

1 Introduction
The incorporation of main group elements into the π-conjugated framework is a powerful strategy for the design of new π-electron materials with unusual electronic structures [1]. Among various main group elements, the group 15 phosphorus is particularly interesting, because phosphanyl groups can be readily modified by oxidation to phosphine oxides or sulfides, formation of phosphonium salts, and complexation with Lewis acids or transition metals. This diversity in the chemical modification enables us to tune the electronic and structural properties over a wide range, and thus, has attracted many researchers to develop a variety of phosphorus-containing π-electron materials [2].
Among the various phosphorus-containing π-conjugated skeletons, phosphole, a phosphorus analogue of pyrrole, is of particular interest, because it is largely different from pyrrole in terms of the geometry, electronic structure, and thus properties, and thereby serves as a unique building unit [3]. Various types of phosphole-containing π-conjugated compounds have been investigated to date, including 2,5-difunctionalized phospholes [4,5], phosphole-based polymers [6], phosphole-containing macrocycles [7], phosphole-cored dendrimers [8], and fused phosphole π systems, such as dibenzophospholes [9,10], dithienophospholes [11], and benzophospholes [12–15].
As a new entity for the fused phosphole derivatives, we have recently succeeded in the synthesis of dibenzo-fused phospholo[3,2-b]phosphole dioxides 1 (R = Ph). The most striking feature of this skeleton is the incorporation of two phosphorus atoms into the skeleton and thereby the accumulative effect of the two phosphoryl groups (> PO) makes the skeleton highly electron-accepting. As a consequence, 1 demonstrated unusual fluorescence properties totally different from the already known bridged stilbenes [16] (Fig. 1).
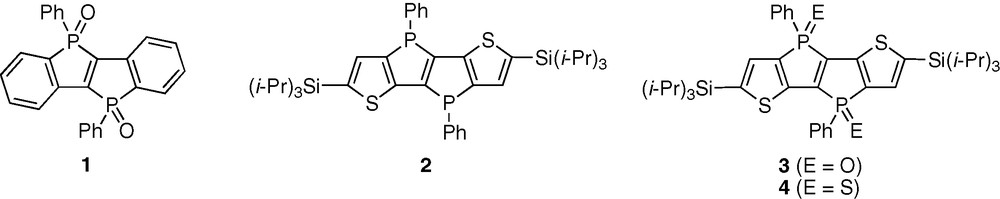
Phospholo[3,2-b]phosphole derivatives.
To elaborate this highly electron-accepting phospholo[3,2-b]phosphole dioxide skeleton, we now replace the stilbene skeleton with a more electron-donating dithienylvinylene skeleton. We envisioned that this structural modification would lead to a narrower HOMO–LUMO gap, and thus, more interesting properties such as absorption and fluorescence at longer wavelengths. Facile functionalization of the thiophene rings would also increase the potential utility of this skeleton as a core for extended π-conjugated materials. This article discloses the syntheses and photophysical properties of a series of thiophene-fused phospholo[3,2-b]phosphole derivatives, including non-oxidized 2, dioxides 3, and disulfides 4. In particular, the effects of the fused rings as well as the oxidation states of the phosphorus atoms on the fluorescence properties are discussed.
2 Results and discussion
2.1 Synthesis
The thiophene-fused phospholophospholes were prepared by a modified procedure of our recently reported PCl3-promoted nucleophilic cascade cyclization, as shown in Scheme 1 [16]. Thus, starting from bis(3-bromo-2-thienyl)acetylene 5, dilithiation using t-BuLi in THF followed by the reaction with PhP(NEt2)Cl produced the corresponding aminophosphanyl intermediate 6 in situ. After replacement of the solvent THF by dioxane, the reaction mixture was treated with an excess amount of PCl3 and refluxed for 18 h in order to accomplish the cyclization. Finally, the produced thiophene-fused phospholophosphole 2 was oxidized with mCPBA to give the corresponding phospholophosphole dioxide 3 as a mixture of cis and trans isomers. These isomers could be separated by silica gel column chromatography and were obtained in 18 and 42% yields for cis-3 and trans-3, respectively. Similarly, the corresponding phospholophosphole disulfide 4 was synthesized by using S8 as an oxidant instead of mCPBA and obtained in 31 and 42% yields for cis-4 and trans-4, respectively. Their structures were verified by NMR spectroscopy and mass spectrometry and finally by X-ray crystallographic analyses (Fig. 2). A noteworthy fact in the reaction is that the cyclization requires a heating condition at high temperature (101 °C), while the cyclization for the synthesis of the benzene analogue 1 spontaneously proceeds even at room temperature [16]. This difference may be ascribed to a more severe ring strain in the fused system consisting only of the five-membered rings and/or the electronic effect of the thiophene rings on the nucleophilicity and electrophilicity of the phosphorus centers (Figs. 3 and 4).
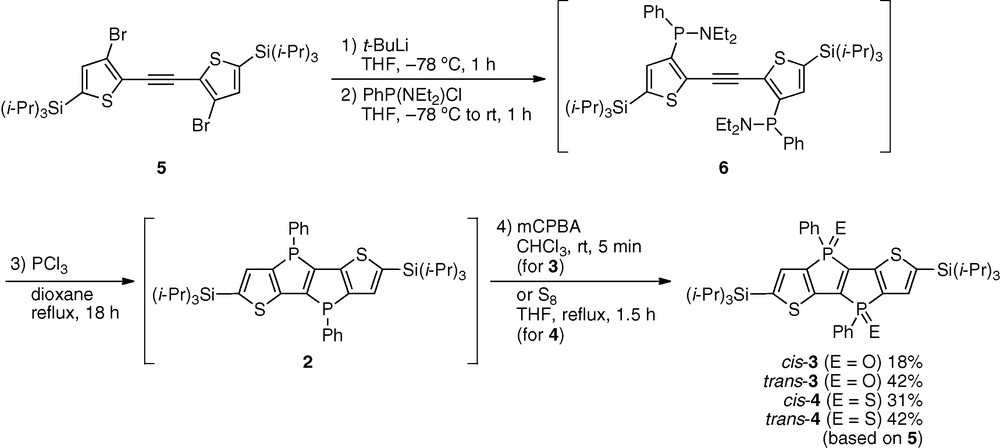
Synthesis of dithiophene-fused phospholo[3,2-b]phosphole derivatives.

ORTEP drawings of (a) cis-3, (b) trans-3, and (c) cis-4 (50% probability for thermal ellipsoids). Hydrogen atoms and solvent molecules are omitted for clarity.

UV-vis absorption spectra of 2 (solid line, in CHCl3), trans-3 (broken dotted line, in CH2Cl2) and trans-4 (dotted line, in CH2Cl2).
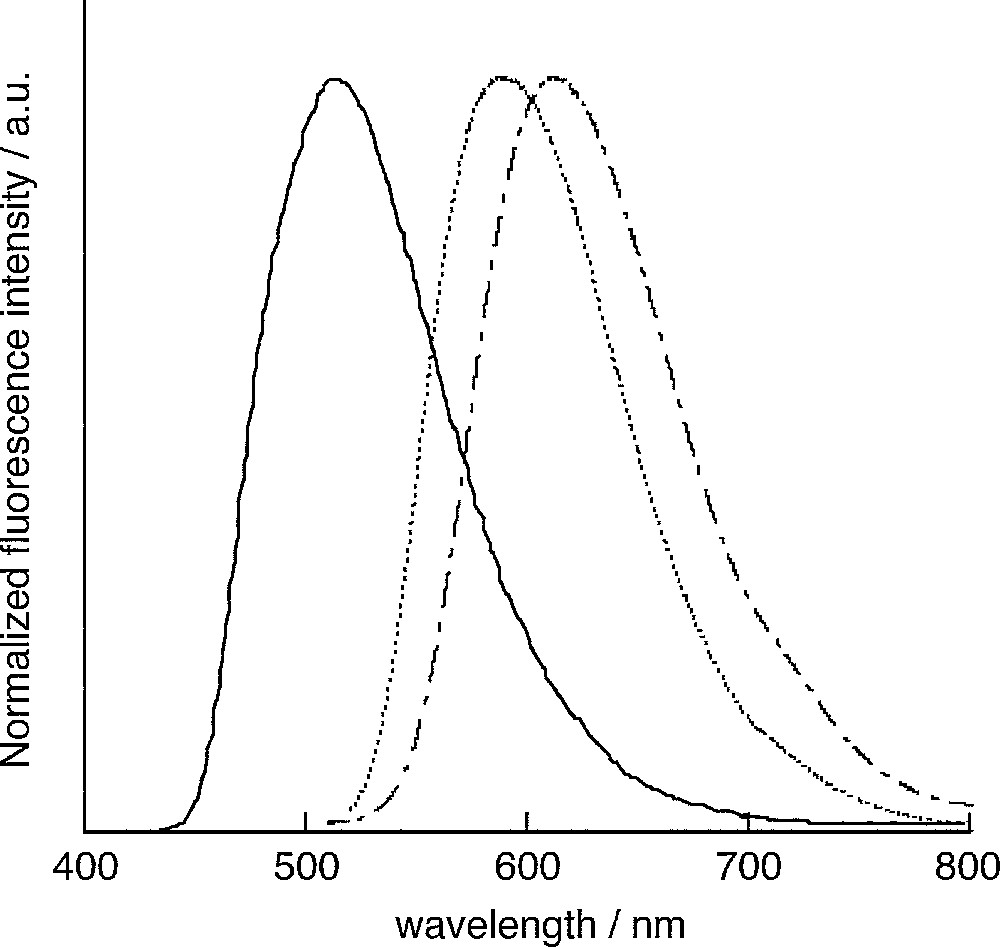
Fluorescence Spectra of 2 (solid line, in CHCl3), trans-3 (broken dotted line, in CH2Cl2) and trans-4 (dotted line, in CH2Cl2).
Although we tried to isolate the non-oxidized phospholophosphole 2 directly from the reaction mixture after the cyclization, all attempts failed due to the contamination of a small amount of the dioxides 3. Therefore, we conducted the reduction of the isolated pure trans-3 with HSiCl3, as shown in Scheme 2, and finally succeeded in obtaining 2 in 54% yield in a pure form. The 1H NMR measurement revealed that this was a mixture of cis and trans isomers in a cis/trans ratio of 1/1.7, despite starting from the pure trans-3. The obtained compound was used for comparisons in the photophyscial properties with compounds 3 and 4, without further separation of the cis and trans isomers.
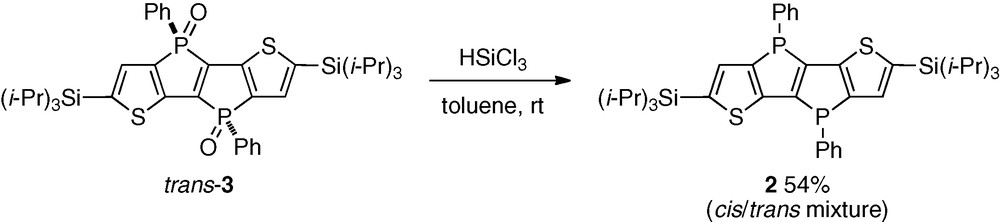
HSiCl3 reduction of phospholo[3,2-b]phosphole dioxide.
2.2 Photophysical properties
Photophysical properties of the thiophene-fused phospholophosphole derivatives 2–4 were investigated, and these data were compared with those of the dibenzo-fused phospholophosphole 7 (1/1.1 cis/trans mixture), its oxides 1, and sulfides 8. Their data are summarized in Table 1. Whereas all the derivatives have cis and trans isomers, their photophysical properties in solutions are essentially comparable. A thiophene-fused phospholophosphole 2 (1/1.7 cis/trans mixture) has an absorption maximum (λabs) at 420 nm and exhibits an intense yellowish green fluorescence with the maximum (λem) at 516 nm.1 On the other hand, by oxidizing the phosphorus atoms to the dioxides trans-3, the λabs and λem are shifted to longer wavelengths by 72–73 nm (3500 cm−1) and 94–95 nm (3000 cm−1), respectively, and thus compounds 3 show a red fluorescence. Similar trends were observed upon oxidation to phosphine sulfides 4, where the λabs and λem are shifted to longer wavelengths by 65–68 nm (3200–3300 cm−1) and 71–76 nm (2400–2500 cm−1), respectively. Whereas the red-shifts upon oxidation of the phosphorus center were also observed for other known phosphorus-containing π-electron systems, the extent of the red-shifts observed for the dithieno-fused phospholophospholes are much larger than those for compounds consisting of a mono-phosphole ring like 2,5-dithienylphospholes [4] and dithienophospholes [11].
The effect of the fused rings on the photophysical properties was investigated next. All the thiophene-fused phospholophosphole derivatives 2, 3, and 4 exhibited longer absorption and fluorescence maxima compared with their dibenzo-fused counterparts 7, 1, and 8, respectively, reflecting the narrower HOMO–LUMO energy gap in the thiophene-fused system. The most noteworthy fact is that in the thiophene series, the non-oxidized compound 2 shows a very high quantum yield of 0.95, although their oxidized derivatives show only faint fluorescence with a ΦF of 0.04 and 0.21–0.33 for 3 and 4, respectively. This trend is totally opposite to that for the benzene-fused analogues as well as other phosphorus-containing π-conjugated compounds. In general, while trivalent phosphanes are weakly fluorescent due to the quenching effect by lone-pair electrons, their fluorescence quantum yields significantly increase upon oxidation [17]. To the best of our knowledge, the only exceptions are Baumgartner's dithienophosphole derivatives, which exhibit intense fluorescence regardless of the oxidation states of the phosphorus atoms [11].
To obtain deeper insights into the origin of these differences in ΦF between the thiophene series and the benzene series, we investigated the excited-state dynamics. According to the equations ΦF = kr × τs and , the radiative (kr) and nonradiative (knr) decay rate constants were determined (Table 1). In both of the series, the oxidation of the phosphorus atoms to the phosphine oxides or sulfides results in a decrease in the kr value. This is consistent with the fact that the oxidized compounds have longer absorption wavelengths and smaller molar absorption coefficients ɛ compared to the non-oxidized derivatives 2 and 7. This different tendency is that, in the thiophene-fused series, the non-oxidized phosphine 2 has a significantly small knr value of 6.7 × 106 s−1, while the phosphine oxide 1 has a small knr value in the benzene series. Obviously, these retarded nonradiative decay processes for 2 and 1 are responsible for their high fluorescence quantum yields. These facts demonstrate that the fluorescence efficiency highly depends not on the oxidation state of the phosphorus atom but on the π-conjugated skeleton. Because both series of compounds have rigid and flat π-conjugated skeletons, their rate constants for the internal conversion may not be significantly different. Instead, the rate constant of the intersystem crossing process may be dependent on the π-conjugated skeletons.
2.3 Theoretical calculations
To elucidate the effect of the fused rings and the oxidation states of the phosphorus atoms on the electronic structures, we performed the TD-DFT calculations on model compounds trans-2′, trans-3′, and trans-4′, where trimethylsilyl groups are used in place of triisopropylsilyl, at the B3LYP/6-31G(d) level of theory. The calculations of the benzene-fused trans-1 were also conducted for a comparison. As shown in Fig. 5, the oxidation of the thiophene-fused phospholophosphole 2′ to the phosphine oxide 3′ results in a significant decrease in the LUMO level (trans-2′: −1.73 eV, trans-3′: −2.55 eV), which is much greater than the decrease in the HOMO level (trans-2′: −5.01 eV, trans-3′: −5.48 eV). The oxidation to the phosphine sulfide 4′ also leads to a decrease in the LUMO level (−2.59 eV for trans-4′), the extent of which is almost comparable to that for the phosphine oxide. In a comparison between the thiophene-fused trans-3′ and the benzene-fused trans-1, the thiophene derivative has a much higher HOMO level as well as a slightly lower LUMO level. This result demonstrates that the high electron-donating character of the dithienylvinylene skeleton compared to the stilbene skeleton is mainly responsible for the lower transition energies for the thiophene derivatives. In addition, the thiophene derivative 3′ has a larger oscillator strength compared to that of 1 (trans-1: 0.1508, trans-3′: 0.3121). The thiophene derivative 3′ may have a more significant intramolecular charge transfer character in the excited state that leads to the stronger oscillator strength.

Energy diagrams and pictorial representations of the frontier MOs for thiophene-fused phospholo[3,2-b]phospholes trans-2′, trans-3′, and trans-4′, together with those of benzene-fused phospholo[3,2-b]phosphole dioxide trans-1, calculated at the B3LYP/6-31G(d) level. Their lowest-energy transitions were estimated by TD-DFT calculations at the same level.
3 Conclusion
We have synthesized a series of thiophene-fused phospholo[3,2-b]phosphole derivatives as new fused phosphole derivatives on the basis of the PCl3-induced intramolecular cascade cyclization. The obtained thiophene-fused phospholophosphole derivatives exhibit significantly red-shifted absorptions and fluorescences compared with their benzene-fused analogues, mainly due to the highly electron-donating character of the dithienylvinylene skeleton. The study on the structure–photophysical properties relationship demonstrated significantly different trends between the thiophene series and the benzene series. Although the thiophene-fused phospholophosphole dioxide 3 and disulfide 4 show only faint fluorescences, the non-oxidized derivative 2 exhibits an intense fluorescence. This is in contrast to the fact that a dioxidized derivative shows the highest quantum yield among the benzene-fused derivatives. These facts demonstrate that the relationship between the fluorescence efficiency and the oxidation states of the phosphorus atoms is highly influenced by the character of the π-conjugated skeletons.
4 Experimental
General. Melting points (mp) were determined with a Yanaco MP-S3 or a Stanford Research System OptiMelt MPA100 instrument (MPA100). 1H and 13C NMR spectra were recorded with a JEOL AL-400 spectrometer (400 MHz for 1H and 100 MHz for 13C) in CDCl3 and chemical shifts are reported in δ ppm using CHCl3 for 1H (7.26 ppm) and 13C (77.16 ppm) for CDCl3 as an internal standard. 31P NMR spectra were recorded with a JEOL AL-400 spectrometer (162 MHz) using H3PO4 (0.0 ppm) as an external standard. Mass spectra were measured with a JEOL JMS 700 (FAB) or Bruker micrOTOF Focus (APCI). Thin layer chromatography (TLC) was performed on plates coated with a 0.25-nm thickness of Silica Gel 60 F254 (Merck). Column chromatography was performed using silica gel PSQ100B (Fuji Silysia Chemical). Recycle preparative gel permeation chromatography (GPC) was performed using LC-918 with polystyrene gel columns (JAIGEL 1H and 2H, Japan Analytical Industry) with chloroform as an eluent. Anhydrous Et2O, THF, and toluene were purchased from Kanto Chemicals. PhP(NEt2)Cl [18] and bis(3-bromo-5-triisopropylsilyl-2-thienyl)acetylene 5 [19] were synthesized according to the literature method. All reactions were performed under an argon atmosphere, unless stated otherwise.
Thiophene-fused phospholo[3,2-b]phosphole dioxide 3. To a solution of bis(3-bromo-5-triisopropylsilyl-2-thienyl)acetylene 5 (0.50 g, 0.76 mmol) in anhydrous THF (12.0 mL) was added t-BuLi in n-pentane (1.57 M, 2.00 mL, 3.14 mmol) dropwise over 10 min at −78 °C, and the mixture was stirred for 1 h. PhP(NEt2)Cl (0.29 mL, 326 mg, 3.14 mmol) was added to the mixture dropwise over 5 min at the same temperature. After stirring for 10 min, the reaction mixture was allowed to warm to room temperature and stirred for 1 h. All volatiles were then removed under reduced pressure, and the residual mixture was dissolved in anhydrous dioxane (12.0 mL). PCl3 (0.40 mL, 628 mg, 4.57 mmol) was added to the mixture in one portion at room temperature, and the reaction mixture was stirred at reflux temperature for 18 h. After concentration of the reaction mixture under reduced pressure, degassed anhydrous toluene (72 mL) was added. The resulting precipitate was removed by filtration under an argon atmosphere and the filtrate was concentrated under reduced pressure to give 765 mg of crude 2 as brown waxy solids. Without further purification, this crude 2 was dissolved in CHCl3 (12.0 mL), and mCPBA (341 mg, 1.52 mmol) was added at room temperature, resulting in an immediate color change of the reaction mixture from dark brown to bright red. After stirring for 1 h, the mixture was washed with 10% Na2SO3 aq., and the aqueous layer was extracted with CHCl3 for three times. The combined organic layer was washed with brine, dried over Na2SO4, and filtered. After concentration of the filtrate under reduced pressure, the resulting crude products were subjected to a silica gel column chromatography (CHCl3 as eluent) to give almost pure trans-3 (Rf = 0.40) and cis-3 (Rf = 0.06) both as red solids. These products were thoroughly purified by a preparative GPC to afford spectroscopically pure trans-3 (240 mg, 0.32 mmol, 42% yield) and cis-3 (106 mg, 0.14 mmol, 18% yield) both as red solids.
cis-3. mp: 208.0–209.0 °C. 1H NMR (400 MHz, CDCl3) δ 1.06 (d, J = 6.8 Hz, 36H), 1.29 (sept, J = 6.8 Hz, 6H), 7.34 (t, J = 1.5 Hz, 2H), 7.45 (t, J = 7.3 Hz, 4H), 7.58 (t, J = 7.3 Hz, 2H), 7.80 (dd, J = 7.3, 1.5 Hz, 4H). 13C {1H} NMR (100 MHz, CDCl3) δ 11.8 (s), 18.6 (s), 128.5 (dd, JCP = 105, 6.6 Hz), 129.4 (t, JCP = 6.6 Hz), 130.8 (t, JCP = 5.8 Hz), 133.2 (t, J = 7.8 Hz), 137.3 (dd, JCP = 122, 2.5 Hz), 143.7 (d, JCP = 5.7 Hz), 144.4 (d, JCP = 119 Hz), 151.2 (pseudo t, JCP = 20 Hz). 31P {1H} NMR (162 MHz, CDCl3) δ 22.1. HRMS (FAB) m/z Calcd. for C40H55O2P2S2Si2: 749.2657. Found: 749.2659 ([M + H]+).
trans-3. mp > 300 °C. 1H NMR (CDCl3) δ 1.05 (dd, J = 7.6 Hz, 36H), 1.29 (sept, J = 7.2 Hz, 6H), 7.33 (t, J = 1.4 Hz, 2H), 7.50 (t, J = 6.8 Hz, 4H), 7.61 (t, J = 7.2 Hz, 2H), 7.80 (dd, J = 6.8, 1.4 Hz, 4H). 13C {1H} NMR (CDCl3) δ 11.6 (s), 18.6 (s), 127.7 (dd, JCP = 113, 6.6 Hz), 129.5 (t, JCP = 6.2 Hz), 131.1 (t, JCP = 5.7 Hz), 133.5 (s), 137.2 (dd, JCP = 122, 1.7 Hz), 143.7 (t, JCP = 5.8 Hz), 144.1 (dd, JCP = 107, 1.7 Hz), 151.2 (pseudo t, JCP = 20 Hz). 31P {1H} NMR (CDCl3) δ 23.2. HRMS (FAB) m/z Calcd. for C40H55O2P2S2Si2: 749.2657. Found: 749.2674 ([M + H]+).
Thiophene-fused phospholo[3,2-b]phosphole disulfide 4. To a solution of bis(3-bromo-5-triisopropylsilyl-2-thienyl)acetylene 5 (200 mg, 0.30 mmol) in anhydrous THF (5.0 mL) was added t-BuLi in n-pentane (1.59 M, 0.80 mL, 1.25 mmol) dropwise over 10 min at –78 °C, and the mixture was stirred for 2.5 h. PhP(NEt2)Cl (0.12 mL, 139 mg, 0.645 mmol) was then added to the mixture dropwise over 5 min at the same temperature. After stirring for 10 min, the reaction mixture was allowed to warm to room temperature and stirred for 1.5 h. All volatiles were removed under reduced pressure, and the residual mixture was dissolved in anhydrous dioxane (5 mL). PCl3 (0.27 mL, 416 mg, 3.03 mmol) was then added in one portion at room temperature, and the resulting reaction mixture was stirred at reflux temperature for 18 h. After concentration of the reaction mixture under reduced pressure, degassed anhydrous THF (20 mL) and S8 (933 mg, 3.64 mmol) were added. After stirring at reflux temperature for 1.5 h, the reaction mixture was allowed to cool to room temperature. The resulting precipitates were removed by filtration, the filtrate was washed with water, and the aqueous layer was then extracted with CHCl3 three times. The combined organic layer was washed with brine, dried over Na2SO4, and filtered. After concentration of the filtrate under reduced pressure, the resulting crude products were subjected to a silica gel column chromatography (2/1 hexane/CHCl3) to give 99 mg (0.13 mmol, 43% yield) of trans-4 (Rf = 0.58) and 73 mg (0.093 mmol, 31% yield) of cis-4 (Rf = 0.38) as orange solids, respectively.
cis-4. mp 126.8–127.7 °C. 1H NMR (CDCl3) δ 1.08 (dd, J = 7.6, 2.0 Hz, 36H), 1.31 (sept, J = 7.6 Hz, 6H), 7.32 (t, J = 2.0 Hz, 2H), 7.42 (pseudo t, J = 6.8 Hz, 4H), 7.54 (t, J = 7.2 Hz, 2H), 7.69–7.74 (m, 4H). 13C{1H} NMR (CDCl3) δ 11.9 (s, CH), 18.7 (s, CH3), 128.8 (dd, JCP = 89, 6.6 Hz, C), 129.3 (pseudo t, JCP = 6.6 Hz, CH), 130.7 (pseudo t, JCP = 6.2 Hz, CH), 132.8 (s, CH), 133.3 (pseudo t, JCP = 7.9 Hz, CH), 141.1 (d, JCP = 102 Hz, C), 144.1 (pseudo t, JCP = 5.8 Hz, C), 145.3 (d, JCP = 88 Hz, C), 149.5 (pseudo t, JCP = 19 Hz, C). 31P{1H} NMR (CDCl3) δ 29.3. HRMS (APCI) m/z Calcd. for C40H55P2S4Si2: 781.2195. Found: 781.2224 ([M + H]+).
trans-4. mp 254.3–254.8 °C. 1H NMR (CDCl3) δ 1.06 (d, J = 7.6 Hz, 18H), 1.09 (d, J = 7.6 Hz, 18H), 1.31 (sept, J = 7.6 Hz, 6H), 7.32 (t, J = 2.0 Hz, 2H), 7.47 (pseudo t, J = 6.4 Hz, 4H), 7.55 (t, J = 6.4 Hz, 2H), 7.81–7.87 (m, 4H). 13C{1H} NMR (CDCl3) δ 11.9 (s, CH), 18.66 (s, CH3), 18.70 (s, CH3), 127.9 (dd, JCP = 100, 8.7 Hz, C), 129.4 (pseudo t, JCP = 6.6 Hz, CH), 131.0 (pseudo t, JCP = 6.2 Hz, CH), 133.0 (s, CH), 133.2 (pseudo t, JCP = 7.8 Hz, CH), 141.2 (d, JCP = 101 Hz, C), 144.1 (pseudo t, JCP = 5.4 Hz, C), 145.1 (d, JCP = 88 Hz, C), 149.4 (pseudo t, JCP = 19 Hz, C). 31P{1H} NMR (CDCl3) δ 29.9. HRMS (APCI) m/z Calcd. for C40H55P2S4Si2: 781.2195. Found: 781.2217 ([M + H]+).
Thiophene-fused phospholo[3,2-b]phosphole 2. To a solution of trans-3 (30.8 mg, 0.041 mmol) in degassed anhydrous toluene (6.0 mL) was added HSiCl3 (41 μL, 55 mg, 0.406 mmol) in one portion at room temperature, and the mixture was stirred for 5 min, resulting in an immediate color change from red to yellow green. After removal of volatiles under reduced pressure, 26 mg of crude 2 was obtained as yellow solids. After reprecipitation using degassed hexane at 4 °C, 15.8 mg (0.022 mmol) of 2 was obtained in 54% yield as yellow solids. According to the integral ratio of the signals in 31P NMR measurement, the ratio of the cis and trans isomers was determined to be 1:1.7: mp 261–262 °C. 1H NMR (CDCl3) δ 1.08–1.33 (m, 36H), 1.27–1.37 (m, 6H), 7.30–7.31 (m, 8H), 7.43–7.47 (m, 4H). 31P{1H} NMR (CDCl3) δ –18.5, –16.2. HRMS (APCI) m/z Calcd. for C40H55P2S2Si2: 717.2753. Found: 717.2772 ([M + H]+).
Benzene-fused phospholo[3,2-b]phosphole disulfide 8. A solution of trans-1 (306 mg, 0.72 mmol) and Lawesson reagent (295 mg, 0.73 mmol) in anhydrous toluene (14 mL) was stirred at reflux temperature for 4 h. All volatiles were removed under reduced pressure, and the residual mixture was subjected to silica gel column chromatography (1/2 hexane/CH2Cl2, Rf = 0.55) to give 332 mg of crude mixture of trans-8 containing a considerable amount of impurities. This mixture was thoroughly purified by recrystallization from a hexane/CHCl3 mixed solvent to give pure trans-8 as yellow crystals (93 mg, 0.20 mmol, 28% yield): mp > 300 °C. 1H NMR (CDCl3) δ 7.37–7.42 (m, 2H), 7.44–7.48 (m, 6H), 7.54 (d, J = 7.6 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.66 (dd, J = 12, 7.6 Hz, 2H), 7.87–7.92 (m, 4H). 13C{1H} NMR (CDCl3) δ 124.5 (t, JCP = 4.5 Hz, CH), 125.2 (d, JCP = 92 Hz, C), 127.2 (d, JCP = 12 Hz, CH), 127.9 (d, JCP = 40 Hz, CH), 129.4 (d, JCP = 13, 6.2 Hz, CH), 130.2 (t, JCP = 6.6 Hz, CH), 131.0 (t, JCP = 6.2 Hz, CH), 133.0 (s, CH), 136.9 (t, JCP = 16 Hz, C), 139.9 (dd, JCP = 106, 4.5 Hz, C), 149.2 (d, JCP = 87 Hz, C). 31P{1H} NMR (CDCl3) δ 37.6. HRMS (APCI) m/z Calcd. for C26H19P2S2: 457.0398. Found: 457.0407 ([M + H]+).
X-ray crystallographic analysis of cis-3. Single crystals of cis-3 suitable for X-ray crystallographic analysis were obtained by slow diffusion of hexane into a solution of cis-3 in chloroform. Intensity data were collected at 123 K with Mo Kα radiation (λ = 0.71070 Å) and a graphite monochromator. A total of 15,120 reflections was measured at a maximum 2θ angle of 50.0°, of which 7713 were independent reflections (Rint = 0.0513). The structure was solved by a direct method (SIR97) [20] and refined by the full-matrix least squares on F2 (SHELXL-97) [21]. All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were placed using AFIX instructions. The crystal data are as follows: Formula C41H55Cl3O2P2S2Si2, FW = 868.50, crystal size 0.16 mm × 0.16 mm × 0.02 mm, triclinic, P-1 (#2), a = 8.512(3) Å, b = 16.002(6) Å, c = 16.865(6) Å, α = 83.469(12)°, β = 79.632(11)°, γ = 84.845(12)°, V = 2239.4(14) Å3, Z = 2, Dcalcd = 1.288 g cm−3, μ = 0.456 mm−1, R1 = 0.0728 (I > 2σ(I)), (all data), GOF = 1.141.
X-ray crystallographic analysis of trans-3. Single crystals of trans-3 suitable for X-ray crystallographic analysis were obtained by slow diffusion of hexane into a solution of trans-3 in chloroform. Intensity data were collected at 123 K with Mo Kα radiation (λ = 0.71070 Å) and graphite monochromator. A total of 13,745 reflections was measured at a maximum 2θ angle of 50.0°, of which 3763 were independent reflections (Rint = 0.0593). The structure was solved by a direct method (SIR97) [20] and refined by the full-matrix least squares on F2 (SHELXL-97) [21]. All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were placed using AFIX instructions. The crystal data are as follows: Formula C40H54O2P2S2Si2, FW = 749.07, crystal size 0.20 mm × 0.10 mm × 0.05 mm, monoclinic, P21/a (#14), a = 16.26(5) Å, b = 8.03(2) Å, c = 16.33(9) Å, β = 91.44(4)°, V = 2132(14) Å3, Z = 2, Dcalcd = 1.167 g cm−3, μ = 0.287 mm−1, R1 = 0.0784 (I > 2σ(I)), (all data), GOF = 1.178.
X-ray crystallographic analysis of cis-4. Single crystals of cis-4 suitable for X-ray crystallographic analysis were obtained by slow diffusion of hexane into a solution of cis-4 in dichloromethane. Intensity data were collected at 123 K with Mo Kα radiation (λ = 0.71070 Å) and graphite monochromator. A total of 18,960 reflections was measured at a maximum 2θ angle of 55.0°, of which 5498 were independent reflections (Rint = 0.0518). The structure was solved by a direct method (SIR97) [20] and refined by the full-matrix least squares on F2 (SHELXL-97) [21]. All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were placed using AFIX instructions. The crystal data are as follows: Formula C42H58Cl4P2S4Si2, FW = 951.04, crystal size 0.14 mm × 0.07 mm × 0.02 mm, monoclinic, C2/c (#15), a = 29.333(5) Å, b = 8.9161(14) Å, c = 18.380(3) Å, β = 91.6565(8)°, V = 4805.1(14) Å3, Z = 4, Dcalcd = 1.315 g cm−3, μ = 0.566 mm−1, R1 = 0.0616 (I > 2σ(I)), (all data), GOF = 1.108.
Photophysical Properties Measurements. UV/Vis absorption spectra were recorded on a Shimadzu UV-3510 spectrometer with a resolution of 0.5 nm. Emission spectra of 1–3 were measured with a Hitachi F-4500 spectrometer with a resolution of 1 nm. Dilute solutions in degassed spectral grade solvents in a 1-cm square quartz cell were used for the absorption and fluorescence measurements. Absolute fluorescence quantum yields were determined with a calibrated integrating sphere system C9920-02 (Hamamatsu Photonics). Fluorescence lifetimes were measured on a Picosecond Fluorescence Lifetime Measurement System C4780 (Hamamatsu Photonics) equipped with a PLP laser (377 nm for cis- and trans-4, and 405 nm for 2) or a dye laser (coumarin 307 in ethanol, for cis- and trans-2). All solvents (purchased from Nakarai Tesque) were degassed by Ar bubbling for 30 min before preparation of the sample solution.
Computational Method. The geometry optimizations of model compounds 1, trans-2′, trans-3′, and trans-4′ were performed using the Gaussian 03 program [22] at the B3LYP/6-31G(d) level of theory. The lowest energy transitions of 1, trans-2′, trans-3′, and trans-4′ were estimated by TD-DFT calculations at the B3LYP/6-31G(d)//B3LYP/6-31G(d) level of theory.
Acknowledgements
This work was partially supported by Grant-in-Aid (Nos. 17069011, 19675001, and 20750029) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Appendix A Supplementary data
Supplementary information for this article is available with the electronic version, or from the author. CCDC-764165, 764167, and 764166 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.uk/datarequest.cis/Fachinformationzentrum Karisruhe, D-76344 Eggenstein-Leopoldshafen, Germany, on quoting the depository numbers CSD-764165, 764167, and 764166, for cis-3, trans-3, and cis-4, respectively.
1 TD-DFT calculations for compounds 2, 3, and 4 demonstrated that the transition energies for the lowest-energy excited states are nearly identical to each other between the cis and trans isomers. Such an identity between geometrical isomers were experimentally and theoretically also confirmed in the benzene-fused phospholo[3,2-b]phospholes [16].
| Compound | Absorption | Fluorescence | |||||
| λabs/nma | ɛ/104 M−1 cm−1 | λem/nmb | ΦFLc | τs/nsd | kr/s−1 | knr/s−1 | |
| 2e,g | 420 | 1.67 | 516 | 0.95 | 7.5 | 1.3 × 108 | 6.7 × 106 |
| cis-3f | 492 | 1.13 | 610 | 0.04 | 1.0 | 4.0 × 107 | 9.6 × 108 |
| trans-3f | 493 | 1.12 | 611 | 0.04 | 1.0 | 4.0 × 107 | 9.6 × 108 |
| cis-4f | 488 | 1.07 | 592 | 0.21 | 5.2 | 4.0 × 107 | 1.5 × 108 |
| trans-4f | 485 | 1.11 | 587 | 0.33 | 6.5 | 5.1 × 107 | 1.0 × 108 |
| 7f,h | 351 | 1.27 | 415 | 0.07 | 1.4 | 5.0 × 107 | 6.6 × 108 |
| trans-1f | 395 | 0.69 | 480 | 0.98 | 15.7 | 6.2 × 107 | 1.3 × 106 |
| trans-8f | 395(sh) | 0.51 | –i | –i | –i | –i | –i |
a Only the longest absorption maxima are shown.
b Emission maxima upon excitation at the absorption maximum wavelengths.
c Absolute fluorescence quantum yields determined by a calibrated integrating sphere system within ± 3% errors.
d Fluorescence lifetimes within ± 0.5 ns errors.
e In CHCl3.
f In CH2Cl2.
g A mixture of 1/1.7 cis/trans isomers.
h A mixture of 1/1.1 cis/trans isomers.
i No fluorescence was observed. .