acetate 222a as donor in order to obtain β-glycoside 224 as the sole product in 90% yield (Scheme 37). The 1H-NMR spectra suggests that the 222a donor exists in a twist-boat conformation in order to minimize the gauche interaction between the bulky C3- and C4-O-TBDPS substituents (Scheme 37). This unusual conformation of the donor is probably the reason for the high reactivity and the excellent selectivity of the reaction. The donor 222a has been prepared in 99% yield (α:β = 9:1) from the glycal 221 which exists in a trans-diaxial 5H4 conformation due to the repulsions between the bulky silyl substituents from the 3- and 4-positions [113].
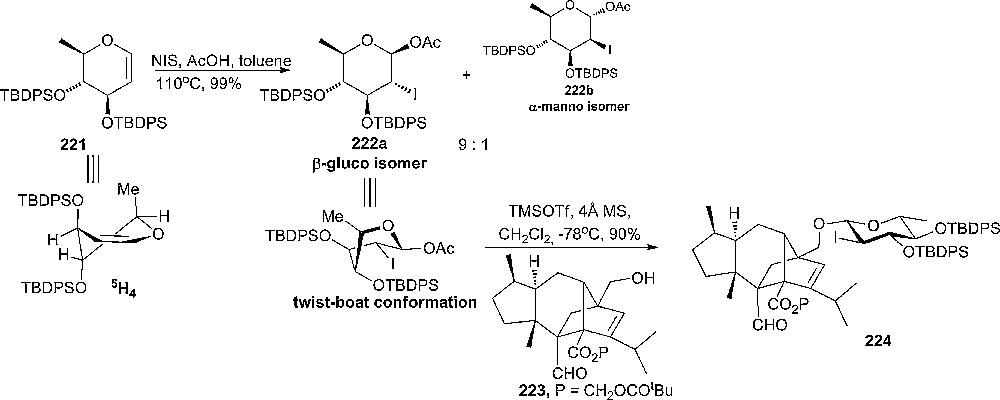
Selective synthesis of β-glycoside 224.
Conformational changes of D-glucopyranosides donors have been used as tools for achieving β-stereoselectivity up to α/β = 3:97 in O-glucosylation reactions [114]. When the pyranose ring is flipped to a 1C4-form and the 6-position has a fairly big substituent, the O-glucosylation reaction can give anomeric-mixtures of products due to sterical hindrance from both ring-faces (Fig. 15, top). Therefore, it was suggested that the β-selectivity in these examples occurs from an oxocarbenium ion intermediate restricted in a twist-boat conformation where the C-6 hinders the β-face less than in the conventional chair conformation (Fig. 15, bottom) [114]. It is worth mentioning that the anomeric selectivity was not solvent dependent, the reaction was efficient with various acceptors and independent on donors stereochemistry at the anomeric center. The reactivity of the donors, however, was influenced by solvents.
α-Attack inhibited by the substituent at 2-position.
Since the α-attack was inhibited by the bulky silyl substituent from the 2-position, the method was extended to 2-O-glycosylated donors that also keep a twist boat conformation (Fig. 15) [115]. The authors reported three disaccharide donors with 3S1-constrained conformation on the glucose component, that produced highly β-selective glycosylation, up to α/β = 2:98 using MeOTf as activator, 2,6-lutidine as additive and cholesterol or 1-adamantanol as acceptor.
When building up a strategy for the stereoselective synthesis of an oligosaccharide, it is important to have full control over the stereoselectivity of each glycosylation step, but also over the reactivity difference between donors.
The work summarized until now in this subchapter was mainly focused on the glycosylation stereoselectivity when donors in restricted conformations were employed. Only few papers described the reactivity changes when varying the reaction conditions.
A study of the influence of stereochemistry on carbohydrate reactivity was made recently by Bols and coworkers [41,116]. Studying the substituent effects on the pKa of piperidines, it was observed that axial hydroxyl groups were less electron-withdrawing than the corresponding equatorial ones [10,41]. The concept of positive charge stabilization by an axial polar substituent being better than an equatorial one, has been observed in glycosylation reactions, where benzylated tolylthiogalactoside reacts 6.4 times faster than the corresponding thioglycoside with MeOH under NIS activation [70]. Glycosyl donors having TBS protecting groups on 2-,3- and 4-O were prepared and their reactivity compared with the corresponding benzylated analogues. It was observed that the silylated donors are more reactive in glycosylations than their benzylated analogues. By forcing the donor to a more axial-rich conformation, it becomes significantly more reactive, “super armed”, since the axial substituents are less electron withdrawing. Competition experiment with equal amounts of armed 225 and super armed 226 donors and the same acceptor 227 gave 75% yield of 228 (α:β = 1:4), but none of the coupling products between 225 and 228 was isolated (Scheme 38).
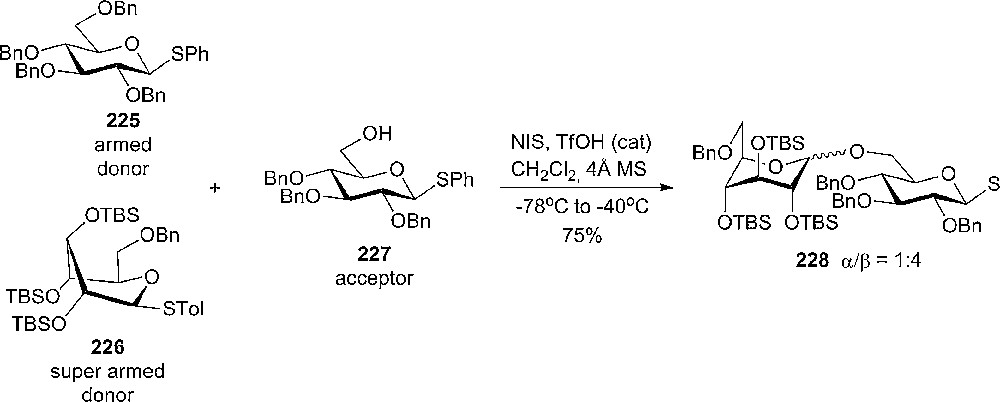
Competition experiment between armed and super armed donors.
To demonstrate that the reactivity increase is not a simple silicon effect [117], the authors prepared a silyl-protected glycosyl donor restricted to 4C1 conformation which proved to be significantly less reactive than the axial-rich silyl protected donors [116]. Therefore, the high reactivity of these silylated donors is intrinsically linked to their conformation.
The authors expanded the “armed – disarmed” principle of Fraser-Reid [118] with the conformationally armed (or so called “super armed”) glycosyl donors. The work was continued by quantifying the relative reactivity of these super armed glycosyl donors comparing them with the disarmed or armed donors. A quantitative method to measure the rate constant for glycosylation reactions was developed. Kinetic studies have been made on a series of donors and a clear difference in reactivity was observed between the disarmed, armed and super armed donors [69]. The results of the kinetic studies underlined once again that a more axial rich conformation gives more reactive donors. It is possible to disarm a silicon rich donor by locking it into 4C1 conformation, but also to arm a donor by tethering into an axial rich 1C4 conformation.
By knowing the activity of different donors, it has been possible to finetune the reaction conditions in order to perform a well-defined oligosaccharide synthesis. An example of a “one pot – one addition” trisaccharide synthesis is shown in Scheme 39.
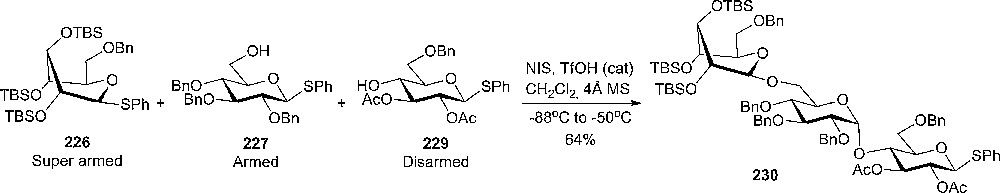
One-pot, one-addition synthesis of trisaccharide 230.
This reaction is carried as a true one-pot, one-addition reaction, with the reactants being present simultaneously and not just added sequentially. The trisaccharide 230 was obtained in good yield by a sequential temperature dependent activation of the donors [116].
5 Conclusion
It is clear from the above that glycosyl donors and derivatives in “unusual” conformations behave differently in two ways: firstly, stereoselectivity in glycosylation reactions is, not very surprisingly, profoundly altered by a change in donor conformation. After all, a change in donor conformation is also likely to lead to a change transition state conformation, and the sterical and electronic effects of both ground and transition state are altered. While the selectivity observed has frequently been unpredicted, Woerpel's work suggesting a relationship between transition state conformation and stereoselectivity could mean that conformational control may led to the much desired stereocontrol in glycosylation. Secondly, it has been shown the reactivity of carbohydrate derivatives for reaction at the anomeric center depends much on conformation. Among pyranose chair conformers, it is clear that the conformer with more axial electronegative groups (normally hydroxyl or other oxygroups) is the more reactive conformer towards reactions generating positive charge at the anomeric position, i.e., most glycosylation and glycoside reactions. The reactivity changes are caused by the different electronic interactions between charge and oxygroups existing in different carbohydrate conformers.
1 Hans Paulsen's statement: “Although we have now learned to synthesize oligosaccharides, it should be emphasized that each oligosaccharide synthesis remains an independent problem, whose resolution requires considerable systematic research and a good deal of know-how. There are no universal reaction condition for oligosaccharide synthesis”.
2 For the use in glycosylation, see e.g.: [35].
3 Orthoesters as glycosyl donor has been review recently: [43].
4 First example of glycosylation with 1,2-di-O-cyanoethyllidene donor: [57].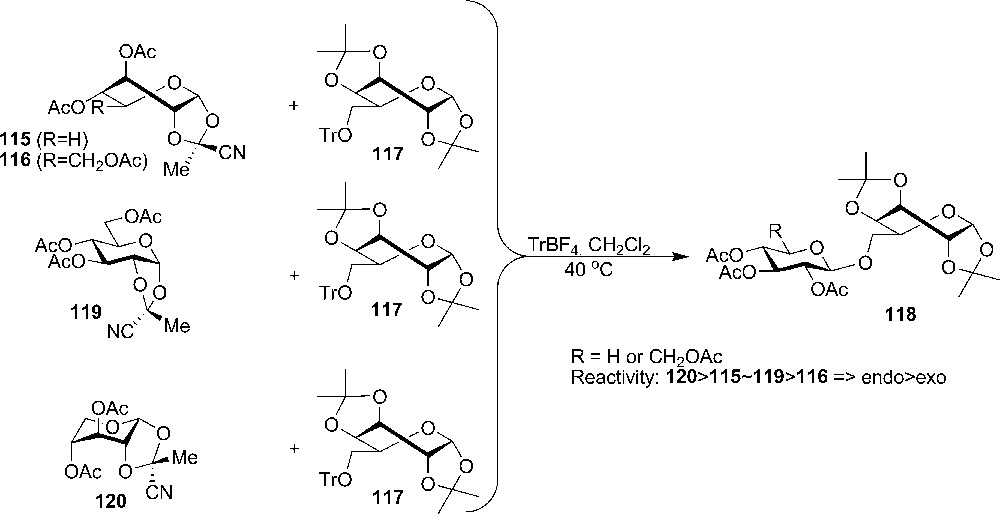
5 Oxazolines have been analyses earlier by: [64].
6 See the section “Radicalic C-glycosylation” for discussion about this topic.