

1 Introduction
It is often said [1] that the founding moment of “radiation element” chemistry was in February 1896, when Antoine Henri Becquerel, a physicist and member of the French Academy of Sciences, reported his results concerning the mysterious radiation coming from uranium salts. From that date (or even going back to the discovery of X-rays by Wilhelm Conrad Röntgen the previous year) through to the decade immediately after the Second World War, the chemistry of radiating elements, still called radioelements, expanded considerably, concomitant with the amazing progress in quantum and atomic physics [2]. Certainly the work of Pierre and Marie Curie from 1898 onwards, whose public aura largely exceeded the purely scientific world, ensured that the chemistry of the radioelements, commonly called radiochemistry, took off and became a separate discipline in itself. We are therefore looking at a late xixth century discovery, which led to fierce international rivalry, remarkably concentrated in time and with considerable strategic interests at stake, particularly on the threshold of World War II. At the heart of these interests, linked to military strategy until very recently, but also, and more particularly, linked to present and future energy production, actinide element chemistry has played a special role.
Today, actinide chemistry is involved in the growing demand in nuclear energy and related processes, environmental as well as toxicological issues. From a more fundamental point of view, actinide chemistry involves the heaviest elements available at a weighable amount for the chemist; their electronic configuration involves the highest orbitals also available, namely the 5f orbitals (and to a lesser extend the 6d orbitals), that are at the origin of interesting properties, as will be pointed out in this article.
For this special issue on actinide chemistry, we focused on oxidation +III coordination chemistry and, more specifically, on some aspects of trans-plutonium +III coordination chemistry. This contribution is an invitation to discuss some of the perspectives in transuranium molecular chemistry, given the advances of Michel Ephritikhine and his group in the science of uranium(III) chemistry. The subject is also particularly relevant to nuclear reprocessing issues since the comparison between trans-plutonium elements and corresponding lanthanide elements is still a subject of major debate [3]. From a structural point of view, very little is known about trans-plutonium molecular chemistry. As in September 2009, less than 10 structures involving americium were reported in the Cambridge Structural Database, although many structures are probably still unpublished. This is the reason why we propose here to discuss from a structural point of view, some new americium and related lanthanide complexes.
After a brief review of the occurrence of typical coordination polyhedra of the solid state hydrates and aquo species of actinide(III) and lanthanide(III), the article focuses on two chemical systems that are both characterized by their hard acid character, namely the aminocarboxylic example with nitrilotriacetic acid and the hydroxycarboxylic example with citric acid (Scheme 1). Both ligands bear three carboxylic groups plus an additional chemical function that often plays a very important structural role: the nitrogen atom for the first one, the hydroxy group for the second one. Furthermore, both ligands are relevant to the more general problematic of lanthanide – minor actinide selectivity. They also give the opportunity to directly compare both parent families in isostructural complexes.
2 A borderline electronic structure between transition metals and lanthanides
In their general electronic configuration [Rn]5fn6dm7s2, the actinide elements (the actinides, put simply) begin with thorium (Z = 90) and end with lawrencium (Z = 103). They appear in the periodic table when the 4f and 5d orbitals are totally filled. They are thus considered to make up an internal transition family of elements, corresponding to the filling of the 5f atomic shell (for atoms in their fundamental states). Since their discovery, these elements have been considered as an analogous family for the 14 lanthanides (whose electronic configuration is [Xe]4fn5dm6s2), and their physicochemical properties have often been compared. However, regarding their chemical behavior, though the lanthanide family is relatively homogeneous, this cannot be said for the actinides. It is now agreed that the first elements of the series up to americium possess properties identical to those of uranium, and completely different from those of the first lanthanides. However, the second part of the series’ elements, beyond americium, has properties similar to the lanthanides (from neodymium to lutetium).
The comparison of the physicochemical behavior of the actinides with that of the lanthanides can be justified by the analogy of their electronic structure, as each of the series is made up of elements corresponding to the filling of a given (n)f atomic shell. These orbitals (4f for the lanthanides, 5f for the actinides) present a weak radial extension and are thus protected from interactions with the ligands by the saturated shells −5s2 and 5p6 in the case of the lanthanides, and 6s2 and 6p6 for the actinides. Consequently, these (n)f electrons only weakly interact with the electrons of neighboring ligands, and their electronic properties are very little influenced by the environment. However, there is an important difference between the two series: the 4f orbitals are more localized than the 5f orbitals, and can thus be considered as core orbitals. This weak extension of the 4f electrons perfectly explains why the lanthanides’ degree of oxidation is mainly +3. On the contrary, the 5f electrons, less tightly localized, participate a little more in the chemical binding, and the interactions between the 5f electrons and the ligands are stronger than is the case for the lanthanides. This relative distance is significant for the elements in the first half of the actinides, from Pa to Am. Indeed, for these, the energies of the 5f, 6d, 7s and 7p orbitals are close enough to hybridize, making a spatial overlap possible between them and the ligands’ valence (or hybrid) orbitals. It is therefore often difficult to distinguish the ionic or covalent character of this type of binding. This results in a large number of degrees of oxidation and therefore redox properties which are complex and varied, making these elements more akin to transitions metals than lanthanides.
Moreover, within the series, the increase of the atomic number Z means a coulombian attraction which stabilizes the 5f orbitals. This is responsible for the relatively steady decrease in the size of the actinide ions throughout the series, with the increase of Z (actinide contraction). As the 5f electrons are more and more localized, they interact less with the environment, and the chemical behavior of the elements beyond americium becomes similar to the lanthanide ones.
We must emphasize an important point concerning the relativistic effects due to the high value of the atomic number of these actinides (Z > 90). The relativistic corrections on the electronic structure of these elements (other than the spin-orbit coupling) come from direct effects (due to the “mass variation” of the electrons, accompanied by an energetic stabilization) and indirect effects. These must be taken into account for the 5f valence electrons, even if their average speed is not close to that of light. The most important consequence for a high-charge nucleus is the contraction of the distribution of charge (the “orbital radius” decreases when the “mass” of the electron increases with its speed), all the more significant for the shells with a weak angular momentum. The core orbitals, which present a high amplitude near the nucleus, contract, and their energy stabilizes. However, the d or f orbitals, which are more diffused, do not come so close to the nucleus, and are therefore little influenced by this direct effect. However, as the core electrons screen the nuclear charge, the resulting potential, acting on the f and d orbitals is less attractive. These effects, called indirect, are opposed to the direct effects, and it is often difficult to foretell the consequences of the relativistic corrections taken into account. Nevertheless, for the shells with a high angular momentum, like the 5f and 6d shells, the contribution of the indirect effects, due to the redistribution of charge, is the most important. The radial extension of these orbitals will therefore be slightly larger, given these relativistic corrections.
These general considerations are at the basis of differences that might be observed from the 4f to 5f series or from the beginning to the end of a given series.
3 The aquo +III series: an ideal situation for the lanthanide actinide comparison
An interesting point of discussion can be found in the comparison between the lanthanide(III) and heavy actinide(III) coordination spheres in the crystal structure of the hydrates or in aqueous solution. This is an essential and very active area of investigation since most of the processes involving heavy elements are taking place in water, including future aspects in fuel reprocessing, environmental issues, toxicology, etc. Structural aspects of lanthanide(III) hydration have been a matter of debate for long, and subject to numerous communications as detailed bellow. Generally speaking, two models of coordination spheres are most likely [4]: a square prism or antiprism (SP, SA, CN = 8) where all the distances are ideally equivalent, and a tricapped trigonal prism (TTP, CN = 6+3) in which two sets of distances are present. Structural differences have been found as a function of the anion and have been attributed to hydrogen bond networks formed with anions in the second coordination shell. In particular, in the presence of triflates [5–7] and ethyl sulfates [8,9], the coordination polyhedron around lanthanide(III) ions has a C3h symmetry whereas it has an approximate D3h symmetry with bromate anions [10]. Although it is usually believed that a decrease of one unit follows the lanthanide contraction mentioned in the previous paragraph, all the lanthanide(III) hydrates have been described in a tricapped trigonal prism. Recently Persson et al. have proposed that a statistic deficiency of one water molecule in a capping position of the polyhedron may occur as the atomic number increases [11]. In aqueous solution, extended X-ray absorption fine structure (EXAFS), X-ray and neutron scattering techniques (LAXS, HEXS…), spectrophotometry or time-resolved fluorescence studies have been applied on lanthanide aqua ions [12–14]. Metal-oxygen distances and the numbers of coordinated water molecules have been determined for all the lanthanide series except promethium. Various coordination numbers have been found, but in the most recent studies light lanthanide ions from La(III) to Nd(III) are considered as being predominantly nine-coordinated whereas heavier lanthanide ions from Tb(III) to Lu(III) are eight-coordinated. In aqueous solution, EXAFS data are often less conclusive regarding the value of coordination numbers since a difference of one unit over nine in coordination number accounts for an EXAFS amplitude difference of only 11%, of the same order if not bellow uncertainties usually admitted for EXAFS. Nonetheless, D’Angelo et al. have also recently proposed an extensive survey of careful EXAFS data analysis for the lanthanide(III) aquo ions [15] in which the decrease of coordination numbers is discussed in combination with the solid state picture proposed by Persson. As a complement, hydrated lanthanide(III) ions have also been investigated using a number of theoretical approaches and the evolution of the coordination numbers along the series has been recently described through molecular dynamics simulations by Duvail et al. [16].
For the actinide(III) hydrate series, only three crystal structures are available to date: plutonium [17], americium and curium [18,19]. In all of these structures, the actinide cation sits in a TTP polyhedron with An-Oprism distances ranking from 2.48 Å (Pu) to 2.45 Å (Cm) and An-Ocapping distances around 2.57 Å. Interestingly, the capping distances do not follow the actinide contraction as the prismatic ones do. Although the range of investigation here (from Pu to Cm, ΔZ is only equal to 2) is very small, the stability of the capping distances justifies the assumption of a statistical decrease of coordination number as for the lanthanide case. In solution, from optical spectra and EXAFS data, americium and curium were found as mostly nine-coordinated at room temperature [18,20].
Using a combination of EXAFS and Wide Angle X-ray Scattering, Skanthakumar et al. have recently discussed the similarities between the solid state structure of the [Cm(H2O)9]3+ unit and arrangement in solution. They conclude that even in solution, the curium atom prefers a nona-aqua coordination with TTP average configuration. Other examples of transuranium(III) aqua ion have been published in the past, although with less details in the geometrical arrangement and are scattered from 7 to 10 [21]. In a previous article, we have provided a first estimate of the Cf-Oaq average distance with no a priori restriction on the coordination polyhedron [22]. Recently, we have recorded the data with an extended k range [23]. It has been fitted according to two typical distinct models: square antiprism (SA, CN = 8) and tricapped trigonal prism (TTP, CN = 6 + 3) with no clear distinction between the two results: eight oxygen atoms at 2.42 Å for SA, nine oxygen atoms at 2.41 Å for the average of TTP, keeping in mind that a difference of one unit in coordination number over nine (i.e. 11%) is not significant from the EXAFS point of view, especially in the absence of an experimental value. These values are in very good agreement with our former result: 8.5 O at 2.42 Å. In comparison, the Am-Oaq distance has been reported to be 10.3 O at 2.48 Å in HCl solution [24]. In order to obtain additional information on the aqueous coordination sphere arrangement and complete the discussion on the curium3+ aquo ion, the combination of EXAFS and molecular dynamics calculations has been recently carried out [25]. It suggests, although with some care, that the SA model is prefered over the TTP one, in agreement with what might be expected from actinide contraction.
4 Coordination with nitrilotriacetic acid
Polyaminocarboxylates form a promising family of chelating agents of the metal ions. They are particularly considered to trap the actinides cations in solution, offering perspectives of applications in many areas such as synergistic agents in selective extraction processes, decorporation in human detoxicological processes, etc.
Structural investigations of metallic complexes with NTA have been the subject of numerous studies in the solid state. Tetravalent and trivalent metal ions tend to form oligomeric and polymeric structures, but crystal structures of monomeric 1:2 complexes have also been described. The M4+ ions (M = Zr, Sn, Hf, and Hg) [26] generally form eight-coordinate M(NTA)2 complexes where the metal ion is coordinated by six carboxylic oxygen atoms and two nitrogen atoms. With the trivalent M3+ ions (M = Y [27], Bi [28], In [29], lanthanides [30]), the complexes can be eight (6 + 2) or nine (6 + 3) coordinate with an additional water molecule. In several papers, Wang et al. have described the various coordinating modes of NTA with lanthanides: the Ln3+ ion often retain water in their first coordination sphere as in K3[Nd(NTA)2(H2O)]·6H2O [31] or [Pr(NTA)(H2O)2]·H2O [32]. In all the cases, the three carboxylic oxygen and nitrogen atoms interact with the metal center. In the latter case, NTA is a tetradentate ligand coordinating one of the Pr3+ ion with the nitrogen atom and three oxygen atoms of the carboxylic groups and the second Pr3+ with one oxygen atom from the same carboxylic group which acts as a bridging ligand. In [Nd(NTA)(H2O)]n, each of the three carboxylic group of the same NTA molecule are bridging between the two Nd3+ cations [33]. This demonstrates the versatility of NTA ligand that can behave either as a tetradentate chelate or as a single to triple bridging ligand. The size of the lanthanide cations also play a key role as the coordination number tends to decrease from 9 for Pr to 8 for Yb.
In this article, we have synthesized the Nd(III) (compound I) and Yb(III) (compound II) NTA complexes of composition [Co(NH3)6][M(NTA)2(H2O)]·8H2O. In these compounds, and also in the isostructural gadolinium equivalent [34], the lanthanide cations are nine-coordinated. Both NTA3− anions act as tetradentate ligands, coordinated by the central N atom and three O atoms of different carboxylate groups as shown in Fig. 1a and b for I and II. The ninth coordination site is occupied by water molecule. All three compounds are isostructural as shown by the crystal data reported in Table 1. Within the actinide(III) series, only the americium adduct has been isolated with an identical structure to the lanthanide (compound III) (Fig. 2a and b). The coordination polyhedra of Am and Ln atoms are distorted tricapped trigonal prisms with capping positions occupied by the N atoms of the nitrilotriacetate anions and one oxygen atom (O1) of the water molecule (Fig. 2b). Bond distances for the Nd, Gd, Yb and Am complexes are given in Table 2. The coordinated and crystallization water molecules in compound III act as proton donors in a number of H-bonds (Table 3), the acceptors being the terminal O atoms of the NTA3− anions and O atoms of the other water molecules. The patterns of H-bonding in Nd and Yb compounds seem to be the same, although the major part of the H atoms was not located.

(a) a view of [Co(NH3)6][Nd(NTA)2(H2O)]·8H2O (I). Displacement ellipsoids are drawn at the 30% probability level, H atoms omitted for clarity. Dashed lines indicate H-bonding interactions; (b) a view of [Co(NH3)6][Yb(NTA)2(H2O)]·8H2O (II). Displacement ellipsoids are drawn at the 30% probability level, H atoms omitted for clarity. Dashed lines indicate H-bonding interactions.
Crystal data for for compounds I, II and III, [Co(NH3)6][M(NTA)2(H2O)]·8H2O (M = Nd, Yb, Am).
| Am | Nd | Yb | |
| Compound | III | I | II |
| Chemical formula | C12H48AmCoN8O21 | C12H48CoN8NdO21 | C12H48CoN8O21Yb |
| Formula weight | 942.51 | 843.75 | 872.55 |
| System | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n | P21/n |
| a, Ǻ | 11.9057 (6) | 11.7924 (3) | 11.6357 (2) |
| b, Ǻ | 20.7618 (11) | 20.5883 (5) | 20.4762 (4) |
| с, Ǻ | 13.9663 (6) | 14.1204 (3) | 14.1041 (3) |
| β,° | 113.4174 (13) | 113.4593 (12) | 113.5528 (11) |
| V, Ǻ3 | 3167.9 (3) | 3144.86 (13) | 3080.43 (10) |
| Z | 4 | 4 | 4 |
| R1 | 0.0214 | 0.0336 | 0.0396 |
| 0.0478 | 0.0855 | 0.0976 | |
| Temperature, K | 100 | 298 | 298 |

(a) view of [Co(NH3)6][Am(NTA)2(H2O)]·8H2O (III). Displacement ellipsoids are drawn at the 50% probability level, H atoms omitted for clarity. Dashed lines indicate H-bonding interactions; (b) coordination polyhedron of the Am atom showing distorted tricapped trignal prism.
M-O bond lengths in compounds I, II and III: [Co(NH3)6][M(NTA)2(H2O)]·8H2O (M = Nd, Yb, Am). Atom numbering corresponds to Figs. 1 and 2.
| Bond (Å) | Nd | Gd [34] | Yb | Am | Δ(Am-Nd) |
| Compound | I | II | III | ||
| M-O(1) | 2.492 (4) | 2.449 (6) | 2.366 (4) | 2.4780 (15) | −0.014 |
| M-O(11) | 2.445 (3) | 2.403 (5) | 2.324 (3) | 2.4512 (13) | +0.006 |
| M-O(13) | 2.456 (3) | 2.399 (5) | 2.333 (3) | 2.4497 (14) | −0.006 |
| M-O(15) | 2.459 (3) | 2.412 (5) | 2.346 (3) | 2.4749 (13) | +0.016 |
| M-O(21) | 2.452 (3) | 2.399 (5) | 2.331 (3) | 2.4553 (14) | +0.003 |
| M-O(23) | 2.436 (3) | 2.342 (6) | 2.323 (3) | 2.4235 (13) | −0.012 |
| M-O(25) | 2.455 (3) | 2.411 (5) | 2.345 (3) | 2.4619 (13) | +0.007 |
| M-N(1) | 2.716 (4) | 2.682 (6) | 2.638 (4) | 2.7053 (16) | −0.011 |
| M-N(2) | 2.715 (4) | 2.674 (6) | 2.630 (4) | 2.6997 (16) | −0.015 |
H-bonding geometry in III [Co(NH3)6][Am(NTA)2(H2O)]·8H2O (Å,°).
| D-H…A | D-H | H…A | D…A | D-H…A | Symmetry code for A |
| O(1)-H(1A)…O(2) | 0.830 (17) | 1.943 (17) | 2.764 (2) | 170 (3) | 1 1/2-x, -1/2+y, 1 1/2-z |
| O(1)-H(1B)…O(4) | 0.834 (17) | 1.955 (17) | 2.787 (2) | 176 (3) | |
| O(2)-H(2A)…O(12) | 0.857 (17) | 1.942 (17) | 2.789 (2) | 170 (3) | |
| O(2)-H(2B)…O(16) | 0.838 (17) | 1.922 (17) | 2.759 (2) | 176 (3) | 1-x, 1-y, 1-z |
| O(3)-H(3D)…O(14) | 0.858 (17) | 1.826 (18) | 2.671 (2) | 168 (3) | |
| O(3)-H(3E)…O(7) | 0.853 (16) | 1.897 (17) | 2.748 (2) | 175 (3) | |
| O(4)-H(4D)…O(26) | 0.858 (16) | 1.877 (17) | 2.731 (2) | 174 (3) | -1/2+x, 1/2-y, -1/2+z |
| O(4)-H(4E)…O(22) | 0.832 (17) | 1.989 (16) | 2.818 (2) | 175 (3) | 1 1/2-x, -1/2+y, 1 1/2-z |
| O(5)-H(5D)…O(9) | 0.872 (18) | 1.93 (2) | 2.786 (3) | 167 (4) | |
| O(5)-H(5E)…O(12) | 0.883 (18) | 1.91 (2) | 2.762 (3) | 162 (4) | |
| O(6)-H(6D)…O(9) | 0.864 (18) | 2.08 (2) | 2.918 (3) | 162 (4) | 1-x, 1-y, 2-z |
| O(6)-H(6E)…O(12) | 0.865 (18) | 2.03 (2) | 2.841 (3) | 157 (4) | |
| O(7)-H(7D)…O(8) | 0.899 (18) | 1.926 (19) | 2.797 (3) | 163 (3) | |
| O(7)-H(7E)…O(22) | 0.891 (18) | 1.949 (18) | 2.826 (2) | 168 (3) | 1-x, 1-y, 1-z |
| O(8)-H(8D)…O(9) | 0.868 (18) | 2.22 (3) | 3.002 (3) | 149 (4) | |
| O(8)-H(8E)…O(16) | 0.871 (18) | 2.31 (3) | 2.856 (3) | 121 (3) | 1-x, 1-y, 1-z |
| O(9)-H(9A)…O(22) | 0.872 (17) | 1.884 (19) | 2.747 (2) | 170 (3) | -1+x, y, z |
| O(9)-H(9B)…O(3) | 0.899 (17) | 1.815 (19) | 2.706 (3) | 170 (4) |
The isostructurality of the Am and lanthanide adducts gives a unique opportunity to compare the influence of 5f-electrons on the bonding of the central cation with O and N atoms. In Fig. 3, the average M-N and M-ONTA distances versus ionic radii of the central atoms (ionic radii for coordination number 8 were taken from [35]) are presented. In a first approximation, the Am-N and Ln-N distances may be considered equivalent, as already observed in some other An and Ln complexes with N-bearing ligands [36,37]. However a closer inspection of Fig. 3 and Table 2 shows that the average contraction of the M-O bond distances from Nd to Am is −0.0012 Å, whereas it is of −0.013 Å for the M-N bond. Although these differences are all equally small in the absolute scale, it appears that the nitrogen bonding is more affected by the americium cation than the carboxylate one. This is an interesting observation related to ligand selectivity considering the structural similarities that are often observed between americium and neodymium complexes.

Average M-Onta (circles) and M-N (squares) distances in [Co(NH3)6][M(NTA)2(H2O)]·8H2O (M = Nd, Gd, Yb, Am) compounds.
In the past, the structures of An(IV)/NTA2 complexes have been investigated through spectrophotometry, EXAFS and quantum chemistry calculations [38]. We have considered eight-coordinate structures and nine-coordinate structures obtained by adding a water molecule in the inner coordination sphere. Average calculated An–O distances were equal to 2.40–2.41 Å for Th(IV) and comprised between 2.30 and 2.34 Å for U(IV), Np(IV) and Pu(IV). An-N distances were 0.3 to 0.4 Å longer than An–O distances. For all the cations, a structural change of geometry does not imply significant variations in the actinide-oxygen distance; this is not the case for the weaker actinide-nitrogen bonds, which are far more sensitive to the geometry and coordination number. Interestingly, the americium coordination sphere in III, [Co(NH3)6][Am(NTA)2(H2O)]·8H2O might be compared to that of the nine-coordinate structure calculated for the An(IV)/NTA2 series and shown in Fig. 4. For this geometry, the calculated An-O distances rank from 2.40 Å for Th(IV) to 2.34 Å for Pu(IV). The An-N distances are however calculated with much less accuracy given the weak M-N interaction. In III, the average Am-O distance is equal to 2.456 Å. The difference between the average Pu(IV)-O distance (2.34 Å) and the average Am(III)-O one (2.46 Å) is a proper trend considering the decrease of oxidation state from +IV to +III as well as the actinide ionic size contraction between plutonium and americium (however expected to negligible from Z to Z + 1).
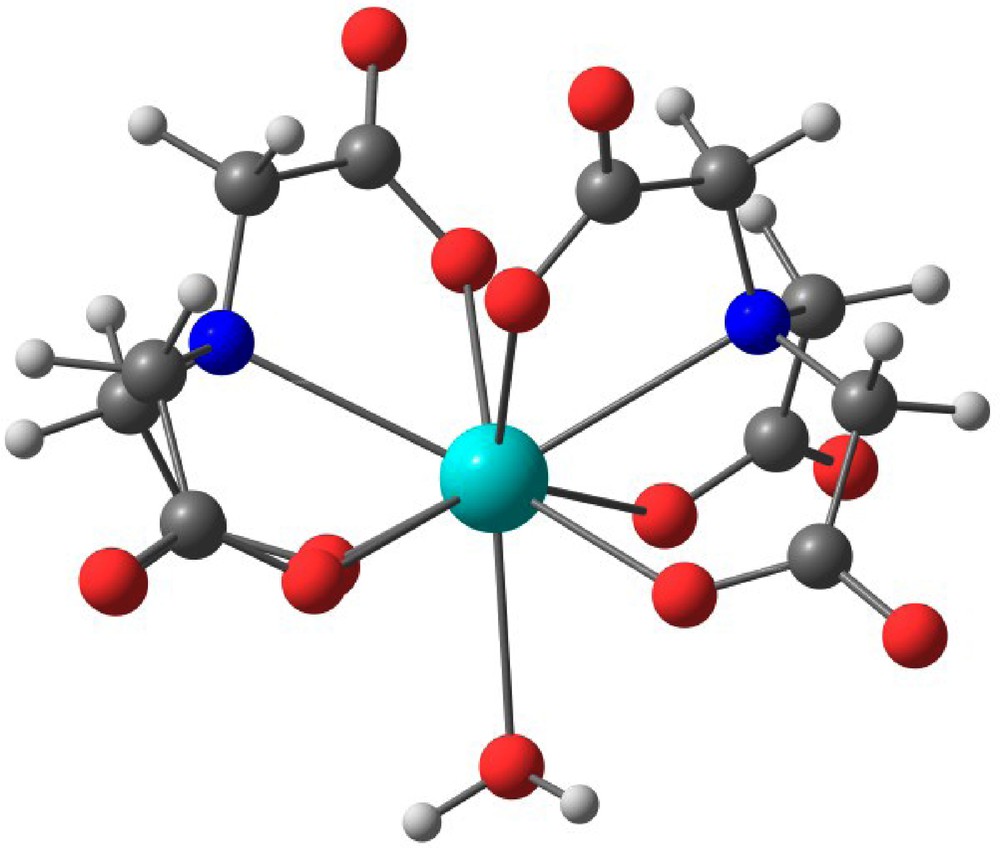
Structures of [An(IV)(NTA)2H2O] complexes optimized at the DFT level of theory with Amsterdam Density Functional (ADF) program package. Details in [38].
5 Coordination with citric acid
Citric acid has three carboxylic functions in common with nitrilotriacetic acid. In addition, citric acid possesses one hydroxy group that plays a major role in coordination modes. Citric acid is a very important natural component. It is present in plants, produced by bacteria, and plays a significant role in the physiological cycle of tricarboxylic acids in human body. Furthermore, citrate anions are widely present in various kinds of radioactive wastes. It is therefore necessary to understand the interactions which could take place in liquid and solid media involving citrate ligands. In the presence of strong ligands such as EDTA, carbonate or citrate anions could play an important role by stabilizing one preferred oxidation state and consequently favor oxido-reduction reactions during partitioning of neptunium and plutonium. Citric acid is a typical ligand for d- and f-elements and the possibility for interaction of the actinide-ions with naturally-occurring acid is almost ubiquitous in biosphere [39]. Among many the alfa- hydroxycarboxy acids, citric acid can coordinate to metal ions as a potential multidentate ligand. It has the ability to act as a versatile ligand, i.e. with three carboxylic acid and one alcoholic group potentially involved in bonding. Very few structural studies of citric acid with actinides have been reported [40]. Indeed, Thuéry has produced an extensive study on complexation of uranyl ion by citric acid [41,42].
For lanthanide cations, crystal structures of this kind are not known. However there are several Ln(III)-citrate structures reported in the literature. Two isostructural compounds, [Ln(HCitr)(H2O)]n (Ln = Nd [43], La [44]), have polymeric structures formed by chains of formula [Ln(HCitr)(H2O)]n. Each Ln atom is coordinated by one water molecule and eight O atoms of five different HCitr ions. Complexes of the same formula, [Ln(HCitr)(H2O)] (Ln = Eu, Tb [45], Sm [46]) have 2D structures with Ln atoms surrounded by eight O atoms of four HCitr ions and one water molecule. Each HCitr ion functions as a μ4-bridging ligand, forming a layered structure. Another series of Ln(III) citrates, [Ln(HCitr)(H2O)2]·H2O (Ln = Gd, Nd [47], La [48]), have 1D topology. In that case, the coordination polyhedron of the Ln cation is monocapped square antiprism. The basic unit is a binuclear entity consisting of two Ln coordination polyhedra sharing one edge. These units are connected by citrate anions. The carboxylate groups of the citrate are coordinated in monodentate, bidentate and a bridging manner. The hydroxyl group is also coordinated to the metal cation. Two water molecules complete the coordination sphere. A trimeric Nd citrate complex [Co(NH3)6]2(NH4)[Nd3(HCit)4(OH)4]·18H2O has been described in the past [49]. According to spectroscopic data, it contains two different types of Nd atoms with CN 8 and 9.
For this study, we have synthesized trimeric Am and Nd citrate complexes of the form [Co(NH3)6]2K[M3(Cit)4(H2O)3]·18H2O with Nd (compound IV) and Am (compound V). Again, both compounds are isostructural as shown by the crystallographic parameters that are displayed in Table 4. The trimeric complex anion [Am3(Cit)4(H2O)3]7− has a C2 symmetry (Fig. 5a). A two-fold axis passes through Am(1) atom and O(1) atom of bridging coordination water molecule. Due to the bridging functions of O atoms of deprotonated OH-groups of the citrate anions and one water molecule, the coordination polyhedra of the three cations differ: dodecahedron for Am(1) (CN = 8), capped square antiprism for Am(2, 2A) (CN = 9). They share three faces as shown in Fig. 5b. The Am-O distances reported in Table 5 vary from 2.373(8) Å for μ3-O atom of citrate anion to 2.609(8) Å for μ2-O atom of bridging water molecule. The average Am-O distances are 2.45(4) and 2.50(2) Å for Am(1) and Am(2), respectively. The corresponding values for the Nd compound are 2.45(2) and 2.49(2) Ǻ, showing again the remarkable similarity between those two cations. The H atoms of coordination water molecules were located from difference Fourier maps (showing absence of OH-groups in the complex anions) and refined with restrained O-H distances and H-O-H angles. The H-atoms of Cit4− anions were placed in geometrically calculated positions. The H atoms of [Co(NH3)6]3+ cations were located and placed in idealized positions, only orientation of NH3 groups was refined. The H atoms of crystallization water molecules were not located. It was not possible to locate all crystallization water molecules, some of them are, probably disordered, but the number of crystallization water molecules per formula unit is at least 18.
Crystal data for compounds IV and V: [Co(NH3)6]2K[Nd3(Cit)4(H2O)3]·18H2O and [Co(NH3)6]2K[Am3(Cit)4(H2O)3]·18H2O.
| Am | Nd | |
| Compound | V | IV |
| Chemical formula | C24H94Am3Co2KN12O49 | C24H94Co2KN12Nd3O49 |
| Formula weight | 2221.07 | 1924.79 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c |
| a, Å | 15.1687 (17) | 15.0898 (2) |
| b, Å | 37.852 (4) | 38.2494 (3) |
| c, Å | 14.1296 (16) | 14.2267 (2) |
| β, deg | 111.017 (2) | 110.9989 (5) |
| V, Z | 7573.0 (15), 4 | 7665.97 (16), 4 |
| R1 | 0.0468 | 0.0531 |
| 0.1454 | 0.1500 | |
| Temperature K | 100 | 120 |
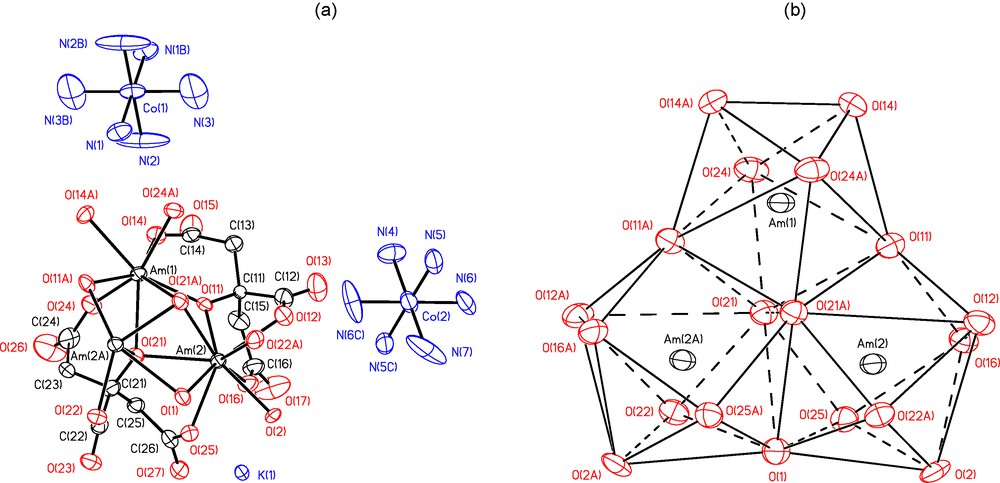
(a) a view of [Co(NH3)6]2K[Am3(Cit)4(H2O)3]·18H2O (V). Displacement ellipsoids are drawn at the 50% probability level, H atoms and coordination water molecules omitted for clarity; (b) coordination polyhedra of Am atoms. Symmetry transformations: A – (1-x, y, 0.5-z); B – (1-x, -y, 1-z); C – (-x, y, 0.5-z).
Selected bond lengths (Å) in the structures of compounds IV and V [Co(NH3)6]2K[M3(Cit)4(H2O)3]·18H2O (M = Nd, Am).
| Bond distances | Am | Nd |
| Compound | V | IV |
| M(1)-O(11) | 2.372 (8) | 2.374 (4) |
| M(1)-O(14) | 2.459 (8) | 2.462 (4) |
| M(1)-O(21) | 2.542 (8) | 2.541 (4) |
| M(1)-O(24) | 2.421 (9) | 2.427 (4) |
| M(1)…M(2) | 3.6902 (7) | 3.7049 (4) |
| M(2)-O(1) | 2.609 (8) | 2.604 (4) |
| M(2)-O(2) | 2.452 (8) | 2.455 (4) |
| M(2)-O(11) | 2.373 (8) | 2.361 (4) |
| M(2)-O(12) | 2.510 (8) | 2.479 (4) |
| M(2)-O(16) | 2.539 (9) | 2.533 (4) |
| M(2)-O(21) | 2.537 (8) | 2.544 (4) |
| M(2)-O(21a) | 2.519 (8) | 2.512 (4) |
| M(2)-O(22a) | 2.465 (8) | 2.468 (4) |
| M(2)-O(25) | 2.482 (9) | 2.467 (4) |
| M(2)…M(2a) | 3.8417 (9) | 3.8449 (6) |
In addition, UV-VIS spectra of polycrystalline IV [Co(NH3)6]2K[Nd3(Cit)4(H2O)3]·nH2O and V [Co(NH3)6]2K[Am3(Cit)4(H2O)3]·18H2O were recorded at ambiant temperature. The spectrum of IV (not shown) exhibits several absorption bands, as reported in Table 6. For comparison, the data on the spectra of Nd3+(aq) and Nd3+ in citric acid solution (pH 13) are also reported. The spectrum of complex IV does not exhibit all the transitions that are characteristic of Nd3+ in solution. One shall note that the coordination of Nd by citrate-ion is accompanied by long-wave shift. The spectrum of V is shown in Fig. 6. One can identify the strong bands of f-f transitions (Table 6): a doublet at 509 and 516 nm.
Band positions in the optical spectra of Nd3+ and Am3+ solid complexes IV and V and related systems.
| System | Wavenumber, nm |
| Complex IV | 462; 583; 743.5; 804.6; 873.7 |
| Nd3+ in citric acid | 464; 515; 526; 581.5; 686; 746; 803; 878.9 [57] |
| Nd3+ (aquo ion) | 461.8; 511; 521.6; 579; 681.9; 738; 795.9; 871.9 [57] |
| Complex V | 458; 509, 516sh, 817.4 |
| Am3+ (aquo ion) | 454; 502.7; 812.2 [58] |

UV-Vis absorption spectrum of the solid compound V K[Co(NH3)6]2[Am3(Citr)4(H2O)3] (∼0.5 mg dispersed in nujol).
6 Conclusion
The relative physicochemical behavior of the actinides versus the lanthanides has always been a matter of intensive research, both as a problematic of fundamental science but also for possible applications in the nuclear fuel cycle. This comparison may be justified by the analogy of their electronic structure, as each of the series is made up of elements corresponding to the filling of a given (n)f atomic shell. Although the chemical properties of the actinides versus the lanthanides differ significantly for the first part of the series, they become much closer after plutonium. This may be explained by a combination of several phenomena including the increasing influence of the nucleus charge, relativistic effects and filling of the 5f electrons.
Nevertheless, relatively few points of comparison between the two families are available, given the lack of published structures for trans-plutonium(III) elements and the additional difficulty to stabilize coordination complexes of uranium(III) to plutonium(III). After a brief review on the occurrence of typical coordination polyhedra of the solid state hydrates and aquo species of actinide(III) and lanthanide(III), the article focuses on two chemical systems that are both characterized by their hard acid character, namely the aminocarboxylic example with nitrilotriacetic acid and the hydroxycarboxylic example with citric acid. Both ligands bear three carboxylic groups plus an additional chemical function that often plays a very important structural role: the nitrogen atom for the first one, the hydroxy group for the second one. Furthermore, both ligands are relevant to the more general problematic of lanthanide – minor actinide selectivity.
Five new complexes of either americium or lanthanide were described: [Co(NH3)6][M(NTA)2(H2O)]·8H2O where M = Nd, Yb and Am, and [Co(NH3)6]2K[M3(Cit)4(H2O)3]·18H2O where Nd and Am. In all of these complexes, but as well in the hydrate series, coordination numbers oscillate from 8 to 9, showing a remarkable stability over the series in comparison with the actinide(IV) compounds (where coordination numbers oscillate from 8 to 12). The coordination polyhedra also show some limited variety, both trigonal tricapped prism and square antiprism being typical representative of coordination numbers 9 and 8 respectively. This is the case for complexes I to III which all exhibit a distorted TTP geometry, in resemblance with most of the hydrate species. In IV and V, a mixing of dodecahedron and square monocapped antiprism form the trimeric structure. The complexes are isostructural between the americium and their lanthanide analogues.
Interestingly, a direct comparison of the M-O(carboxylate) in the NTA and citric molecules shows a slight evolution from Am to Nd for the NTA complexes (2.47 Å for Am and 2.45 Å for Nd) and none for the citrate complexes (2.50 Å for Am(2) and 2.50 Å for Nd(2)). Similarly, the M-N(amine) evolution in the NTA complexes (2.70 Å for Am and 2.72 Å for Nd) is slightly larger than the M-O(hydroxy) evolution in the citrate complexes (2.37 Å for Am, 2.36 Å for Nd). Overall, these differences are small and must be taken cautiously.
This fascinating chemistry shows a remarkable stability in terms of geometry although subtle differences in polyhedron distortion occur between the americium and lanthanide equivalents. These distortions should in turn be interpreted as possible differences of reactivity. Although difficult to quantify in energetic terms, these structural differences are fundamental in order to better understand the trans-plutonium versus lanthanide chemistry.
7 Experimental
All starting materials (except Am(NO3)3nH2O) were purchased from Sigma Aldrich, reagent grade and used without further purification.
7.1 Spectroscopy
IR spectra were recorded at ambient temperature on a Specord M-80 spectrometer controlled by a PC using NaCl discs.
UV-vis-NIR spectra were obtained at ambient temperature on a Shimadzu UVPC 3100 spectrophotometer with 1 mL quartz cells. Solid samples were prepared in the same way as for IR spectroscopy but with larger amounts of actinide compounds. Therefore the characteristic bands were visible but the disc of pressed NaCl remained sufficiently transparent.
7.2 Synthesis
7.2.1 [Co(NH3)6][M(NTA)2(H2O)]·8H2O (M = Nd, Yb), I and II
0.8 mL of 1 M Na2HNTA water solution was added to 0.5 mL of 0.5 M Ln(NO3)3 (Ln = Nd or Yb) solution. After dissolution, 1 mL of 0.25 M [Co(NH3)6]Cl3 was added to the solution. After several days, light orange plates appeared at room temperature. Crystals were filtered off washed with cold water and dried at ambient temperature.
7.2.2 [Co(NH3)6][Am(NTA)2(H2O)]·8H2O, III
All synthesis were performed with 243Am(NO3)3 stock material of the Institute of Physical Chemistry and Electrochemistry. 243Am is a higly radioactive radioelement that has to be handled in dedicated facilities with appropriate equipment for radioactive materials.
The complex was synthesized according to the procedure described in [34] for the gadolinium complex with NTA3−. 1 mL of 10−2 M Am(NO3)3nH2O in 0.01 M HNO3 was evaporated to dryness at temperature around 100–120 °C. The residue was then dissolved in a minimum quantity of water and 2 mL of ∼0.02 M aqueous solution of the disodium salt of nitrilotriacetic acid (H3NTA). After dissolution, 0.2 mL of 0.1 M [Co(NH3)6]Cl3 was added. Storage for 2 days of the solution at about 6–10 °C resulted in crystallisation of light orange plates. Crystals were filtered off washed with cold water and dried at ambient temperature.
7.2.3 [Co(NH3)6]2K[Nd3(Cit)4(H2O)3]·18H2O, IV
1 mL of 10−2 M neodimium nitrate in 0.01 M HNO3 was evaporated to dryness at about 100–120 °C. The residue was dissolved in a minimum quantity of water and 0.3 mL of 0.1 M of citric acid aqueous solution was added. After complete dissolution, 1 M KOH was added dropwise up to pH of about 7–9. Equimolar quantity of [Co(NH3)6]Cl3 0.1 M solution was finally added. After few hours of storage at 6–10 °C, crystals were obtained. Crystals were filtered off washed with cold water and dried at ambient temperature.
7.2.4 [Co(NH3)6]2K[Am3(Cit)4(H2O)3]·18H2O, V
1 mL of 10−2 M Am(NO3)3nH2O in 0.01 M HNO3 was evaporated to dryness at 100–120 °C. The residue was dissolved in a minimum quantity of water and 0.3 mL of 0.1 M of citric acid aqueous solution was added. After complete dissolution, 1 M KOH was added dropwise up to pH of about 7–9. Equimolar quantity of [Co(NH3)6]Cl3 0.1 M solution was finally added. In few hours of storage at about 6–10 °C, orange plate crystals were obtained. Crystals were filtered off washed with cold water and dried at ambient temperature.
The infrared spectrum of complex V has been registered and found to be identical of that of complex IV. The high frequency 4000–2000 cm−1 region contains some intense overlapping absorption bands corresponding to the valence vibrations ν(HOH) of coordination water molecules (3504, 3456, 3442 cm−1), vibrations of amine groups of outer sphere cations [Co(NH3)6]3+ (3290, 3250, 3056 cm−1) and vibrations of (C-H) methylene groups of citrate-ion (2968, 2926, 2856 cm−1). The frequency interval 1700–1500 cm−1 is characteristic of the deformation vibration modes δ(H2O) of the coordinated water molecules and degenerate vibration modes of the amine groups δ(NH3) (1680, 1644, 1620 cm−1) [50]. Moreover, the frequencies of antisymmetric vibrations of the carboxylic groups νas(COO−) (1588, 1574, 1556 cm−1) are significantly decreased in comparison with the free citric ligand [51]. In the 1500–500 cm−1 region, the spectrum exhibits some narrow well resolved absorption bands which may tentatively be attributed to the symmetric vibrations of the carboxylic groups νs(COO−) (1404, 1392, 1360sh cm−1), vibration of the carboxy group ν(CO) (1080, 1048 cm−1) and finaly ρ(NH3) (760 cm−1).
7.3 X-ray single crystal diffraction
The X-ray diffraction experiment for Am compound was carried out on a Bruker KAPPA APEX II autodiffractometer (MoKα radiation, graphite monochromator). The crystal was sealed in glass capillary. First 20 frames were remeasured at the end of the experiment to check for possible self-radiolysis. After 19 hours of measurements, average loss of diffraction intensities was less than 2%. Data reduction was made using SAINT-Plus program [52]. Absorption correction was made using SADABS program [53]. The X-ray diffraction experiments for Nd and Yb compounds were carried out on a Nonius KappaCCD diffractometer (Mo–Kα radiation, graphite monochromator). The data were processed with HKL program [54]. The experimental intensities were corrected for absorption using the MULABS procedure in the PLATON suite [55]. The structures were solved by direct method (SHELXS97) and refined on F2 with the full-matrix least-squares procedure (SHELXL97 [56]) using all reflections.
CCDC-766051-766055 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request.cif.